
SGLT2 Inhibitors as a Therapeutic Option for Diabetic Nephropathy


 ,
,  and
and
Abstract
:1. Introduction
2. Pathogenesis and Current Therapeutic Strategies of DN
3. Renal Glucose Handling by SGLT2
4. Regulation of SGLT2 and Glucose Reabsorption in Diabetes
5. Experimental Studies
6. Clinical Studies
7. Mechanisms Underlying the Renoprotection Exerted by SGLT2 Inhibitors beyond Glucose Reduction
7.1. BP Reduction
7.2. Glomerular Hyperfiltration
7.3. Erythropoietin
7.4. RAAS
7.5. Uric Acid Levels
8. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Nicola, L.; Gabbai, F.B.; Liberti, M.E.; Sagliocca, A.; Conte, G.; Minutolo, R. Sodium/glucose cotransporter 2 inhibitors and prevention of diabetic nephropathy: Targeting the renal tubule in diabetes. Am. J. Kidney Dis. 2014, 64, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.G.; Perkovic, V.; Chalmers, J.; Woodward, M.; Li, Q.; Cooper, M.E.; Hamet, P.; Harrap, S.; Heller, S.; MacMahon, S.; et al. Long-term benefits of intensive glucose control for preventing end-stage kidney disease: ADVANCE-ON. Diabetes Care 2016, 39, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Arah, O.A.; Goto, M.; Terauchi, Y.; Noda, M. Severe hypoglycaemia and cardiovascular disease: Systematic review and meta-analysis with bias analysis. BMJ 2013, 347, f4533. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Lowering plasma glucose concentration by inhibiting renal sodium-glucose cotransport. J. Intern. Med. 2014, 276, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Matoba, K.; Utsunomiya, K. Signaling pathways in diabetic nephropathy. Histol. Histopathol. 2016, 31, 1059–1067. [Google Scholar] [PubMed]
- Kawanami, D.; Matoba, K.; Sango, K.; Utsunomiya, K. Incretin-based therapies for diabetic complications: Basic mechanisms and clinical evidence. Int. J. Mol. Sci. 2016, 17, 1223. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; Norton, L.; DeFronzo, R.A. Renal sodium-glucose cotransporter inhibition in the management of type 2 diabetes mellitus. Am. J. Physiol. Ren. Physiol. 2015, 309, F889–F900. [Google Scholar] [CrossRef] [PubMed]
- Zanoli, L.; Granata, A.; Lentini, P.; Rastelli, S.; Fatuzzo, P.; Rapisarda, F.; Castellino, P. Sodium-glucose linked transporter-2 inhibitors in chronic kidney disease. Sci. World J. 2015, 2015, 317507. [Google Scholar] [CrossRef] [PubMed]
- Vrhovac, I.; Balen Eror, D.; Klessen, D.; Burger, C.; Breljak, D.; Kraus, O.; Radovic, N.; Jadrijevic, S.; Aleksic, I.; Walles, T.; et al. Localizations of Na+-d-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflug. Arch. 2015, 467, 1881–1898. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed]
