
Regulation of G Protein-Coupled Receptors by Ubiquitination

Abstract
:1. Introduction
1.1. GPCR Signaling
1.2. Ubiquitination
2. Functional Role of GPCR Ubiquitination
2.1. Regulation of GPCR Cell-Surface Expression by Ubiquitination
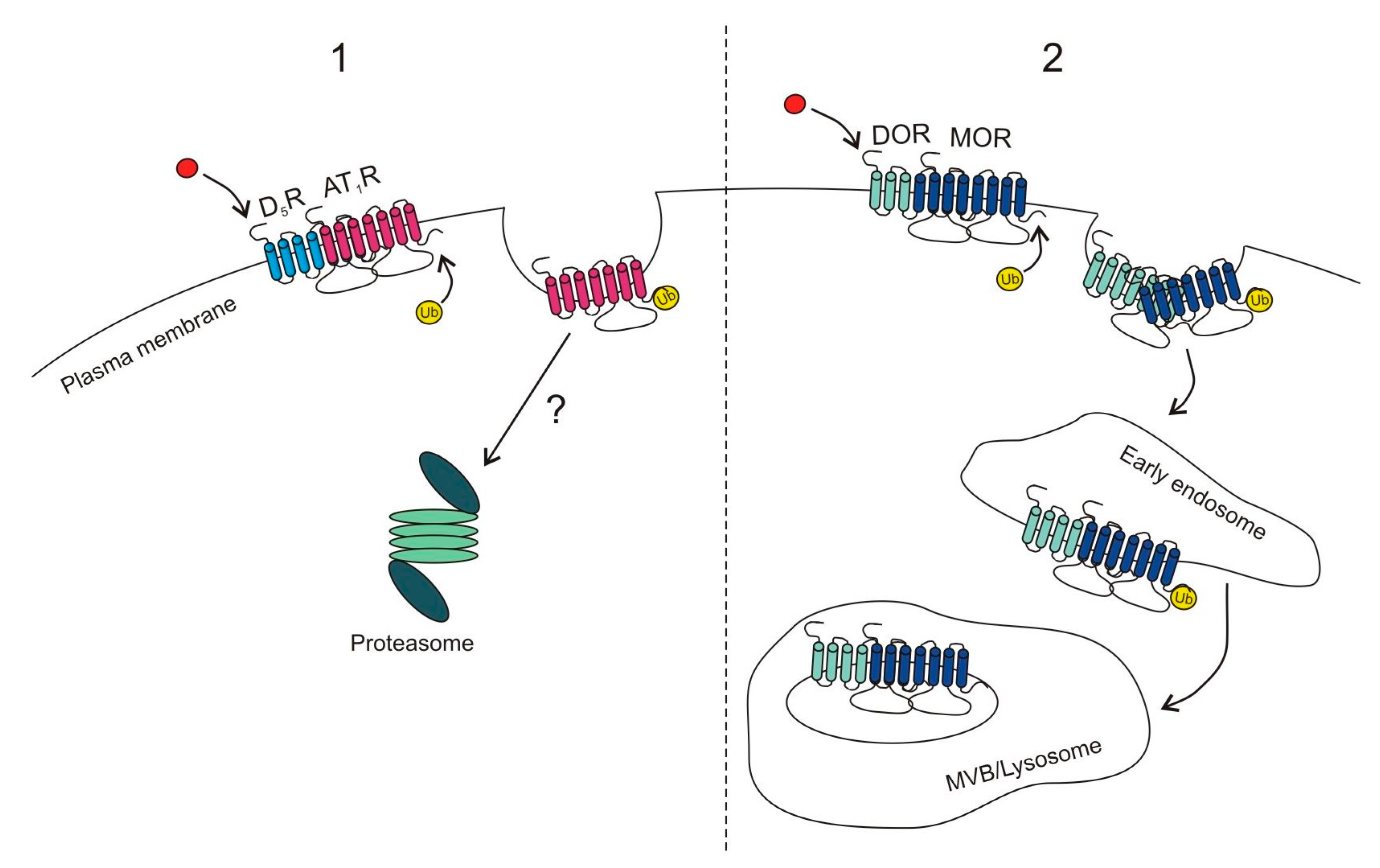
2.1.1. Proteasomal Degradation
2.1.2. Lysosomal Degradation
2.1.3. Deubiquitination and GPCR Cell-Surface Expression
2.1.4. β-Arrestins, Ubiquitination and GPCR Trafficking
2.2. Importance of Ubiquitination in GPCR Signaling and Biased Agonism
2.2.1. GPCR Signaling
2.2.2. Biased Agonism
2.3. Transubiquitination
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Congreve, M.; Langmead, C.J.; Mason, J.S.; Marshall, F.H. Progress in structure based drug design for G protein-coupled receptors. J. Med. Chem. 2011, 54, 4283–4311. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.S.; Bortolato, A.; Congreve, M.; Marshall, F.H. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 2011, 336, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Xiao, K.; Lefkowitz, R.J. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 2011, 36, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Gurevich, V.V.; Vishnivetskiy, S.A.; Sigler, P.B.; Schubert, C. Crystal structure of β-arrestin at 1.9 A: Possible mechanism of receptor binding and membrane translocation. Structure 2001, 9, 869–880. [Google Scholar] [CrossRef]
- Hirsch, J.A.; Schubert, C.; Gurevich, V.V.; Sigler, P.B. The 2.8 A crystal structure of visual arrestin: A model for arrestin’s regulation. Cell 1999, 97, 257–270. [Google Scholar] [CrossRef]
- Zhan, X.; Gimenez, L.E.; Gurevich, V.V.; Spiller, B.W. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 2011, 406, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Lefkowitz, R.J. Classical and new roles of β-arrestins in the regulation of G-protein-coupled receptors. Nat. Rev. Neurosci. 2001, 2, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.S.; Tian, X.; Benovic, J.L. Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr. Opin. Cell Biol. 2014, 27, 63–71. [Google Scholar] [CrossRef] [PubMed]
- DeFea, K.A. Beta-arrestins as regulators of signal termination and transduction: How do they determine what to scaffold? Cell. Signal. 2011, 23, 621–629. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. β-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Gesty-Palmer, D. Beyond desensitization: Physiological relevance of arrestin-dependent signaling. Pharmacol. Rev. 2010, 62, 305–330. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Ahn, S.; Shukla, A.K.; Lefkowitz, R.J. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.D.; Clarke, W.P.; Von Zastrow, M.; Nichols, D.E.; Kobilka, B.; Weinstein, H.; Javitch, J.A.; Roth, B.L.; Christopoulos, A.; Sexton, P.M. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007, 320, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, A.C.; Dunn, H.; Ferguson, S.S.G. Regulation of G protein-coupled receptor activity, trafficking and localization by GPCR-interacting proteins. Br. J. Pharmacol. 2012, 165, 1717–1736. [Google Scholar] [CrossRef] [PubMed]
- Jean-Alphonse, F.; Hanyaloglu, A. Regulation of GPCR signal networks via membrane trafficking. Mol. Cell. Endocrinol. 2011, 331, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Hanyaloglu, A.C.; Zastrow, M.v. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 537–568. [Google Scholar] [CrossRef] [PubMed]
- Chini, B.; Parenti, M. G-protein-coupled receptors, cholesterol and palmitoylation: Facts about fats. J. Mol. Endocrinol. 2009, 42, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. Ring domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. The HECT family of E3 ubiquitin ligases: Multiple players in cancer development. Cancer Cell 2008, 14, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Coscoy, L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Sci. STKE 2005, 309, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Herr, R.A.; Hansen, T.H. Ubiquitination of substrates by esterification. Traffic 2012, 13, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Ben-Saadon, R. N-terminal ubiquitination: More protein substrates join in. Trends Cell Biol. 2004, 14, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Okuda-Shimizu, Y.; Hendershot, L.M. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol. Cell. 2010, 40, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Herr, R.A.; Chua, W.J.; Lybarger, L.; Wiertz, E.J.H.J.; Hansen, T.H. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase MK3. J. Cell Biol. 2007, 177, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Skieterska, K.; Rondou, P.; Lintermans, B.; Van Craenenbroeck, K. KLHL12 promotes non-lysine ubiquitination of the dopamine receptors D4.2 and D4.4, but not of the ADHD-associated D4.7 variant. PLoS ONE 2015, 10, e014565. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 2004, 1695, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Turcu, F.E.R.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed]
