
Neurotrauma: The Crosstalk between Neurotrophins and Inflammation in the Acutely Injured Brain

Abstract
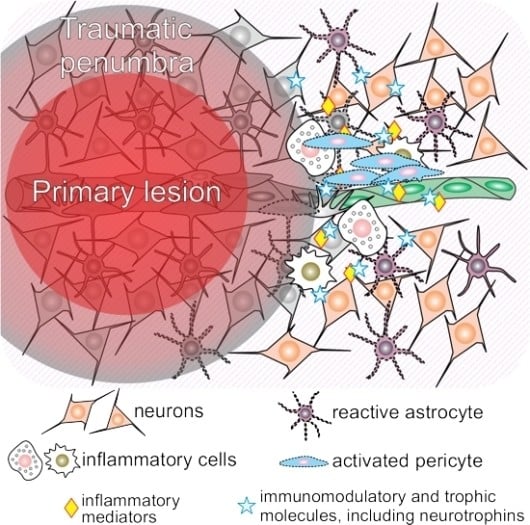
1. What Happens to the Acutely Traumatized Neural Tissue?
1.1. Traumatic Penumbra
1.2. Mechanisms Involved in Ongoing Secondary Injury Shortly After Traumatic Brain Injury (TBI)
1.3. Neuroinflammation Shortly after Traumatic Brain Injury (TBI)
2. What Are the Roles of Neurotrophins after Acute Neural Injury?
2.1. Neurotrophins
2.2. Roles of Neurotrophins Shortly after TBI
3. What Happens to Neural Tissue during Recovery after Acute Neural Injury?
3.1. Neuroinflammation and Pericytes
3.2. The Crosstalk between Neurotrophins and Pericytes
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| APOE | Apolipoprotein E |
| ATMSCs | Adipose Tissue-Derived MSCs |
| BBB | Blood Brain Barrier |
| BDNF | Brain-Derived Neurotrophic Factor |
| CCI | Controlled Cortical Impact |
| cPCs | Cultured Pericytes |
| CSF | Cerebrospinal Fluid |
| CNS | Central Nervous System |
| EDNRA | Endothelin Receptor A |
| EDNRB | Endothelin Receptor B |
| GCS | Glasgow Coma Scale |
| GDNF | Glial Cell-Derived Neurotrophic Factor |
| GEO | Gene Expression Omnibus |
| HUVECs | Human Umbilical Vein Endothelial Cells |
| ICP | Intracranial Pressure |
| IFN-λ | Interferon-λ |
| IL | Interleukin |
| MSC | Mesenchymal Stromal Cell |
| MMP | Matrix Metalloproteinase |
| ncPCs | non-cultured Pericytes |
| NT-3 | Neurotrophin-3 |
| NT-4 | Neurotrophin-4 |
| NGF | Nerve Growth Factor |
| NVU | Neurovascular Unit |
| PBWBCs | Peripheral Blood White Blood Cells |
| PSPN | Persephin |
| ROS | Reactive Oxygen Species |
| TBI | Traumatic Brain Injury |
| TGFβ | Transforming Growth Factor 1β |
| TNFα | Tumor Necrosis Factor α |
References
- Fleminger, S.; Ponsford, J. Long term outcome after traumatic brain injury. BMJ 2005, 331, 1419–1420. [Google Scholar] [CrossRef] [PubMed]
- Bruns, J., Jr.; Hauser, W.A. The epidemiology of traumatic brain injury: A review. Epilepsia 2003, 44, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Leibson, C.L.; Brown, A.W.; Ransom, J.E.; Diehl, N.N.; Perkins, P.K.; Mandrekar, J.; Malec, J.F. Incidence of traumatic brain injury across the full disease spectrum: A population-based medical record review study. Epidemiology 2011, 22, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Llompart-Pou, J.A.; Chico-Fernandez, M.; Sanchez-Casado, M.; Alberdi-Odriozola, F.; Guerrero-Lopez, F.; Mayor-Garcia, M.D.; Gonzalez-Robledo, J.; Ballesteros-Sanz, M.A.; Herran-Monge, R.; Leon-Lopez, R.; et al. Age-related injury patterns in Spanish trauma ICU patients. Results from the RETRAUCI. Injury 2016, 47, S61–S65. [Google Scholar] [CrossRef]
- Chieregato, A.; Martino, C.; Pransani, V.; Nori, G.; Russo, E.; Noto, A.; Simini, B. Classification of a traumatic brain injury: The Glasgow Coma scale is not enough. Acta Anaesthesiol. Scand. 2010, 54, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Faul, M.; Xu, L.; Wald, M.M.; Coronado, V.G. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006; Centers for Disease Control and PreventionNational Center for Injury: Atlanta, GA, USA, 2010. [Google Scholar]
- Langlois, J.A.; Rutland-Brown, W.; Wald, M.M. The epidemiology and impact of traumatic brain injury: A brief overview. J. Head Trauma Rehabil. 2006, 21, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Agrawal, D.; Ahmed, S.; Khan, S.; Gupta, D.; Sinha, S.; Satyarthee, G.D. Outcome in 2068 patients of head injury: Experience at a level 1 trauma centre in India. Asian J. Neurosurg. 2016, 11, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Evans, D.; Hameed, S.M.; Yanchar, N.L.; Stelfox, H.T.; Simons, R.; Kortbeek, J.; Bourgeois, G.; Clement, J.; Lauzier, F.; et al. Mortality in Canadian Trauma Systems: A Multicenter Cohort Study. Ann. Surg. 2017, 265, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, F.; Compagnone, C.; Korsic, M.; Servadei, F.; Kraus, J. A systematic review of brain injury epidemiology in Europe. Acta Neurochir. 2006, 148, 255–268. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.J.; Sun, M.; Agoston, D.V.; Shultz, S.R. The effect of concomitant peripheral injury on traumatic brain injury pathobiology and outcome. J. Neuroinflamm. 2016, 13, 90. [Google Scholar] [CrossRef]