- Farber, S.J.; Berger, E.Y.; Earle, D.P. Effect of diabetes and insulin of the maximum capacity of the renal tubules to reabsorb glucose. J. Clin. Investig. 1951, 30, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; Norton, L.; Defronzo, R.A. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr. Rev. 2011, 32, 515–531. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat. Rev. Nephrol. 2017, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, C.E. Maximum tubular reabsorption capacity for glucose and renal hemodynamcis during rapid hypertonic glucose infusion in normal and diabetic subjects. Scand. J. Clin. Lab. Investig. 1971, 28, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Novikov, A.; Vallon, V. Sodium glucose cotransporter 2 inhibition in the diabetic kidney: An update. Curr. Opin. Nephrol. Hypertens. 2016, 25, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Ren. Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Rose, M.; Gerasimova, M.; Satriano, J.; Platt, K.A.; Koepsell, H.; Cunard, R.; Sharma, K.; Thomson, S.C.; Rieg, T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Ren. Physiol. 2013, 304, F156–F167. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, K.; Ishihara, T.; Oku, A.; Nawano, M.; Ueta, K.; Kitamura, K.; Matsumoto, M.; Saito, A. Improved diabetic syndrome in C57BL/KsJ-db/db mice by oral administration of the Na+-glucose cotransporter inhibitor T-1095. Br. J. Pharmacol. 2001, 132, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 protein expression is increased in human diabetic nephropathy: SGLT2 protein inhibition decreases renal lipic accumulation, inflammation, and the development of nephropathy in diabetic mice. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. Renal function in diabetic disease models: The tubular system in the pathophysiology of the diabetic kidney. Annu. Rev. Physiol. 2012, 74, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Rieg, T.; Masuda, T.; Gerasimova, M.; Mayoux, E.; Platt, K.; Powell, D.R.; Thomson, S.C.; Koepsell, H.; Vallon, V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol. Ren. Physiol. 2014, 306, F188–F193. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V. The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu. Rev. Med. 2015, 66, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.R.; DaCosta, C.M.; Gay, J.; Ding, Z.M.; Smith, M.; Greer, J.; Doree, D.; Jeter-Jones, S.; Mseeh, F.; Rodriguez, L.A.; et al. Improved glycemic control in mice lacking Sglt1 and Sglt2. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E117–E130. [Google Scholar] [CrossRef] [PubMed]
- Malatiali, S.; Francis, I.; Barac-Nieto, M. Phlorizin prevents glomerular hyperfiltration but not hypertrophy in diabetic rats. Exp. Diabetes Res. 2008, 2008, 305403. [Google Scholar] [CrossRef] [PubMed]