- Milojevic, T.; Reiterer, V.; Stefan, E.; Korkhov, V.M.; Dorostkar, M.M.; Ducza, E.; Ogris, E.; Boehm, S.; Freissmuth, M.; Nanoff, C. The ubiquitin-specific protease USP4 regulates the cell surface level of the A2A receptor. Mol. Pharmacol. 2006, 69, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Berthouze, M.; Venkataramanan, V.; Li, Y.; Shenoy, S.K. The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 2009, 28, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; McDonald, P.H.; Kohout, T.A.; Lefkowitz, R.J. Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 2001, 294, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Modi, A.S.; Shukla, A.K.; Xiao, K.; Berthouze, M.; Ahn, S.; Wilkinson, K.D.; Miller, W.E.; Lefkowitz, R.J. β-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase MDM2. Proc. Natl. Acad. Sci. USA 2009, 106, 6650–6655. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Xiao, K.; Venkataramanan, V.; Snyder, P.M.; Freedman, N.J.; Weissman, A.M. Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the β2-adrenergic receptor. J. Biol. Chem. 2008, 283, 22166–22176. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Shenoy, S.K. β2-adrenergic receptor lysosomal trafficking is regulated by ubiquitination of lysyl residues in two distinct receptor domains. J. Biol. Chem. 2011, 286, 12785–12795. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-o.; Xiao, K.; Kim, J.; Wu, J.-H.; Wisler, J.W.; Nakamura, N.; Freedman, N.J.; Shenoy, S.K. MARCH2 promotes endocytosis and lysosomal sorting of carvedilol-bound β2-adrenergic receptors. J. Cell Biol. 2012, 199, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Armando, I.; Yu, P.; Escano, C.; Mueller, S.C.; Asico, L.; Pascua, A.; Lu, Q.; Wang, X.; Villar, V.A.M. Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells. J. Clin. Investig. 2008, 118, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Leclair, H.M.; Dubois, S.M.; Azzi, S.; Dwyer, J.; Bidere, N.; Gavard, J. Control of CXCR2 activity through its ubiquitination on k327 residue. BMC Cell Biol. 2014, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Berlin, I.; Higginbotham, K.M.; Dise, R.S.; Sierra, M.I.; Nash, P.D. The deubiquitinating enzyme USP8 promotes trafficking and degradation of the chemokine receptor 4 at the sorting endosome. J. Biol. Chem. 2010, 285, 37895–37908. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, D.; Trejo, J.; Benovic, J.L.; Marchese, A. Arrestin-2 interacts with the E3 ubiquitin ligase AIP4 and mediates endosomal sorting of the chemokine receptor CXCR4. J. Biol. Chem. 2007, 282, 36971–36979. [Google Scholar] [CrossRef] [PubMed]
- Holleman, J.; Marchese, A. The ubiquitin ligase deltex-3l regulates endosomal sorting of the G protein-coupled receptor CXCR4. Mol. Biol. Cell 2014, 25, 1892–1904. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Marchese, A. Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol. Biol. Cell 2010, 21, 2529–2541. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Soh, U.J.K.; Trejo, J.A.; Marchese, A. Novel roles for the E3 ubiquitin ligase atrophin-interacting protein 4 and signal transduction adaptor molecule 1 in G protein-coupled receptor signaling. J. Biol. Chem. 2012, 287, 9013–9027. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Benovic, J.L. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR44 mediates lysosomal sorting. J. Biol. Chem. 2001, 276, 45509–45512. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Raiborg, C.; Santini, F.; Keen, J.H.; Stenmark, H.; Benovic, J.L. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 2003, 5, 709–722. [Google Scholar] [CrossRef]
- Mines, M.A.; Goodwin, J.S.; Limbird, L.E.; Cui, F.-F.; Fan, G.-H. Deubiquitination of CXCR4 by USP14 is critical for both CXCL12-induced CXCR4 degradation and chemotaxis but not ERK activation. J. Biol. Chem. 2009, 284, 5742–5752. [Google Scholar] [CrossRef] [PubMed]
- Canals, M.; Scholten, D.J.; de Munnik, S.; Han, M.K.L.; Smit, M.J.; Leurs, R. Ubiquitination of CXCR7 controls receptor trafficking. PLoS ONE 2012, 7, e34192. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin. Cell 2001, 105, 891–902. [Google Scholar] [CrossRef]