- Papurica, M.; Rogobete, A.F.; Sandesc, D.; Dumache, R.; Cradigati, C.A.; Sarandan, M.; Nartita, R.; Popovici, S.E.; Bedreag, O.H. Advances in Biomarkers in Critical Ill Polytrauma Patients. Clin Lab. 2016, 62, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Kawata, K.; Liu, C.Y.; Merkel, S.F.; Ramirez, S.H.; Tierney, R.T.; Langford, D. Blood biomarkers for brain injury: What are we measuring? Neurosci. Biobehav. Rev. 2016, 68, 460–473. [Google Scholar] [CrossRef]
- Da Rocha, A.B.; Schneider, R.F.; de Freitas, G.R.; Andre, C.; Grivicich, I.; Zanoni, C.; Fossa, A.; Gehrke, J.T.; Pereira Jotz, G.; Kaufmann, M.; et al. Role of serum S100B as a predictive marker of fatal outcome following isolated severe head injury or multitrauma in males. Clin. Chem. Lab. Med. 2006, 44, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Regner, A.; Kaufman, M.; Friedman, G.; Chemale, I. Increased serum S100β protein concentrations following severe head injury in humans: A biochemical marker of brain death? Neuroreport 2001, 12, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Nascimento, R.I.; Filho, E.M.; Bencke, J.; Regner, A. Plasma brain-derived neurotrophic factor levels after severe traumatic brain injury. Brain Inj. 2016, 30, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Nicol, J.M.; Sabino da Silva, S.; Graziottin, C.; Silveira, P.C.; Ikuta, N.; Regner, A. Serum ferritin correlates with Glasgow coma scale scores and fatal outcome after severe traumatic brain injury. Brain Inj. 2015, 29, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.C.; Regner, A.; Miotto, K.D.; Moura, S.; Ikuta, N.; Vargas, A.E.; Chies, J.A.; Simon, D. Increased levels of interleukin-6, -8 and -10 are associated with fatal outcome following severe traumatic brain injury. Brain Inj. 2014, 28, 1311–1316. [Google Scholar] [CrossRef]
- Rodrigues Filho, E.M.; Simon, D.; Ikuta, N.; Klovan, C.; Dannebrock, F.A.; Oliveira de Oliveira, C.; Regner, A. Elevated cell-free plasma DNA level as an independent predictor of mortality in patients with severe traumatic brain injury. J. Neurotrauma 2014, 31, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, C.O.; Reimer, A.G.; Da Rocha, A.B.; Grivicich, I.; Schneider, R.F.; Roisenberg, I.; Regner, A.; Simon, D. Plasma von Willebrand factor levels correlate with clinical outcome of severe traumatic brain injury. J. Neurotrauma 2007, 24, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Crespo, A.R.; da Rocha, A.B.; Jotz, G.P.; Schneider, R.F.; Grivicich, I.; Pinheiro, K.; Zanoni, C.; Regner, A. Increased serum sFas and TNFα following isolated severe head injury in males. Brain Inj. 2007, 21, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, A.B.; Zanoni, C.; de Freitas, G.R.; Andre, C.; Himelfarb, S.; Schneider, R.F.; Grivicich, I.; Borges, L.; Schwartsmann, G.; Kaufmann, M.; et al. Serum Hsp70 as an early predictor of fatal outcome after severe traumatic brain injury in males. J. Neurotrauma 2005, 22, 966–977. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, C.E.; de Sousa Filho, J.L.; Dourado, J.C.; Gontijo, P.A.; Dellaretti, M.A.; Costa, B.S. Traumatic Brain Injury Epidemiology in Brazil. World Neurosurg. 2016, 87, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Hawryluk, G.W.; Bullock, M.R. Past, Present, and Future of Traumatic Brain Injury Research. Neurosurg. Clin. N. Am. 2016, 27, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Fountain, D.M.; Kolias, A.G.; Laing, R.J.; Hutchinson, P.J. The financial outcome of traumatic brain injury: A single centre study. Br. J. Neurosurg. 2016, 1–6. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 45–66. [Google Scholar] [PubMed]
- Kinoshita, K. Traumatic brain injury: Pathophysiology for neurocritical care. J. Intensive Care 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, J. Traumatic brain injury. Lancet 2000, 356, 923–929. [Google Scholar] [CrossRef]