- Osorio, H.; Coronel, I.; Arellano, A.; Pacheco, U.; Bautista, R.; Franco, M.; Escalante, B. Sodium-glucose cotransporter inhibition prevents oxidative stress in the kidney of diabetic rats. Oxid. Med. Cell. Longev. 2012, 2012, 542042. [Google Scholar] [CrossRef] [PubMed]
- Gembardt, F.; Bartaun, C.; Jarzebska, N.; Mayoux, E.; Todorov, V.T.; Hohenstein, B.; Hugo, C. The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in BTBR ob/ob type 2 diabetic mice with and without hypertension. Am. J. Physiol. Ren. Physiol. 2014, 307, F317–F325. [Google Scholar] [CrossRef] [PubMed]
- Ojima, A.; Matsui, T.; Nishino, Y.; Nakamura, N.; Yamagishi, S. Empagliflozin, an inhibitor of sodium-glucose cotransporter 2 exerts anti-inflammatory and antifibrotic effects on experimental diabetic nephropathy partly by suppressing AGEs-receptor axis. Horm. Metab. Res. 2015, 47, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Panchapakesan, U.; Pegg, K.; Gross, S.; Komala, M.G.; Mudaliar, H.; Forbes, J.; Pollock, C.; Mather, A. Effects of SGLT2 inhibition in human kidney proximal tubular cells—Renoprotection in diabetic nephropathy? PLoS ONE 2013, 8, e54442. [Google Scholar] [CrossRef] [PubMed]
- Kojima, N.; Williams, J.M.; Takahashi, T.; Miyata, N.; Roman, R.J. Effects of a new SGLT2 inhibitor, luseogliflozin, on diabetic nephropathy in T2DN rats. J. Pharmacol. Exp. Ther. 2013, 345, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Terami, N.; Ogawa, D.; Tachibana, H.; Hatanaka, T.; Wada, J.; Nakatsuka, A.; Eguchi, J.; Horiguchi, C.S.; Nishii, N.; Yamada, H.; et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS ONE 2014, 9, e100777. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, T.; Ogawa, D.; Tachibana, H.; Eguchi, J.; Inoue, T.; Yamada, H.; Takei, K.; Makino, H.; Wada, J. Inhibition of SGLT2 alleviates diabetic nephropathy by suppressing high glucose-induced oxidative stress in type 1 diabetic mice. Pharmacol. Res. Perspect. 2016, 4, e00239. [Google Scholar] [CrossRef] [PubMed]
- Nagata, T.; Fukuzawa, T.; Takeda, M.; Fukazawa, M.; Mori, T.; Nihei, T.; Honda, K.; Suzuki, Y.; Kawabe, Y. Tofogliflozin, a novel sodium-glucose co-transporter 2 inhibitor, improves renal and pancreatic function in db/db mice. Br. J. Pharmacol. 2013, 170, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Gangadharan Komala, M.; Gross, S.; Mudaliar, H.; Huang, C.; Pegg, K.; Mather, A.; Shen, S.; Pollock, C.A.; Panchapakesan, U. Inhibition of kidney proximal tubular glucose reabsorption does not prevent against diabetic nephropathy in type 1 diabetic eNOS knockout mice. PLoS ONE 2014, 9, e108994. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.A.; Ward, M.S.; Fotheringham, A.K.; Zhuang, A.; Borg, D.J.; Flemming, N.B.; Harvie, B.M.; Kinneally, T.L.; Yeh, S.M.; McCarthy, D.A.; et al. Once daily administration of the SGLT2 inhibitor, empagliflozin, attenuates markers of renal fibrosis without improving albuminuria in diabetic db/db mice. Sci. Rep. 2016, 6, 26428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thai, K.; Kepecs, D.M.; Gilbert, R.E. Sodium-glucose linked cotransporter-2 inhibition does not attenuate disease progression in the rat remnant kidney model of chronic kidney disease. PLoS ONE 2016, 11, e0144640. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Steiger, S.; Anders, H.J. Sodium glucose transporter-2 inhibition has no renoprotective effects on non-diabetic chronic kidney disease. Physiol. Rep. 2017, 5, e13228. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E. Sodium-glucose linked transporter-2 inhibitors: Potential for renoprotection beyond blood glucose lowering? Kidney Int. 2014, 86, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.; Desai, M.; Jardine, M.; Balis, D.; Meininger, G.; Perkovic, V. Canagliflozin slows progression of renal function decline independently of glycemic effects. J. Am. Soc. Nephrol. 2016, 28, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; Fulcher, G.; Stein, P.; Desai, M.; Shaw, W.; Jiang, J.; Vercruysse, F.; et al. Rationale, design, and baseline characteristics of the Canagliflozin Cardiovascular Assessment Study (CANVAS)—A randomized placebo-controlled trial. Am. Heart J. 2013, 166, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Baker, W.L.; Smyth, L.R.; Riche, D.M.; Bourret, E.M.; Chamberlin, K.W.; White, W.B. Effects of sodium-glucose co-transporter 2 inhibitors on blood pressure: A systematic review and meta-analysis. J. Am. Soc. Hypertens. 2014, 8, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: Cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef] [PubMed]
- Sjostrom, C.D.; Hashemi, M.; Sugg, J.; Ptaszynska, A.; Johnsson, E. Dapagliflozin-induced weight loss affects 24-week glycated haemoglobin and blood pressure levels. Diabetes Obes. Metab. 2015, 17, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Cefalu, W.T.; Stenlof, K.; Leiter, L.A.; Wilding, J.P.; Blonde, L.; Polidori, D.; Xie, J.; Sullivan, D.; Usiskin, K.; Canovatchel, W.; et al. Effects of canagliflozin on body weight and relationship to HbA1c and blood pressure changes in patients with type 2 diabetes. Diabetologia 2015, 58, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Har, R.; Fagan, N.; Johansen, O.E.; Woerle, H.J.; von Eynatten, M.; Broedl, U.C. The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc. Diabetol. 2014, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Chilton, R.; Tikkanen, I.; Cannon, C.P.; Crowe, S.; Woerle, H.J.; Broedl, U.C.; Johansen, O.E. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Brenner, B.M. Pathogenesis of diabetic glomerulopathy: Hemodynamic considerations. Diabetes Metab. Rev. 1988, 4, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Sochett, E.B.; Cherney, D.Z.; Curtis, J.R.; Dekker, M.G.; Scholey, J.W.; Miller, J.A. Impact of renin angiotensin system modulation on the hyperfiltration state in type 1 diabetes. J. Am. Soc. Nephrol. 2006, 17, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.C.; Vallon, V.; Blantz, R.C. Kidney function in early diabetes: The tubular hypothesis of glomerular filtration. Am. J. Physiol. Ren. Physiol. 2004, 286, F8–F15. [Google Scholar] [CrossRef] [PubMed]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Maione, M.; Lai, V.; Lee, A.; Fagan, N.M.; Woerle, H.J.; Johansen, O.E.; Broedl, U.C.; et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 2014, 129, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Schnermann, J.; Levine, D.Z. Paracrine factors in tubuloglomerular feedback: Adenosine, ATP, and nitric oxide. Annu. Rev. Physiol. 2003, 65, 501–529. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, M.; Yang, G.K.; Perkins, B.A.; Soleymanlou, N.; Lytvyn, Y.; von Eynatten, M.; Woerle, H.J.; Johansen, O.E.; Broedl, U.C.; Hach, T.; et al. Characterisation of glomerular haemodynamic responses to SGLT2 inhibition in patients with type 1 diabetes and renal hyperfiltration. Diabetologia 2014, 57, 2599–2602. [Google Scholar] [CrossRef] [PubMed]
- Cherney, D.Z.; Miller, J.A.; Scholey, J.W.; Bradley, T.J.; Slorach, C.; Curtis, J.R.; Dekker, M.G.; Nasrallah, R.; Hebert, R.L.; Sochett, E.B. The effect of cyclooxygenase-2 inhibition on renal hemodynamic function in humans with type 1 diabetes. Diabetes 2008, 57, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Ikuma-Suwa, E.; Inoue, K.; Kaneko, K.; Yanagisawa, M.; Inukai, K.; Noda, M.; Shimada, A. Effects of ipragliflozin on diabetic nephropathy and blood pressure in patients with type 2 diabetes: An open-label study. J. Clin. Med. Res. 2017, 9, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Petrykiv, S.I.; Laverman, G.D.; Zeeuw, D.; Heerspink, H.J. The albuminuria lowering response to dapagliflozin is variable and reproducible between individual patients. Diabetes Obes. Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lambers Heerspink, H.J.; de Zeeuw, D.; Wie, L.; Leslie, B.; List, J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes. Metab. 