- Omura, T.; Kaneko, M.; Okuma, Y.; Orba, Y.; Nagashima, K.; Takahashi, R.; Fujitani, N.; Matsumura, S.; Hata, A.; Kubota, K. A ubiquitin ligase HRD1 promotes the degradation of Pael receptor, a substrate of parkin. J. Neurochem. 2006, 99, 1456–1469. [Google Scholar] [CrossRef] [PubMed]
- Rondou, P.; Haegeman, G.; Vanhoenacker, P.; Van Craenenbroeck, K. BTB protein KLHL1212 targets the dopamine D4 receptor for ubiquitination by a CUL3-based E3 ligase. J. Biol. Chem. 2008, 283, 11083–11096. [Google Scholar] [CrossRef] [PubMed]
- Rondou, P.; Skieterska, K.; Packeu, A.; Lintermans, B.; Vanhoenacker, P.; Vauquelin, G.; Haegeman, G.; Van Craenenbroeck, K. KLHL12-mediated ubiquitination of the dopamine D4 receptor does not target the receptor for degradation. Cell. Signal. 2010, 22, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.D.; Bariteau, J.T.; Magenis, L.M.; Dias, J.A. Regulation of follitropin receptor cell surface residency by the ubiquitin-proteasome pathway. Endocrinology 2003, 144, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.-T.; Lai, Y.-J.; Makarova, N.; Tigyi, G.; Lin, W.-C. The lysophosphatidic acid 2 receptor mediates down-regulation of SIVA-1 to promote cell survival. J. Biol. Chem. 2007, 282, 37759–37769. [Google Scholar] [CrossRef] [PubMed]
- Oo, M.L.; Chang, S.-H.; Thangada, S.; Wu, M.-T.; Rezaul, K.; Blaho, V.; Hwang, S.-I.; Han, D.K.; Hla, T. Engagement of s1p1-degradative mechanisms leads to vascular leak in mice. J. Clin. Investig. 2011, 121, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Oo, M.L.; Thangada, S.; Wu, M.-T.; Liu, C.H.; Macdonald, T.L.; Lynch, K.R.; Lin, C.-Y.; Hla, T. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J. Biol. Chem. 2007, 282, 9082–9089. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S.N.; Guasti, L.; Clark, A.J. The E3 ubiquitin ligase mahogunin ubiquitinates the melanocortin 2 receptor. Endocrinology 2011, 152, 4224–4231. [Google Scholar] [CrossRef] [PubMed]
- Henry, A.G.; Hislop, J.N.; Grove, J.; Thorn, K.; Marsh, M.; von Zastrow, M. Regulation of endocytic clathrin dynamics by cargo ubiquitination. Dev. Cell 2012, 23, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Tanowitz, M.; von Zastrow, M. Ubiquitination-independent trafficking of G protein-coupled receptors to lysosomes. J. Biol. Chem. 2002, 277, 50219–50222. [Google Scholar] [CrossRef] [PubMed]
- Hislop, J.N.; Henry, A.G.; Marchese, A.; von Zastrow, M. Ubiquitination regulates proteolytic processing of G protein-coupled receptors after their sorting to lysosomes. J. Biol. Chem. 2009, 284, 19361–19370. [Google Scholar] [CrossRef] [PubMed]
- Henry, A.G.; White, I.J.; Marsh, M.; von Zastrow, M.; Hislop, J.N. The role of ubiquitination in lysosomal trafficking of δ-opioid receptors. Traffic 2011, 12, 170–184. [Google Scholar] [CrossRef] [PubMed]
- He, S.Q.; Zhang, Z.N.; Guan, J.S.; Liu, H.R.; Zhao, B.; Wang, H.B.; Li, Q.; Yang, H.; Luo, J.; Li, Z.Y. Facilitation of µ-opioid receptor activity by preventing δ-opioid receptor-mediated codegradation. Neuron 2010, 69, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Petaja-Repo, U.E.; Hogue, M.; Laperriere, A.; Bhalla, S.; Walker, P.; Bouvier, M. Newly synthesized human δ-opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J. Biol. Chem. 2001, 276, 4416–4423. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-G.; Benovic, J.L.; Liu-Chen, L.-Y. Mechanisms of agonist-induced down-regulation of the human κ-opioid receptor: Internalization is required for down-regulation. Mol. Pharmacol. 2000, 58, 795–801. [Google Scholar] [PubMed]
- Li, J.-G.; Haines, D.S.; Liu-Chen, L.-Y. Agonist-promoted lys63-linked polyubiquitination of the human κ-opioid receptor is involved in receptor down-regulation. Mol. Pharmacol. 2008, 73, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Groer, C.E.; Schmid, C.L.; Jaeger, A.M.; Bohn, L.M. Agonist-directed interactions with specific β-arrestins determine µ-opioid receptor trafficking, ubiquitination, and dephosphorylation. J. Biol. Chem. 2011, 286, 31731–31741. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, K.; Bandari, P.; Chinen, N.; Howells, R.D. Proteasome involvement in agonist-induced down-regulation of µ and δ opioid receptors. J. Biol. Chem. 2001, 276, 12345–12355. [Google Scholar] [CrossRef] [PubMed]