- Krishnamurthy, K.; Laskowitz, D.T. Cellular and Molecular Mechanisms of Secondary Neuronal Injury. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2016. [Google Scholar]
- Plummer, S.; Van den Heuvel, C.; Thornton, E.; Corrigan, F.; Cappai, R. The Neuroprotective Properties of the Amyloid Precursor Protein Following Traumatic Brain Injury. Aging Dis. 2016, 7, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Y.; Jenkins, L.W.; Kochanek, P.M.; Clark, R.S. Bench-to-bedside review: Apoptosis/programmed cell death triggered by traumatic brain injury. Crit Care 2005, 9, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Baeza, A.; Reina-de la Torre, F.; Poca, A.; Marti, M.; Garnacho, A. Morphological features in human cortical brain microvessels after head injury: A three-dimensional and immunocytochemical study. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2003, 273, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Vajtr, D.; Benada, O.; Kukacka, J.; Prusa, R.; Houstava, L.; Toupalik, P.; Kizek, R. Correlation of ultrastructural changes of endothelial cells and astrocytes occurring during blood brain barrier damage after traumatic brain injury with biochemical markers of BBB leakage and inflammatory response. Physiol. Res. 2009, 58, 263–268. [Google Scholar] [PubMed]
- Da Fonseca, A.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell Neurosci. 2014, 8, 362. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Minter, D.; Yue, J.K.; Manley, G.T. Cerebral Edema in Traumatic Brain Injury: Pathophysiology and Prospective Therapeutic Targets. Neurosurg. Clin. N. Am. 2016, 27, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg. Focus 2007, 22, E1. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, V.N.; Lifshitz, J.; Adelson, P.D.; Kodibagkar, V.D.; Stabenfeldt, S.E. Temporal assessment of nanoparticle accumulation after experimental brain injury: Effect of particle size. Sci. Rep. 2016, 6, 29988. [Google Scholar] [CrossRef] [PubMed]
- Algattas, H.; Huang, J.H. Traumatic Brain Injury pathophysiology and treatments: Early, intermediate, and late phases post-injury. Int. J. Mol. Sci. 2013, 15, 309–341. [Google Scholar] [CrossRef] [PubMed]
- Buitrago Blanco, M.M.; Prashant, G.N.; Vespa, P.M. Cerebral Metabolism and the Role of Glucose Control in Acute Traumatic Brain Injury. Neurosurg. Clin. N. Am. 2016, 27, 453–463. [Google Scholar] [CrossRef] [PubMed]
- McGinn, M.J.; Povlishock, J.T. Pathophysiology of Traumatic Brain Injury. Neurosurg. Clin. N. Am. 2016, 27, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Baez, E.; Echeverria, V.; Cabezas, R.; Avila-Rodriguez, M.; Garcia-Segura, L.M.; Barreto, G.E. Protection by Neuroglobin Expression in Brain Pathologies. Front. Neurol. 2016, 7, 146. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, J.M.; Morganti-Kossmann, M.C. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Castejon, O.J. Biopathology of astrocytes in human traumatic and complicated brain injuries. Review and hypothesis. Folia Neuropathol. 2015, 53, 173–192. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, M.; Eriskat, J.; Plesnila, M.; Aggarwal, N.; Baethmann, A. The penumbra zone of a traumatic cortical lesion: A microdialysis study of excitatory amino acid release. Acta Neurochir. Suppl. 1997, 70, 91–93. [Google Scholar] [PubMed]
- Harish, G.; Mahadevan, A.; Pruthi, N.; Sreenivasamurthy, S.K.; Puttamallesh, V.N.; Keshava Prasad, T.S.; Shankar, S.K.; Srinivas Bharath, M.M. Characterization of traumatic brain injury in human brains reveals distinct cellular and molecular changes in contusion and pericontusion. J. Neurochem. 2015, 134, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, M.; Rinecker, M.; Graf, R.; Baethmann, A.; Plesnila, N. Nitric oxide in the penumbra of a focal cortical necrosis in rats. Neurosci. Lett. 2002, 324, 201–204. [Google Scholar] [CrossRef]