2013, 15, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nangaku, M. Recent advances and clinical application of erythropoietin and erythropoiesis-stimulating agents. Exp. Cell Res. 2012, 318, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Takei, M.; Shiraishi, Y.; Suzuki, Y. Increased hematocrit during sodium-glucose cotransporter 2 inhibitor therapy indicates recovery of tubulointerstitial function in diabetic kidneys. J. Clin. Med. Res. 2016, 8, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Eto, N.; Wada, T.; Inagi, R.; Takano, H.; Shimizu, A.; Kato, H.; Kurihara, H.; Kawachi, H.; Shankland, S.J.; Fujita, T.; et al. Podocyte protection by darbepoetin: Preservation of the cytoskeleton and nephrin expression. Kidney Int. 2007, 72, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, K.; Takeda, S.; Tashiro, Y.; Yorozu, K.; Hirata, M.; Kanada, H.; Moriguchi, Y.; Endo, K. Renoprotection by continuous erythropoietin receptor activator in puromycin aminonucleoside-induced nephrotic syndrome. Am. J. Nephrol. 2012, 36, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Serizawa, K.; Yogo, K.; Tashiro, Y.; Aizawa, K.; Kawasaki, R.; Hirata, M.; Endo, K. Epoetin β pegol prevents endothelial dysfunction as evaluated by flow-mediated dilation in chronic kidney disease rats. Eur. J. Pharmacol. 2015, 767, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Eren, Z.; Gunal, M.Y.; Ari, E.; Coban, J.; Cakalagaoglu, F.; Caglayan, B.; Beker, M.C.; Akdeniz, T.; Yanikkaya, G.; Kilic, E.; et al. Pleiotropic and renoprotective effects of erythropoietin β on experimental diabetic nephropathy model. Nephron 2016, 132, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Tsuruya, K.; Yoshida, H.; Suehiro, T.; Fujisaki, K.; Masutani, K.; Kitazono, T. Erythropoiesis-stimulating agent slows the progression of chronic kidney disease: A possibility of a direct action of erythropoietin. Ren. Fail. 2016, 38, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Koitka, A.; Tikellis, C. Advances in the renin-angiotensin-aldosterone system: Relevance to diabetic nephropathy. Sci. World J. 2008, 8, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Muskiet, M.H.; van Raalte, D.H.; van Bommel, E.J.; Smits, M.M.; Tonneijck, L. Understanding EMPA-REG OUTCOME. Lancet Diabetes Endocrinol. 2015, 3, 928–929. [Google Scholar] [CrossRef]
- Padda, R.S.; Shi, Y.; Lo, C.S.; Zhang, S.L.; Chan, J.S. Angiotensin-(1–7): A Novel peptide to treat hypertension and nephropathy in diabetes? J. Diabetes Metab. 2015, 6. [Google Scholar] [CrossRef]
- Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes-E-Silva, A.C. The anti-inflammatory potential of ACE2/Angiotensin-(1–7)/Mas receptor axis: Evidence from basic and clinical research. Curr. Drug Targets 2016. [Google Scholar] [CrossRef]