- Hislop, J.N.; Henry, A.G.; von Zastrow, M. Ubiquitination in the first cytoplasmic loop of µ-opioid receptors reveals a hierarchical mechanism of lysosomal down-regulation. J. Biol. Chem. 2011, 286, 40193–40204. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.; Cai, G.-Q.; Zheng, A.; Wang, Y.; Jia, J.; Fang, H.; Yang, Y.; Hu, M.; Ding, Q. Tumor necrosis factor-alpha regulates the hypocretin system via mRNA degradation and ubiquitination. Biochim. Biophys. Acta 2011, 1812, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Grimsey, N.J.; Aguilar, B.; Smith, T.H.; Le, P.; Soohoo, A.L.; Puthenveedu, M.A.; Nizet, V.; Trejo, J. Ubiquitin plays an atypical role in GPCR-induced p38 MAP kinase activation on endosomes. J. Cell Biol. 2015, 210, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Dupre, D.J.; Chen, Z.; Le Gouill, C.; Theriault, C.; Parent, J.-L.; Rola-Pleszczynski, M.; Stankova, J. Trafficking, ubiquitination, and down-regulation of the human platelet-activating factor receptor. J. Biol. Chem. 2003, 278, 48228–48235. [Google Scholar] [CrossRef] [PubMed]
- Donnellan, P.D.; Kinsella, B.T. Immature and mature species of the human prostacyclin receptor are ubiquitinated and targeted to the 26s proteasomal or lysosomal degradation pathways, respectively. J. Mol. Signal. 2009, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Dores, M.R.; Grimsey, N.; Canto, I.; Barker, B.L.; Trejo, J. Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation-and ubiquitination-dependent sorting signals. J. Biol. Chem. 2011, 286, 40760–40770. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.L.; Marchese, A.; Trejo, J. Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J. Cell Biol. 2007, 177, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Hasdemir, B.; Murphy, J.E.; Cottrell, G.S.; Bunnett, N.W. Endosomal deubiquitinating enzymes control ubiquitination and down-regulation of protease-activated receptor 2. J. Biol. Chem. 2009, 284, 28453–28466. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Cottrell, G.S.; Gehringer, D.; Schmidlin, F.; Grady, E.F.; Bunnett, N.W. C-cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J. Biol. Chem. 2005, 280, 16076–16087. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, G.S.; Padilla, B.; Pikios, S.; Roosterman, D.; Steinhoff, M.; Gehringer, D.; Grady, E.F.; Bunnett, N.W. Ubiquitin-dependent down-regulation of the neurokinin-1 receptor. J. Biol. Chem. 2006, 281, 27773–27783. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.B.; Zhu, C.-C.; Hinkle, P.M. Thyrotropin-releasing hormone receptor processing: Role of ubiquitination and proteasomal degradation. Mol. Endocrinol. 2003, 17, 1777–1791. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.P.; Lefkowitz, R.J.; Shenoy, S.K. Regulation of v2 vasopressin receptor degradation by agonist-promoted ubiquitination. J. Biol. Chem. 2003, 278, 45954–45959. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Livak, M.F.; Bernier, M.; Muller, D.C.; Carlson, O.D.; Elahi, D.; Maudsley, S.; Egan, J.M. Ubiquitination is involved in glucose-mediated downregulation of GIP receptors in islets. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E538–E547. [Google Scholar] [CrossRef] [PubMed]
- Alonso, V.; Magyar, C.E.; Wang, B.; Bisello, A.; Friedman, P.A. Ubiquitination-deubiquitination balance dictates ligand-stimulated PTHR sorting. J. Bone Miner. Res. 2011, 26, 2923–2934. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niwa, J.-I.; Sobue, G.; Breitwieser, G.E. Calcium-sensing receptor ubiquitination and degradation mediated by the E3 ubiquitin ligase Dorfin. J. Biol. Chem. 2006, 281, 11610–11617. [Google Scholar] [CrossRef] [PubMed]
- Lahaie, N.; Kralikova, M.; Prezeau, L.; Blahos, J.; Bouvier, M. Post-endocytotic deubiquitination and degradation of the metabotropic g-aminobutyric acid receptor by the ubiquitin-specific protease 14. J. Biol. Chem. 2016, 291, 7156–7170. [Google Scholar] [CrossRef] [PubMed]
- Moriyoshi, K.; Iijima, K.; Fujii, H.; Ito, H.; Cho, Y.; Nakanishi, S. Seven in absentia homolog 1a mediates ubiquitination and degradation of group 1 metabotropic glutamate receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 8614–8619. [Google Scholar] [CrossRef] [PubMed]