- Newcombe, V.F.; Williams, G.B.; Outtrim, J.G.; Chatfield, D.; Gulia Abate, M.; Geeraerts, T.; Manktelow, A.; Room, H.; Mariappen, L.; Hutchinson, P.J.; et al. Microstructural basis of contusion expansion in traumatic brain injury: Insights from diffusion tensor imaging. J. Cereb. Blood Flow Metab. 2013, 33, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Huang, S.C.; Vespa, P.; Hovda, D.A.; Bergsneider, M. Redefining the pericontusional penumbra following traumatic brain injury: Evidence of deteriorating metabolic derangements based on positron emission tomography. Neurotrauma 2013, 30, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Sheriff, F.G.; Hinson, H.E. Pathophysiology and clinical management of moderate and severe traumatic brain injury in the ICU. Semin. Neurol. 2015, 35, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.V.; Maas, A.I.; Bragge, P.; Morganti-Kossmann, M.C.; Manley, G.T.; Gruen, R.L. Early management of severe traumatic brain injury. Lancet 2012, 380, 1088–1098. [Google Scholar] [CrossRef]
- Ding, K.; Wang, H.; Wu, Y.; Zhang, L.; Xu, J.; Li, T.; Ding, Y.; Zhu, L.; He, J. Rapamycin protects against apoptotic neuronal death and improves neurologic function after traumatic brain injury in mice via modulation of the mTOR-p53-Bax axis. J. Surg. Res. 2015, 194, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Kong, R.H.; Zhang, L.M.; Zhang, J.N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef] [PubMed]
- Regner, A.; Alves, L.B.; Chemale, I.; Costa, M.S.; Friedman, G.; Achaval, M.; Leal, L.; Emanuelli, T. Neurochemical characterization of traumatic brain injury in humans. J. Neurotrauma 2001, 18, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.; Zauner, A.; Woodward, J.J.; Myseros, J.; Choi, S.C.; Ward, J.D.; Marmarou, A.; Young, H.F. Factors affecting excitatory amino acid release following severe human head injury. J. Neurosurg. 1998, 89, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Pow, D.V.; Hazell, A.S. Early loss of the glutamate transporter splice-variant GLT-1v in rat cerebral cortex following lateral fluid-percussion injury. Glia 2005, 49, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Katayama, Y.; Hovda, D.A.; Yoshino, A.; Becker, D.P. Administration of excitatory amino acid antagonists via microdialysis attenuates the increase in glucose utilization seen following concussive brain injury. J. Cereb. Blood Flow Metab. 1992, 12, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Bergsneider, M.; Hovda, D.A.; Shalmon, E.; Kelly, D.F.; Vespa, P.M.; Martin, N.A.; Phelps, M.E.; McArthur, D.L.; Caron, M.J.; Kraus, J.F.; et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: A positron emission tomography study. J. Neurosurg. 1997, 86, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Bergsneider, M.; Hovda, D.A.; Lee, S.M.; Kelly, D.F.; McArthur, D.L.; Vespa, P.M.; Lee, J.H.; Huang, S.C.; Martin, N.A.; Phelps, M.E.; et al. Dissociation of cerebral glucose metabolism and level of consciousness during the period of metabolic depression following human traumatic brain injury. J. Neurotrauma 2000, 17, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Bergsneider, M.; Hovda, D.A.; McArthur, D.L.; Etchepare, M.; Huang, S.C.; Sehati, N.; Satz, P.; Phelps, M.E.; Becker, D.P. Metabolic recovery following human traumatic brain injury based on FDG-PET: Time course and relationship to neurological disability. J. Head. Trauma Rehabil. 2001, 16, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Huang, S.C.; Hattori, N.; Glenn, T.C.; Vespa, P.M.; Hovda, D.A.; Bergsneider, M. Subcortical white matter metabolic changes remote from focal hemorrhagic lesions suggest diffuse injury after human traumatic brain injury. Neurosurgery 2004, 55, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, A.; Hovda, D.A.; Kawamata, T.; Katayama, Y.; Becker, D.P. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: Evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991, 561, 106–119. [Google Scholar] [CrossRef]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Vink, R.; Nimmo, A.J. Novel therapies in development for the treatment of traumatic brain injury. Expert Opin. Investig. Drugs 2002, 11, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.; Koopmans, G.; Kobbe, P.; Poeze, M.; Andruszkow, H.; Brink, P.R.; Pape, H.C. Improved fracture healing in patients with concomitant traumatic brain injury: Proven or not? Mediat. Inflamm. 2015, 2015, 204842. [Google Scholar] [CrossRef] [PubMed]