- Shi, Y.; Lo, C.S.; Padda, R.; Abdo, S.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Angiotensin-(1–7) prevents systemic hypertension, attenuates oxidative stress and tubulointerstitial fibrosis, and normalizes renal angiotensin-converting enzyme 2 and Mas receptor expression in diabetic mice. Clin. Sci. 2015, 128, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Bjornstad, P.; Lanaspa, M.A.; Ishimoto, T.; Kosugi, T.; Kume, S.; Jalal, D.; Maahs, D.M.; Snell-Bergeon, J.K.; Johnson, R.J.; Nakagawa, T. Fructose and uric acid in diabetic nephropathy. Diabetologia 2015, 58, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Jalal, D.I.; Maahs, D.M.; Hovind, P.; Nakagawa, T. Uric acid as a mediator of diabetic nephropathy. Semin. Nephrol. 2011, 31, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Skrtic, M.; Yang, G.K.; Yip, P.M.; Perkins, B.A.; Cherney, D.Z. Glycosuria-mediated urinary uric acid excretion in patients with uncomplicated type 1 diabetes mellitus. Am. J. Physiol. Ren. Physiol. 2015, 308, F77–F83. [Google Scholar] [CrossRef] [PubMed]
- Maahs, D.M.; Caramori, L.; Cherney, D.Z.; Galecki, A.T.; Gao, C.; Jalal, D.; Perkins, B.A.; Pop-Busui, R.; Rossing, P.; Mauer, M.; et al. Uric acid lowering to prevent kidney function loss in diabetes: The preventing early renal function loss (PERL) allopurinol study. Curr. Diab. Rep. 2013, 13, 550–559. [Google Scholar] [CrossRef] [PubMed]
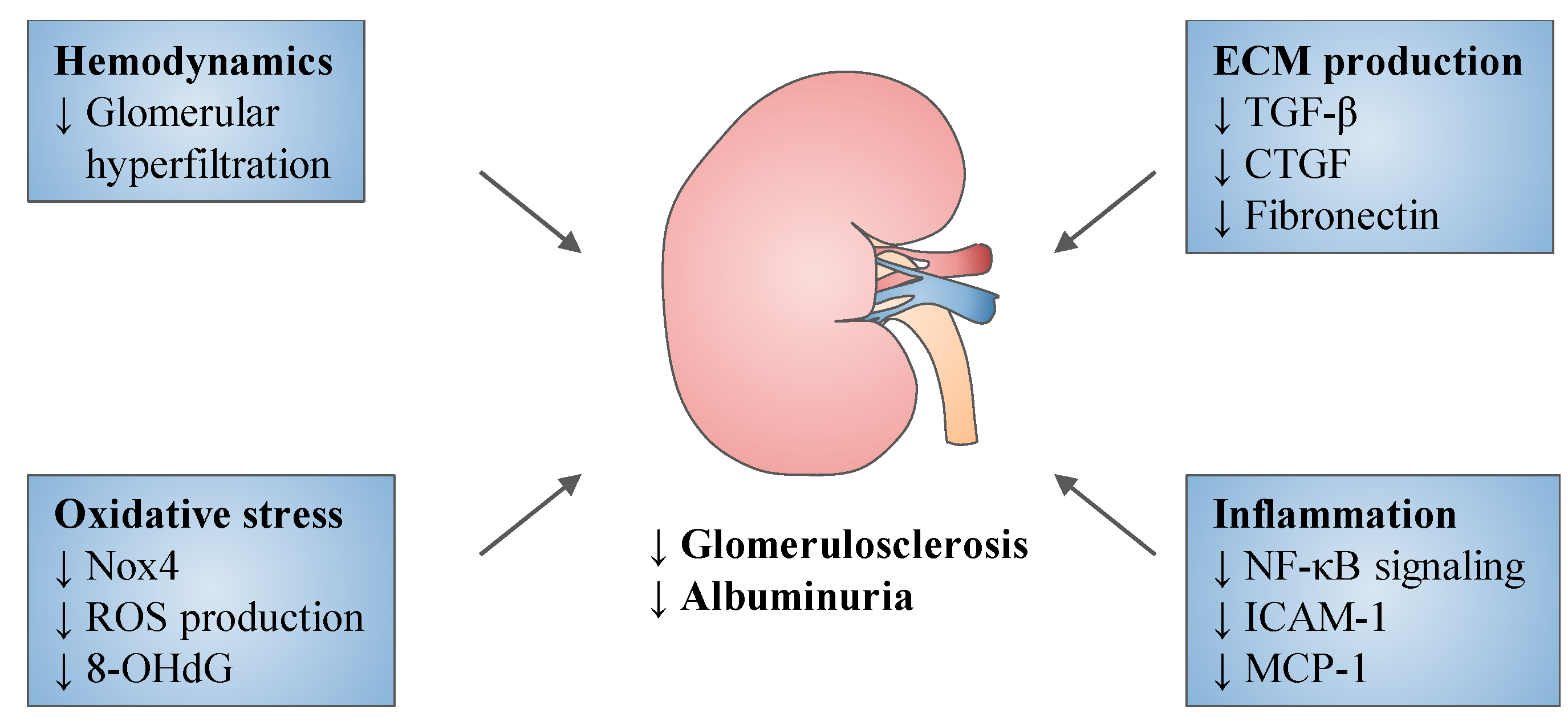

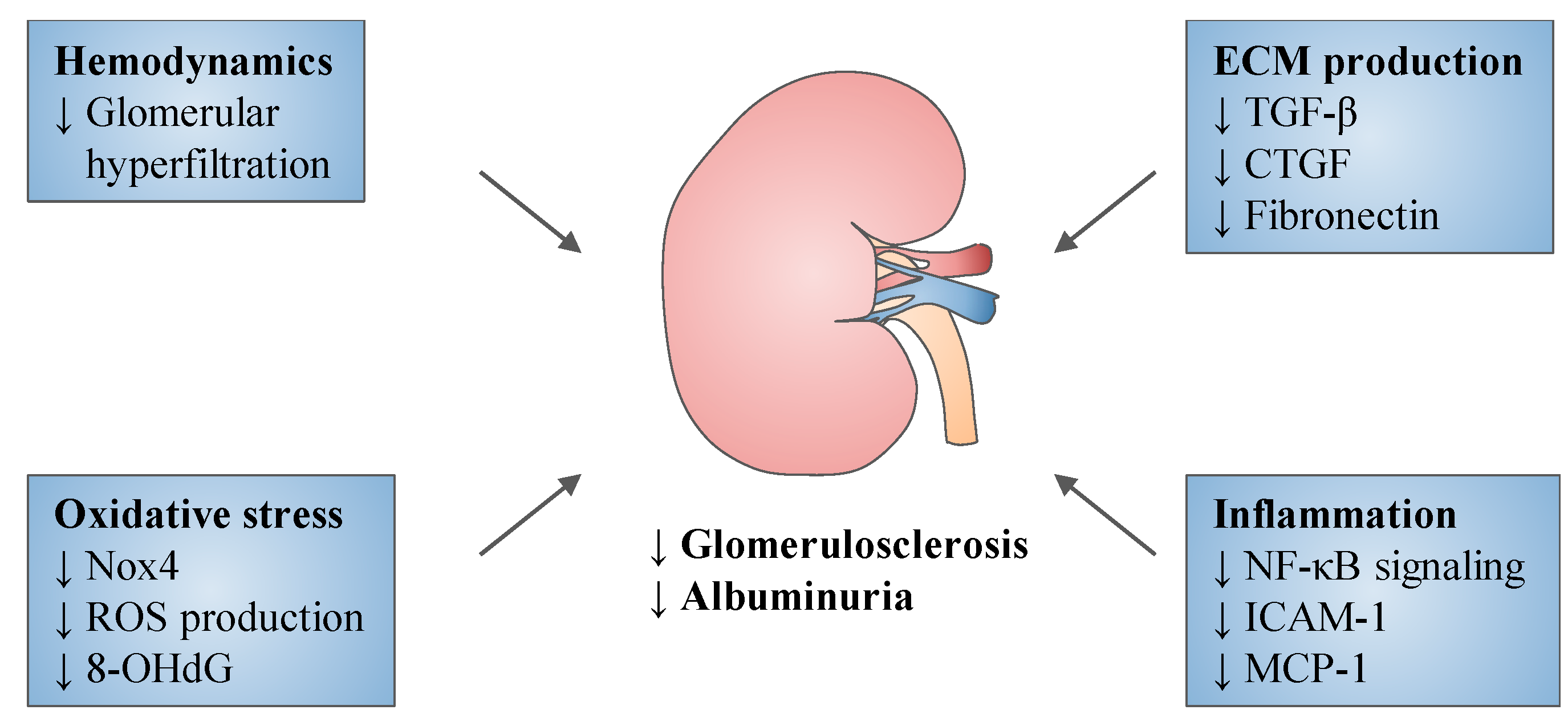{kind=link}
| Study | Model | Drug/Dose/Duration | Major Effects |
|---|---|---|---|
| Malatiali et al. [29] | STZ-diabetic rats | phlorizin, 800 mg/kg, 6 days (initial dose: 400 mg/kg) | ↓ Glomerular hyperfiltration, |
| ↓ Kidney size, | |||
| ↓ Oxidative stress | |||
| Osorio et al. [30] | STZ-diabetic rats | phlorizin, 400 mg/kg, 30 days | ↓ Oxidative stress |
| Vallon et al. [27] | Akita mice | empagliflozin, 300 mg/kg, 15 weeks | ↓ Glomerular hyperfiltration, |
| ↓ Albuminuria, | |||
| ↓ Kidney Weight, | |||
| ↓ Inflammation | |||
| Gembardt et al. [31] | BTBR ob/ob mice | empagliflozin, diet containing 300 ppm of empagliflozin, 12 weeks | ↓ Albuminuria, |
| ↓ Glomerular hypertrophy, | |||
| ↓ Inflammation, | |||
| ↓ Mesangial matrix expansion | |||
| BTBR ob/ob mice with hypertension (angiotensin-II infusion) | empagliflozin, 300 ppm, 12 weeks | ↓ Albuminuria | |
| Ojima et al. [32] | STZ-diabetic rats | empagliflozin, 10 mg/kg, 4 weeks | ↔ Albuminuria, |
| ↓ AGE/RAGE, | |||
| ↓ Oxidative stress, | |||
| ↓ Inflammation, | |||
| ↓ Fibrotic gene markers, | |||
| ↓ Tubular injury | |||
| Gangadharan Komala et al. [38] | eNOS-deficient-STZ-diabetic mice | empagliflozin, 10 mg/kg, 19 weeks | ↔ Albuminuria, |
| ↔ Glomerulosclerosis, | |||
| ↔ Interstitial fibrosis, | |||
| ↔ TGF-β, | |||
| ↔ Fibronectin, | |||
| ↔ MCP-1 | |||
| Gallo et al. [39] | db/db mice | empagliflozin, 10 mg/kg, 10 weeks | ↔ Albuminuria |
| ↓ Profibrotic gene markers, | |||
| ↓ Fibronectin, | |||
| ↓ TGF-β | |||
| Kojima et al. [34] | T2DN rats (McWi strain) | luseogliflozin, 10 mg/kg, 3 weeks | ↔ Proteinuria |
| ↓ Glomerulosclerosis | |||