- Mukai, A.; Yamamoto-Hino, M.; Awano, W.; Watanabe, W.; Komada, M.; Goto, S. Balanced ubiquitylation and deubiquitylation of frizzled regulate cellular responsiveness to Wg/Wnt. EMBO J. 2010, 29, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Trejo, J.A. Ubiquitination of G protein-coupled receptors: Functional implications and drug discovery. Mol. Pharmacol. 2012, 82, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K. Seven-transmembrane receptors and ubiquitination. Circ. Res. 2007, 100, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Alonso, V.; Friedman, P.A. Minireview: Ubiquitination-regulated G protein-coupled receptor signaling and trafficking. Mol. Endocrinol. 2013, 27, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The ubiquitin-proteasome pathway: On protein death and cell life. EMBO J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.A.; Lawson, T.G.; Velayutham, M.; Zweier, J.L.; Pickart, C.M. A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature 2002, 416, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Van Craenenbroeck, K.; Clark, S.D.; Cox, M.J.; Oak, J.N.; Liu, F.; Van Tol, H.H. Folding efficiency is rate-limiting in dopamine D4 receptor biogenesis. J. Biol. Chem. 2005, 280, 19350–19357. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L. Gettin’down with ubiquitin: Turning off cell-surface receptors, transporters and channels. Trends Cell Biol. 1999, 9, 107–112. [Google Scholar] [CrossRef]
- Hicke, L.; Riezman, H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell 1996, 84, 277–287. [Google Scholar] [CrossRef]
- Katzmann, D.J.; Babst, M.; Emr, S.D. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 2001, 106, 145–155. [Google Scholar] [CrossRef]
- Marchese, A.; Paing, M.M.; Temple, B.R.S.; Trejo, J.A. G protein-coupled receptor sorting to endosomes and lysosomes. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 601–629. [Google Scholar] [CrossRef] [PubMed]
- Hislop, J.N.; von Zastrow, M. Role of ubiquitination in endocytic trafficking of G-protein-coupled receptors. Traffic 2011, 12, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Hasdemir, B.; Bunnett, N.W.; Cottrell, G.S. Hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) mediates post-endocytic trafficking of protease-activated receptor 2 and calcitonin receptor-like receptor. J. Biol. Chem. 2007, 282, 29646–29657. [Google Scholar] [CrossRef] [PubMed]
- Malerod, L.; Stuffers, S.; Brech, A.; Stenmark, H. Vps22/eap30 in ESCRT-II mediates endosomal sorting of growth factor and chemokine receptors destined for lysosomal degradation. Traffic 2007, 8, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, G.S.; Padilla, B.; Pikios, S.; Roosterman, D.; Steinhoff, M.; Grady, E.F.; Bunnett, N.W. Post-endocytic sorting of calcitonin receptor-like receptor and receptor activity-modifying protein 1. J. Biol. Chem. 2007, 282, 12260–12271. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Paing, M.M.; Lin, H.; Montagne, W.A.; Marchese, A.; Trejo, J. Ap-3 regulates PAR1 ubiquitin-independent MVB/lysosomal sorting via an ALIX-mediated pathway. Mol. Biol. Cell 2012, 23, 3612–3623. [Google Scholar] [CrossRef] [PubMed]
- Gullapalli, A.; Wolfe, B.L.; Griffin, C.T.; Magnuson, T.; Trejo, J. An essential role for SNX1 in lysosomal sorting of protease-activated receptor-1: Evidence for retromer-, Hrs-, and Tsg101-independent functions of sorting nexins. Mol. Biol. Cell 2006, 17, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Chen, B.; Lin, H.; Soh, U.J.; Paing, M.M.; Montagne, W.A.; Meerloo, T.; Trejo, J. Alix binds a YPX3L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J. Cell Biol. 2012, 197, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Grimsey, N.J.; Mendez, F.; Trejo, J. ALIX regulates the ubiquitin-independent lysosomal sorting of the P2y 1 purinergic receptor via a YPX3Lmotif. PLoS ONE 2016, 11, e0157587. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Lin, H.; Grimsey, N.; Mendez, F.; Trejo, J. The α-arrestin ARRDC3 mediates ALIX ubiquitination and G protein-coupled receptor lysosomal sorting. Mol. Biol. Cell 2015, 26, 4660–4673. [Google Scholar] [CrossRef] [PubMed]
- Mukai, A.; Yamamoto-Hino, M.; Komada, M.; Okano, H.; Goto, S. Balanced ubiquitination determines cellular responsiveness to extracellular stimuli. Cell. Mol. Life Sci. 2012, 69, 4007–4016. [Google Scholar] [CrossRef] [PubMed]
- Kommaddi, R.P.; Jean-Charles, P.-Y.; Shenoy, S.K. Phosphorylation of the deubiquitinase USP20 by protein kinase a regulates post-endocytic trafficking of β2 adrenergic receptors to autophagosomes during physiological stress. J. Biol. Chem. 2015, 290, 8888–8903. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, D.; Robia, S.L.; Marchese, A. The e3 ubiquitin ligase atrophin interacting protein 4 binds directly to the chemokine receptor CXCR4 via a novel WW domain-mediated interaction. Mol. Biol. Cell 2009, 20, 1324–1339. [Google Scholar] [CrossRef] [PubMed]
- Sierra, M.I.; Wright, M.H.; Nash, P.D. Amsh interacts with ESCRT-0 to regulate the stability and trafficking of CXCR4. J. Biol. Chem. 2010, 285, 13990–14004. [Google Scholar] [CrossRef] [PubMed]