- Saatman, K.E.; Creed, J.; Raghupathi, R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics 2010, 7, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.T. Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 2012, 3, 60. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Lewen, A.; Matz, P.; Chan, P.H. Free radical pathways in CNS injury. J. Neurotrauma 2000, 17, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Braughler, J.M.; Hall, E.D. Involvement of lipid peroxidation in CNS injury. J. Neurotrauma 1992, 9, S1–S7. [Google Scholar] [PubMed]
- Halestrap, A.P.; Woodfield, K.Y.; Connern, C.P. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J. Biol. Chem. 1997, 272, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Toklu, H.Z.; Tumer, N. Oxidative Stress, Brain Edema, Blood-Brain Barrier Permeability, and Autonomic Dysfunction from Traumatic Brain Injury. In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; Kobeissy, F.H., Ed.; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2015. [Google Scholar]
- Calabrese, V.; Cornelius, C.; Mancuso, C.; Pennisi, G.; Calafato, S.; Bellia, F.; Bates, T.E.; Giuffrida Stella, A.M.; Schapira, T.; Dinkova Kostova, A.T.; et al. Cellular stress response: A novel target for chemoprevention and nutritional neuroprotection in aging, neurodegenerative disorders and longevity. Neurochem. Res. 2008, 33, 2444–2471. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, B.; Signoretti, S.; Lazzarino, G.; Amorini, A.M.; Delfini, R.; Cimatti, M.; Marmarou, A.; Vagnozzi, R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 2005, 56, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Maciel, E.N.; Vercesi, A.E.; Castilho, R.F. Oxidative stress in Ca2+-induced membrane permeability transition in brain mitochondria. J. Neurochem. 2001, 79, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Rockswold, S.B.; Rockswold, G.L.; Defillo, A. Hyperbaric oxygen in traumatic brain injury. Neurol. Res. 2007, 29, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Mathew, P.; Graham, D.I.; Bullock, R.; Maxwell, W.; McCulloch, J.; Teasdale, G. Focal brain injury: Histological evidence of delayed inflammatory response in a new rodent model of focal cortical injury. Acta Neurochir. Suppl. 1994, 60, 428–430. [Google Scholar] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia activation as a biomarker for traumatic brain injury. Front. Neurol. 2013, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Parekkadan, B.; Berdichevsky, Y.; Irimia, D.; Leeder, A.; Yarmush, G.; Toner, M.; Levine, J.B.; Yarmush, M.L. Cell-cell interaction modulates neuroectodermal specification of embryonic stem cells. Neurosci. Lett. 2008, 438, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Liao, Y.E.; Yang, L.Y.; Wang, J.Y.; Tweedie, D.; Karnati, H.K.; Greig, N.H.; Wang, J.Y. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Witcher, K.G.; Eiferman, D.S.; Godbout, J.P. Priming the inflammatory pump of the CNS after traumatic brain injury. Trends Neurosci. 2015, 38, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar] [PubMed]
- Liao, Y.; Liu, P.; Guo, F.; Zhang, Z.Y.; Zhang, Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS ONE 2013, 8, e68963. [Google Scholar] [CrossRef]
- Yu, C.H.; Yhee, J.Y.; Kim, J.H.; Im, K.S.; Kim, N.H.; Jung, D.I.; Lee, H.C.; Chon, S.K.; Sur, J.H. Pro- and anti-inflammatory cytokine expression and histopathological characteristics in canine brain with traumatic brain injury. J. Vet. Sci. 2011, 12, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Csuka, E.; Morganti-Kossmann, M.C.; Lenzlinger, P.M.; Joller, H.; Trentz, O.; Kossmann, T. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: Relationship to IL-6, TNF-α, TGF-β1 and blood-brain barrier function. J. Neuroimmunol. 1999, 101, 211–221. [Google Scholar] [CrossRef]
- Fassbender, K.; Schneider, S.; Bertsch, T.; Schlueter, D.; Fatar, M.; Ragoschke, A.; Kuhl, S.; Kischka, U.; Hennerici, M. Temporal profile of release of interleukin-1β in neurotrauma. Neurosci. Lett. 2000, 284, 135–138. [Google Scholar] [CrossRef]