| Terami et al. [35] | db/db mice | dapagliflozin, 0.1 mg/kg or 1.0 mg/kg, 12 weeks | ↓ Albuminuria, |
| ↓ Oxidative stress, | |||
| ↓ Inflammation | |||
| Hatanaka et al. [36] | Akita mice | dapagliflozin, 1.0 mg/kg, 12 weeks | ↓ Albuminuria, |
| ↓ Oxidative stress, | |||
| ↓ Macrophage infiltration | |||
| Nagata et al. [37] | db/db mice | tofogliflozin, Diet containing 0.005% or 0.015% of tofogliflozin, 8 weeks | ↓ Albuminuria, |
| ↓ Glomerular hypertrophy | |||
| Wang et al. [23] | db/db mice | JNJ39933673, Diet containing 0.07 g/kg of JNJ39933673, 12 weeks | ↓ Albuminuria, |
| ↓ Mesangial expansion, | |||
| ↓ Podocyte injury, | |||
| ↓ Renal lipid accumulation |
| Trial | Drug/Dose | Patients | Major Renal Outcome |
|---|---|---|---|
| EMPA-REG OUTCOME [6,7] (3.1 years) | empagliflozin 10 mg or 25 mg/day (vs. placebo) | T2D patients at high risk of CVD with an eGFR of at least 30 mL/min/1.73 m2 (n = 7020) | ↓ Incidence or worsening of DN (HR: 0.61, 95% CI: 0.53-0.70) |
| ↓ Progression to macroalbuminuria (HR: 0.62, 95% CI: 0.54-0.72) | |||
| ↓ Doubling of serum creatinine level accompanied by eGFR of ≤45 mL/min/1.73 m2 (HR: 0.56, 95% CI: 0.39-0.79) | |||
| ↓ Initiation of renal replacement therapy (HR: 0.45, 95% CI: 0.21-0.97) | |||
| CANTA-SU [38] (2 years) | canagliflozin 100 mg or 300 mg/day (vs. glimepiride 6–8 mg/day) | T2D patients who receive metformin (n = 1450) | ↓ eGFR decline −0.5 (canagliflozin 100 mg), −0.9 (canagliflozin 300 mg), -3.3 (glimepiride) mL/min/1.73 m2 at 2 years |
| ↓ Albuminuria −31.7% (canagliflozin 100 mg), −49.3% (canagliflozin 300 mg) relative to glimepiride |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawanami, D.; Matoba, K.; Takeda, Y.; Nagai, Y.; Akamine, T.; Yokota, T.; Sango, K.; Utsunomiya, K. SGLT2 Inhibitors as a Therapeutic Option for Diabetic Nephropathy. Int. J. Mol. Sci. 2017, 18, 1083. https://doi.org/10.3390/ijms18051083
Kawanami D, Matoba K, Takeda Y, Nagai Y, Akamine T, Yokota T, Sango K, Utsunomiya K. SGLT2 Inhibitors as a Therapeutic Option for Diabetic Nephropathy. International Journal of Molecular Sciences. 2017; 18(5):1083. https://doi.org/10.3390/ijms18051083
Chicago/Turabian StyleKawanami, Daiji, Keiichiro Matoba, Yusuke Takeda, Yosuke Nagai, Tomoyo Akamine, Tamotsu Yokota, Kazunori Sango, and Kazunori Utsunomiya. 2017. "SGLT2 Inhibitors as a Therapeutic Option for Diabetic Nephropathy" International Journal of Molecular Sciences 18, no. 5: 1083. https://doi.org/10.3390/ijms18051083
APA StyleKawanami, D., Matoba, K., Takeda, Y., Nagai, Y., Akamine, T., Yokota, T., Sango, K., & Utsunomiya, K. (2017). SGLT2 Inhibitors as a Therapeutic Option for Diabetic Nephropathy. International Journal of Molecular Sciences, 18(5), 1083. https://doi.org/10.3390/ijms18051083