- Miles, R.; Sluka, J.; Halladay, D.; Santerre, R.; Hale, L.; Bloem, L.; Patanjali, S.; Galvin, R.; Ma, L.; Hock, J. Parathyroid hormone (HPTH 1–38) stimulates the expression of UBP41, an ubiquitin-specific protease, in bone. J. Cell. Biochem. 2002, 85, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Becuwe, M.; Herrador, A.; Haguenauer-Tsapis, R.; Vincent, O.; Leon, S. Ubiquitin-mediated regulation of endocytosis by proteins of the arrestin family. Biochem. Res. Int. 2012, 2012, 242764. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K. Arrestin interaction with E3 ubiquitin ligases and deubiquitinases: Functional and therapeutic implications. Handb. Exp. Pharmacol. 2014, 219, 187–203. [Google Scholar] [PubMed]
- Jean-Charles, P.-Y.; Rajiv, V.; Shenoy, S.K. Ubiquitin-related roles of b-arrestins in endocytic trafficking and signal transduction. J. Cell. Physiol. 2016, 231, 2071–2080. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, J.F.; Pan, H.; Lu, Q. Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the β2-adrenergic receptor. EMBO Rep. 2010, 11, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-O.; Kommaddi, R.P.; Shenoy, S.K. Distinct roles for b-arrestin2 and arrestin-domain-containing proteins in β2 adrenergic receptor trafficking. EMBO Rep. 2013, 14, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.-E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions 1. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Shenoy, S.K.; Wei, H.; Lefkowitz, R.J. Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 2004, 279, 35518–35525. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, D.K.; Luttrell, L.M. Signaling in time and space: G protein-coupled receptors and mitogen-activated protein kinases. Assay Drug Dev. Technol. 2003, 1, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 2007, 213, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by β-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Strungs, E.G.; Luttrell, L.M. Arrestin-dependent activation of ERK and SRC family kinases. Handb. Exp. Pharmacol. 2014, 219, 225–257. [Google Scholar] [PubMed]
- Shenoy, S.K.; Lefkowitz, R.J. Trafficking patterns of b-arrestin and G protein-coupled receptors determined by the kinetics of β-arrestin deubiquitination. J. Biol. Chem. 2003, 278, 14498–14506. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Lefkowitz, R.J. Receptor-specific ubiquitination of β-arrestin directs assembly and targeting of seven-transmembrane receptor signalosomes. J. Biol. Chem. 2005, 280, 15315–15324. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Trejo, J.A. Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell. Signal. 2013, 25, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Saini, V.; Marchese, A.; Majetschak, M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J. Biol. Chem. 2010, 285, 15566–15576. [Google Scholar] [CrossRef] [PubMed]
- Baugher, P.J.; Richmond, A. The carboxyl-terminal PDZ ligand motif of chemokine receptor CXCR2 modulates post-endocytic sorting and cellular chemotaxis. J. Biol. Chem. 2008, 283, 30868–30878. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M. Minireview: More than just a hammer: Ligand “Bias” And pharmaceutical discovery. Mol. Endocrinol. 2014, 28, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Whalen, E.; Rajagopal, S.; Lefkowitz, R. Therapeutic potential of β-arrestin-and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Wisler, J.W.; Xiao, K.; Thomsen, A.R.; Lefkowitz, R.J. Recent developments in biased agonism. Curr. Opin. Cell Biol. 2014, 27, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Schmid, C.L.; Groer, C.E.; Bohn, L.M. Functional selectivity at the µ-opioid receptor: Implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 2011, 63, 1001–1019. [Google Scholar] [CrossRef] [PubMed]
- Wisler, J.W.; DeWire, S.M.; Whalen, E.J.; Violin, J.D.; Drake, M.T.; Ahn, S.; Shenoy, S.K.; Lefkowitz, R.J. A unique mechanism of β-blocker action: Carvedilol stimulates β-arrestin signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 16657–16662. [Google Scholar] [CrossRef] [PubMed]
- Wetzker, R.; Bohmer, F.-D. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat. Rev. Mol. Cell Biol. 2003, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, M. Oligomerization of G-protein-coupled transmitter receptors. Nat. Rev. Neurosci. 2001, 2, 274–286. [Google Scholar] [CrossRef] [PubMed]