- Maier, B.; Schwerdtfeger, K.; Mautes, A.; Holanda, M.; Muller, M.; Steudel, W.I.; Marzi, I. Differential release of interleukines 6, 8, and 10 in cerebrospinal fluid and plasma after traumatic brain injury. Shock 2001, 15, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Lenzlinger, P.M.; Morganti-Kossmann, M.C.; Laurer, H.L.; McIntosh, T.K. The duality of the inflammatory response to traumatic brain injury. Mol. Neurobiol. 2001, 24, 169–181. [Google Scholar] [PubMed]
- Frugier, T.; Morganti-Kossmann, M.C.; O’Reilly, D.; McLean, C.A. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J. Neurotrauma 2010, 27, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.I.; Heyde, C.E.; Ertel, W.; Stahel, P.F. Closed head injury—An inflammatory disease? Brain Res. Brain Res. Rev. 2005, 48, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Ralay Ranaivo, H.; Zunich, S.M.; Choi, N.; Hodge, J.N.; Wainwright, M.S. Mild stretch-induced injury increases susceptibility to interleukin-1β-induced release of matrix metalloproteinase-9 from astrocytes. J. Neurotrauma 2011, 28, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Jenne, C.N.; Leger, C.; Kramer, A.H.; Gallagher, C.N.; Todd, S.; Parney, I.F.; Doig, C.J.; Yong, V.W.; Kubes, P.; et al. Association between the cerebral inflammatory and matrix metalloproteinase responses after severe traumatic brain injury in humans. J. Neurotrauma 2013, 30, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, F.; Campbell, M. Neuroinflammation. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2016. [Google Scholar]
- Lingsma, H.F.; Yue, J.K.; Maas, A.I.; Steyerberg, E.W.; Manley, G.T.; Investigators, T.-T. Outcome prediction after mild and complicated mild traumatic brain injury: External validation of existing models and identification of new predictors using the TRACK-TBI pilot study. J. Neurotrauma 2015, 32, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D. Translational Principles of Neuroprotective and Neurorestorative Therapy Testing in Animal Models of Traumatic Brain Injury. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2016. [Google Scholar]
- Kochanek, P.M.; Jackson, T.C.; Ferguson, N.M.; Carlson, S.W.; Simon, D.W.; Brockman, E.C.; Ji, J.; Bayir, H.; Poloyac, S.M.; Wagner, A.K.; et al. Emerging therapies in traumatic brain injury. Semin. Neurol. 2015, 35, 83–100. [Google Scholar] [PubMed]
- Jablonska, A.; Lukomska, B. Stroke induced brain changes: Implications for stem cell transplantation. Acta Neurobiol. Exp. 2011, 71, 74–85. [Google Scholar]
- Guan, J.; Gluckman, P.D. IGF-1 derived small neuropeptides and analogues: A novel strategy for the development of pharmaceuticals for neurological conditions. Br. J. Pharmacol. 2009, 157, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Madathil, S.K.; Evans, H.N.; Saatman, K.E. Temporal and regional changes in IGF-1/IGF-1R signaling in the mouse brain after traumatic brain injury. J. Neurotrauma 2010, 27, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Thau-Zuchman, O.; Shohami, E.; Alexandrovich, A.G.; Leker, R.R. Vascular endothelial growth factor increases neurogenesis after traumatic brain injury. J. Cereb. Blood Flow Metab. 2010, 30, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, A.; Sun, D. Stem Cell Therapy in Brain Trauma: Implications for Repair and Regeneration of Injured Brain in Experimental TBI Models. In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; Kobeissy, F.H., Ed.; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2015. [Google Scholar]
- Madathil, S.K.; Saatman, K.E. IGF-1/IGF-R Signaling in Traumatic Brain Injury: Impact on Cell Survival, Neurogenesis, and Behavioral Outcome. In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; Kobeissy, F.H., Ed.; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2015. [Google Scholar]
- Conte, V.; Royo, N.C.; Shimizu, S.; Saatman, K.E.; Watson, D.J.; Graham, D.I.; Stocchetti, N.; McIntosh, T.K. Neurotrophic Factors : Pathophysiology and Therapeutic Applications in Traumatic Brain Injury. Eur. J. Trauma 2003, 29, 335–355. [Google Scholar]