- George, S.R.; O’Dowd, B.F. A novel dopamine receptor signaling unit in brain: Heterooligomers of D1 and D2 dopamine receptors. Sci. World J. 2007, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J. Dimerization in GPCR mobility and signaling. Curr. Opin. Pharmacol. 2010, 10, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Prinster, S.C.; Hague, C.; Hall, R.A. Heterodimerization of G protein-coupled receptors: Specificity and functional significance. Pharmacol. Rev. 2005, 57, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, M.; Novi, F.; Schallmach, E.; Lin, R.; Baragli, A.; Colzi, A.; Griffon, N.; Corsini, G.U.; Sokoloff, P.; Levenson, R. D2/D3 dopamine receptor heterodimers exhibit unique functional properties. J. Biol. Chem. 2001, 276, 30308–30314. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Guidolin, D.; Agnati, L.F.; Borroto-Escuela, D.O. Dopamine heteroreceptor complexes as therapeutic targets in parkinson’s disease. Expert Opin. Ther. Targets 2015, 19, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Marcellino, D.; Borroto-Escuela, D.O.; Frankowska, M.; Ferraro, L.; Guidolin, D.; Ciruela, F.; Agnati, L.F. The changing world of G protein-coupled receptors: From monomers to dimers and receptor mosaics with allosteric receptor-receptor interactions. J. Recept. Signal Transduct. 2010, 30, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Marcellino, D.; Guidolin, D.; Woods, A.S.; Agnati, L.F. Heterodimers and receptor mosaics of different types of G-protein-coupled receptors. Physiology 2008, 23, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Borroto-Escuela, D.O.; Marcellino, D.; Romero-Fernandez, W.; Frankowska, M.; Guidolin, D.; Filip, M.; Ferraro, L.; Woods, A.; Tarakanov, A. GPCR heteromers and their allosteric receptor-receptor interactions. Curr. Med. Chem. 2012, 19, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Tarakanov, A.; Fernandez, W.R.; Ferraro, L.; Tanganelli, S.; Filip, M.; Agnati, L.F.; Garriga, P.; Diaz-Cabiale, Z.; Borroto-Escuela, D.O. Diversity and bias through receptor-receptor interactions in GPCR heteroreceptor complexes. Focus on examples from dopamine D2 receptor heteromerization. Front. Endocrinol. 2014, 5, 71. [Google Scholar] [CrossRef] [PubMed]
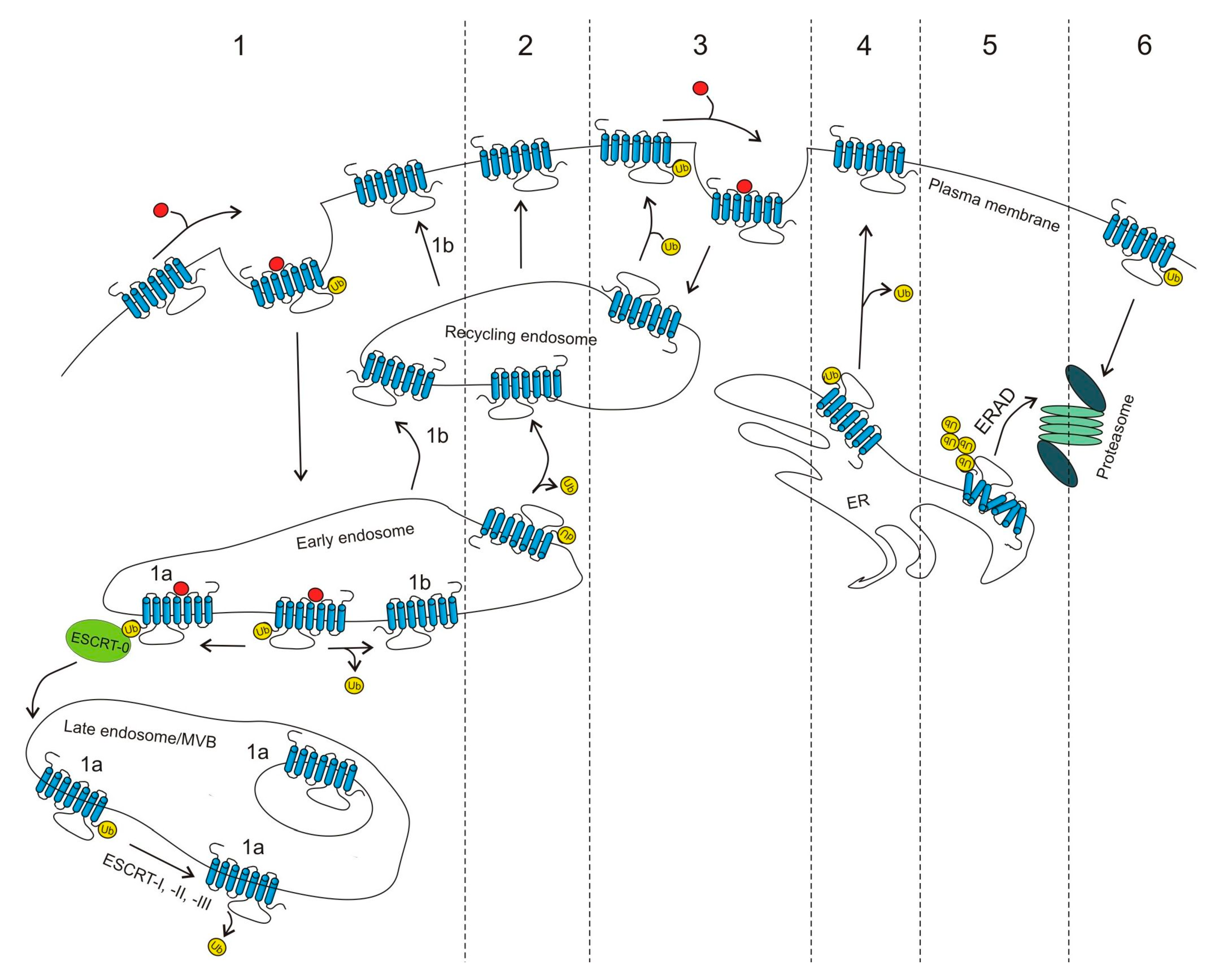

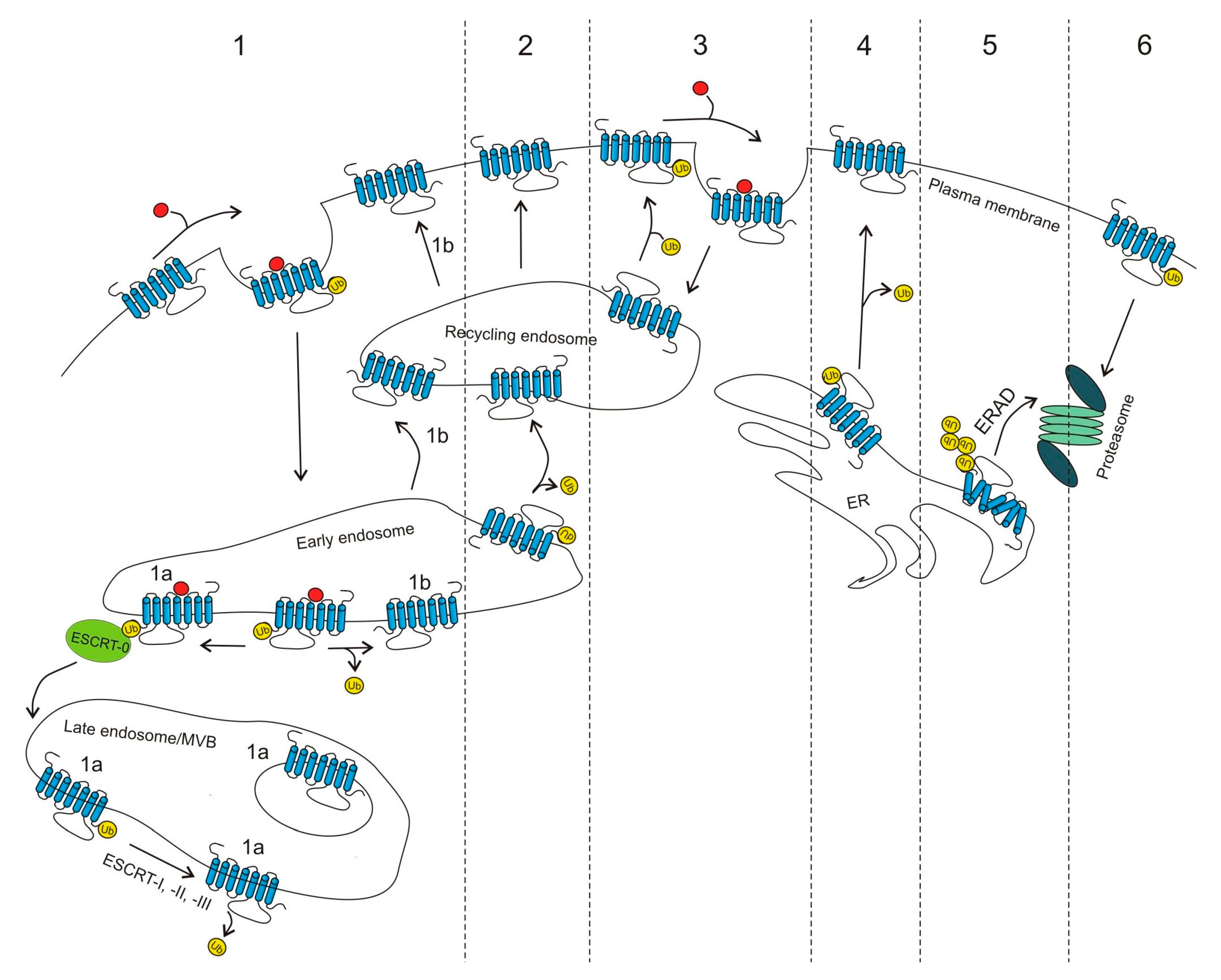{kind=link}
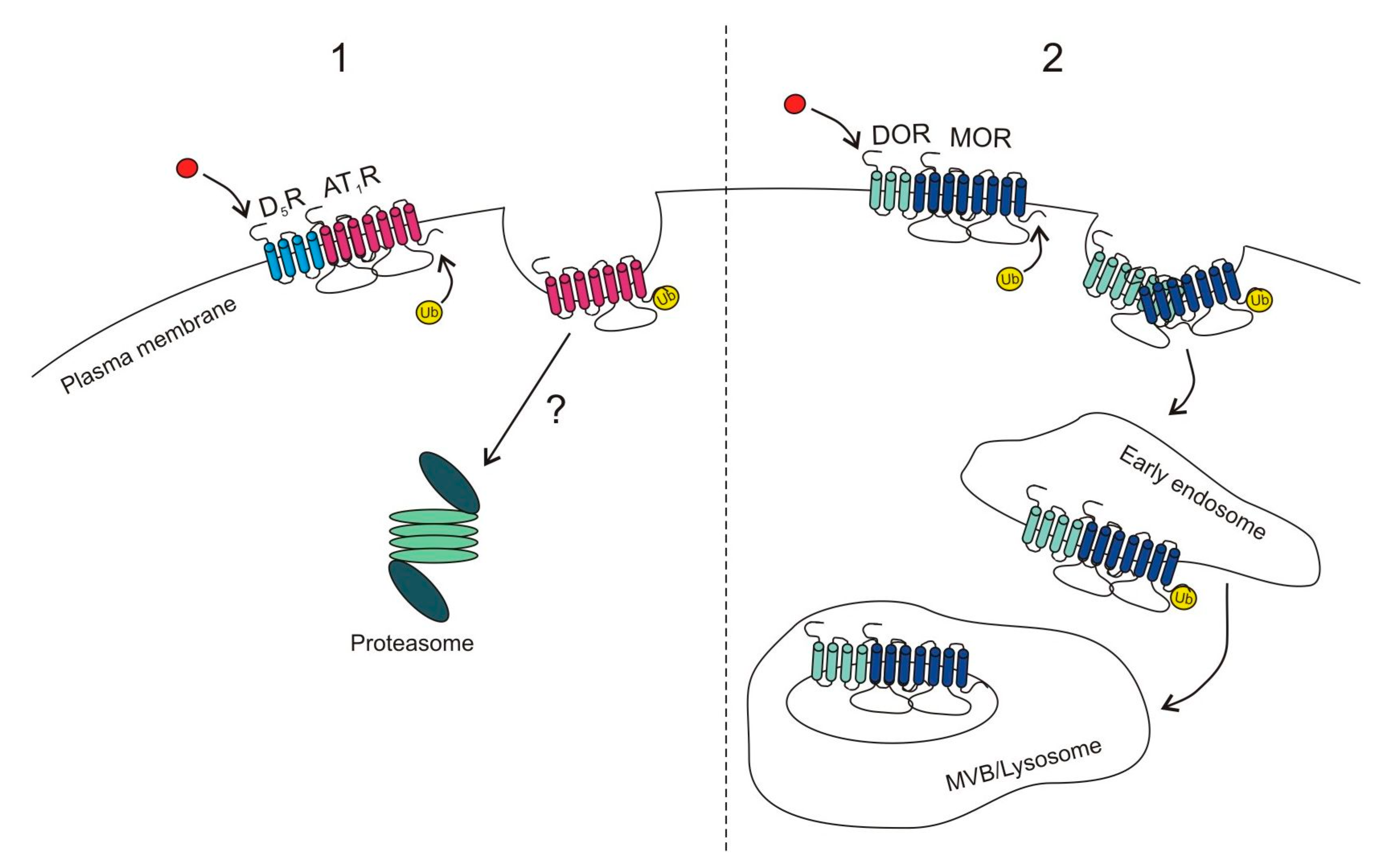{kind=link}
| GPCR | E3 Ligase | DUB | Residues | Induced/Constitutive | Role | Comment | Reference |
|---|---|---|---|---|---|---|---|
| Class A GPCRs | |||||||
| Adenosine receptors | |||||||
| A2A | N.D. | USP4 | N.D. | Constitutive | N.D. | Deubiquitination necessary for surface expression | [32] |
| Adrenoceptors | |||||||
| β2 | Nedd4 | USP20, USP33 | Lys in IC3 and C-term. | Agonist (isoproterenol) | Lysosomal degradation, regulation of arrestin-mediated signaling | β-arrestin 2 involved | [33,34,35,36,37] |
| MARCH2 | N.D. | Non-Lys | β-arrestin biased agonist (carvedilol) | Lysosomal degradation | N.D. | [38] | |
| Angiotensin receptors | |||||||
| AT1 | N.D. | N.D. | N.D. | Activation of D5R (Fenoldopam) | Proteasomal degradation of glycosylated receptor | Polyubiquitination | [39] |
| Chemokine receptors | |||||||
| CXCR2 | N.D. | N.D. | Lys327 | Agonist (IL-8) | Internalization, signaling | Polyubiquitination | [40] |
| CXCR4 | AIP4 | USP14, USP8 (indirectly) | Three Lys in C-term | Agonist (SDF-1α = CXCL12) | Lysosomal degradation; together with STAM-1 role in p44/42 MAPK activation | β-arrestin 1 involved; DTX3L–controls sorting to lysosomes by blocking activity of AIP4 | [41,42,43,44,45,46,47,48] |
| CXCR7 | N.D. | Upon stimulation | Lys in C-term | Constitutive | Ubiquitination required for membrane expression of the receptor | β-arrestin involved | [49] |
| Class A Orphans | |||||||
| GPR37 | Parkin | N.D. | C-term | Constitutive | ERAD | N.D. | [50] |
| HRD1 | N.D. | N.D. | Induced by overexpression of ATF6 | ERAD | Degradation of GPR37 reduces ER stress induced apoptosis | [51] | |
| Dopamine Receptors | |||||||
| D1R, D2R | N.D. | N.D. | N.D. | Constitutive | N.D. | N.D. | [52] |
| D4R | Cullin3 | N.D. | Non-Lys ubiquitination | Constitutive | Does not influence degradation | Polyubiquitination | [28,52,53] |
| D5R | N.D. | N.D. | N.D. | Constitutive | N.D. | N.D. | [52] |
| Glycoprotein hormone receptors | |||||||
| FSH | N.D. | N.D. | Mainly in IC3 | Constitutive | Cell-surface expression | Other residues can also be ubiquitinated | [54] |
| Lysophospholipid receptors | |||||||
| LPA2 | N.D. | N.D. | N.D. | Agonist (LPA) | Proteasomal degradation, cell survival | N.D. | [55] |
| S1P1 | WWP2 | N.D. | N.D. | Functional antagonist (FTY720P) | Proteasomal degradation | Polyubiquitination | [56,57] |
| Melanocortin receptors | |||||||
| MC2 | Mahogunin | N.D. | N.D. | Agonist (ACTH) | N.D. | Multi-monoubiquitination | [58] |
| Opioid receptors | |||||||
| δ (DOR) | AIP4 | N.D. | Lys | Agonist (DADLE) | Proteasomal degradation | Polyubiquitination, stimulates transport to ILVs | [59,60,61,62] |
| N.D. | N.D. | N.D. | Select. agonist (Deltropin I) | Lysosomal degradation | Co-degradation with MOR | [63] | |
| N.D. | N.D. | N.D. | Constitutive | Proteasomal degradation | ER-retained receptor | [64] | |
| κ (KOR) | N.D. | N.D. | Lys338, Lys 349, Lys 378 in C-term | Constitutive but enhanced by agonists | Lysosomal and proteasomal degradation | Lys63 polyubiquitination; β-arrestin involved; enhanced by receptor phosphorylation | [65,66] |
| µ (MOR) | N.D. | N.D. | Residue in IC1 | Agonist (DAMGO, DADLE) | Lysosomal and proteasomal degradation | β-arrestin 1 involved | [67,68,69] |
| Smurf2 | N.D. | Lys94 and Lys96 in IC1 | Non-selective agonist (DADLE) | Internalization by controlling maturation of the receptor-containing CCPs | Polyubiquitination; β-arrestin 2 involved | [59] | |
| N.D. | N.D. | N.D. | DOR activation (Deltropin) | Co-degradation with DOR in lysosomes | N.D. | [63] | |
| Orexin receptors | |||||||
| OX2 | cIAP-1 and -2 are important | N.D. | N.D. | TNF-α | Degradation | N.D. | [70] |
| P2Y receptors | |||||||
| P2Y1 | Nedd4-2 | N.D. | Lys in C-term | Agonist (ADP) | p38 MAPK activation | N.D. | [71] |
| Platelet-activating receptors | |||||||
| PAF receptor | Cbl is important | N.D. | N.D. | Constitutive | Agonist (PAF)-dependent down-regulation in proteasome and lysosome | Monoubiquitination | [72] |
| Prostanoid receptors | |||||||
| IP | N.D. | N.D. | N.D. | Agonist (cicaprost-mature receptor) | Lysosomal degradation of mature receptor; proteasomal degradation of immature receptor | Polyubiquitination | [73] |
| Proteinase-activated receptors | |||||||
| PAR1 | N.D. | Upon stimulation | Lys421, Lys422 in C-term | Constitutive and agonist-induced (SFLLRN-NH2) | Basal ubiquitination blocks constitutive internalization; agonist-dependent ubiquitination is involved in internalization | N.D. | [74,75] |
| Nedd4-2 | Lys | Agonist (α-thrombin) | p38 MAPK activation | Lys63-type polyubiquitination | [71] | ||
| PAR2 | Cbl | AMSH and USP8 | Lys | Agonist (peptide SLIGR-NH2) | Lysosomal degradation | Monoubiquitination; DUBs are essential for lysosomal trafficking | [76,77] |
| Tachykinin receptors | |||||||
| NK1 | N.D. | N.D. | Lys | Agonist (Substance P) | Down-regulation and degradation | N.D. | [78] |
| Thyrotropin-releasing hormone receptor | |||||||
| TRH1 | N.D. | N.D. | N.D. | Constitutive | ERAD | N.D. | [79] |
| Vasopressin and oxytocin receptors | |||||||
| V2 | N.D. | N.D. | Lys268 in IC3 | Agonist (Arg-vasopr.) | Degradation | β-arrestin 2 involved | [80] |
| Class B GPCRs | |||||||
| Glucagon receptors | |||||||
| GIP | N.D. | N.D. | N.D. | Agonist (GIP) | Proteasomal degradation | N.D. | [81] |
| Parathyroid hormone receptors | |||||||
| PTH1 | N.D. | USP2 | N.D. | Activating PTH [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34] and non-activating PTH [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34] ligands | PTH [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]-induced proteasomal degradation | Lys48-type polyubiquitination | [82] |
| Class C GPCRs | |||||||
| Calcium-sensing receptors | |||||||
| CaS | Dorphin | N.D. | Lys | Constitutive | ERAD | N.D. | [83] |
| GABAB receptors | |||||||
| GABAB1 | N.D. | USP14 | Lys | Constitutive and induced by PMA | Internalization and lysosomal degradation | N.D. | [84] |
| Metabotropic glutamate receptors | |||||||
| mGlu1a mGlu5 | Siah1A | N.D. | Lys | Constitutive | Proteasomal degradation | N.D. | [85] |
| Class Frizzled GPCRs | |||||||
| FZD4 | N.D. | USP8 | N.D. | Constitutive | Internalization; lysosomal degradation | N.D. | [86] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skieterska, K.; Rondou, P.; Van Craenenbroeck, K. Regulation of G Protein-Coupled Receptors by Ubiquitination. Int. J. Mol. Sci. 2017, 18, 923. https://doi.org/10.3390/ijms18050923
Skieterska K, Rondou P, Van Craenenbroeck K. Regulation of G Protein-Coupled Receptors by Ubiquitination. International Journal of Molecular Sciences. 2017; 18(5):923. https://doi.org/10.3390/ijms18050923
Chicago/Turabian StyleSkieterska, Kamila, Pieter Rondou, and Kathleen Van Craenenbroeck. 2017. "Regulation of G Protein-Coupled Receptors by Ubiquitination" International Journal of Molecular Sciences 18, no. 5: 923. https://doi.org/10.3390/ijms18050923
APA StyleSkieterska, K., Rondou, P., & Van Craenenbroeck, K. (2017). Regulation of G Protein-Coupled Receptors by Ubiquitination. International Journal of Molecular Sciences, 18(5), 923. https://doi.org/10.3390/ijms18050923