- Sophie Su, Y.R.; Veeravagu, A.; Grant, G. Neuroplasticity after Traumatic Brain Injury. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2016. [Google Scholar]
- Griesbach, G.S.; Hovda, D.A. Cellular and molecular neuronal plasticity. Handb. Clin. Neurol. 2015, 128, 681–690. [Google Scholar] [PubMed]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Josephy-Hernandez, S.; Jmaeff, S.; Pirvulescu, I.; Aboulkassim, T.; Saragovi, H.U. Neurotrophin receptor agonists and antagonists as therapeutic agents: An evolving paradigm. Neurobiol. Dis. 2017, 97, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Schecterson, L.C.; Bothwell, M. Neurotrophin receptors: Old friends with new partners. Dev. Neurobiol. 2010, 70, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Barker, P.A. p75NTR is positively promiscuous: Novel partners and new insights. Neuron 2004, 42, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Pang, P.T.; Woo, N.H. The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 2005, 6, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.; Fujioka, W.; Giarratana, A.; Wissocki, J.; Thakkar, K.; Vuong, P.; Patel, B.; Chakraborty, T.; Elsabeh, R.; Parikh, A.; et al. Genetic and pharmacological intervention of the p75NTR pathway alters morphological and behavioural recovery following traumatic brain injury in mice. Brain Inj. 2016, 30, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, S.G.; Renn, C.L.; Carim-Todd, L.; Barrick, C.A.; Bambrick, L.; Krueger, B.K.; Ward, C.W.; Tessarollo, L. In vivo restoration of physiological levels of truncated TrkB.T1 receptor rescues neuronal cell death in a trisomic mouse model. Neuron 2006, 51, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Shi, Z.; Zhuo, Y.; Liu, J.; Malakhov, A.; Ko, E.; Burgess, K.; Schaefer, H.; Esteban, P.F.; Tessarollo, L.; et al. In glaucoma the upregulated truncated TrkC.T1 receptor isoform in glia causes increased TNF-α production, leading to retinal ganglion cell death. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6639–6651. [Google Scholar] [CrossRef] [PubMed]
- Yanpallewar, S.U.; Barrick, C.A.; Buckley, H.; Becker, J.; Tessarollo, L. Deletion of the BDNF truncated receptor TrkB.T1 delays disease onset in a mouse model of amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e39946. [Google Scholar] [CrossRef] [PubMed]
- Saragovi, H.U.; Gehring, K. Development of pharmacological agents for targeting neurotrophins and their receptors. Trends Pharmacol. Sci. 2000, 21, 93–98. [Google Scholar] [CrossRef]
- Saragovi, H.U.; Hamel, E.; di Polo, A. A neurotrophic rationale for the therapy of neurodegenerative disorders. Curr. Alzheimer Res. 2009, 6, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Moris, G.; Vega, J.A. Neurotrophic factors: Basis for their clinical application. Neurologia 2003, 18, 18–28. [Google Scholar] [PubMed]
- McAllister, A.K.; Katz, L.C.; Lo, D.C. Neurotrophins and synaptic plasticity. Annu. Rev. Neurosci. 1999, 22, 295–318. [Google Scholar] [CrossRef] [PubMed]
- Oyesiku, N.M.; Evans, C.O.; Houston, S.; Darrell, R.S.; Smith, J.S.; Fulop, Z.L.; Dixon, C.E.; Stein, D.G. Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res. 1999, 833, 161–172. [Google Scholar] [CrossRef]
- Kim, D.H.; Zhao, X. BDNF protects neurons following injury by modulation of caspase activity. Neurocrit. Care 2005, 3, 71–76. [Google Scholar] [CrossRef]
- Shulga, A.; Thomas-Crusells, J.; Sigl, T.; Blaesse, A.; Mestres, P.; Meyer, M.; Yan, Q.; Kaila, K.; Saarma, M.; Rivera, C.; et al. Posttraumatic GABA(A)-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J. Neurosci. 2008, 28, 6996–7005. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lu, D.; Jiang, H.; Xiong, Y.; Qu, C.; Li, B.; Mahmood, A.; Zhou, D.; Chopp, M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J. Neurotrauma 2008, 25, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Griesbach, G.S.; Sutton, R.L.; Hovda, D.A.; Ying, Z.; Gomez-Pinilla, F. Controlled contusion injury alters molecular systems associated with cognitive performance. J. Neurosci. Res. 2009, 87, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Felderhoff-Mueser, U.; Sifringer, M.; Pesditschek, S.; Kuckuck, H.; Moysich, A.; Bittigau, P.; Ikonomidou, C. Pathways leading to apoptotic neurodegeneration following trauma to the developing rat brain. Neurobiol. Dis. 2002, 11, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.K.; Rao, M.S.; Hattiangady, B.; Zaman, V.; Shetty, G.A. Hippocampal neurotrophin levels after injury: Relationship to the age of the hippocampus at the time of injury. J. Neurosci. Res. 2004, 78, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, A.; Piastra, M.; Polidori, G.; di Rocco, C.; Caresta, E.; Antonelli, A.; Amendola, T.; Aloe, L. Correlation between neurotrophic factor expression and outcome of children with severe traumatic brain injury. Intensive Care Med. 2003, 29, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Failla, M.D.; Conley, Y.P.; Wagner, A.K. Brain-Derived Neurotrophic Factor (BDNF) in Traumatic Brain Injury-Related Mortality: Interrelationships between Genetics and Acute Systemic and Central Nervous System BDNF Profiles. Neurorehabil. Neural Repair. 2016, 30, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Korley, F.K.; Diaz-Arrastia, R.; Wu, A.H.; Yue, J.K.; Manley, G.T.; Sair, H.I.; Van Eyk, J.; Everett, A.D.; TRACK-TBI investigators; Okonkwo, D.O.; et al. Circulating Brain-Derived Neurotrophic Factor Has Diagnostic and Prognostic Value in Traumatic Brain Injury. J. Neurotrauma 2016, 33, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, A.; Antonelli, A.; Riccardi, R.; Genovese, O.; Pezzotti, P.; di Rocco, C.; Tortorolo, L.; Piedimonte, G. Nerve growth factor expression correlates with severity and outcome of traumatic brain injury in children. Eur. J. Paediatr. Neurol. 2008, 12, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Koshimizu, H.; Hazama, S.; Hara, T.; Ogura, A.; Kojima, M. Distinct signaling pathways of precursor BDNF and mature BDNF in cultured cerebellar granule neurons. Neurosci. Lett. 2010, 473, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Lewin, G.R.; Carter, B.D. Neurotrophic factors. Preface. Handb. Exp. Pharmacol. 2014, 220, v–vi. [Google Scholar] [PubMed]
- Chiaretti, A.; Antonelli, A.; Mastrangelo, A.; Pezzotti, P.; Tortorolo, L.; Tosi, F.; Genovese, O. Interleukin-6 and nerve growth factor upregulation correlates with improved outcome in children with severe traumatic brain injury. J. Neurotrauma 2008, 25, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, A.; Barone, G.; Riccardi, R.; Antonelli, A.; Pezzotti, P.; Genovese, O.; Tortorolo, L.; Conti, G. NGF, DCX, and NSE upregulation correlates with severity and outcome of head trauma in children. Neurology 2009, 72, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, A.; Antonelli, A.; Genovese, O.; Pezzotti, P.; Rocco, C.D.; Viola, L.; Riccardi, R. Nerve growth factor and doublecortin expression correlates with improved outcome in children with severe traumatic brain injury. J. Trauma 2008, 65, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Kromer, L.F. Nerve growth factor treatment after brain injury prevents neuronal death. Science 1987, 235, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Howe, C.L.; Mobley, W.C. Nerve growth factor signaling, neuroprotection, and neural repair. Annu. Rev. Neurosci. 2001, 24, 1217–1281. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Mattson, M.P. NT-3 and BDNF protect CNS neurons against metabolic/excitotoxic insults. Brain Res. 1994, 640, 56–67. [Google Scholar] [CrossRef]
- Yang, J.T.; Lee, T.H.; Weng, H.H.; Chang, C.N.; Chen, W.C.; Cheng, W.C.; Wu, J.H. Dexamethasone enhances NT-3 expression in rat hippocampus after traumatic brain injury. Exp. Neurol. 2005, 192, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, J.H.; Kim, S.H. Therapeutic effects of human mesenchymal stem cells on traumatic brain injury in rats: Secretion of neurotrophic factors and inhibition of apoptosis. J. Neurotrauma 2010, 27, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.M.; Lee, S.M.; Kim, M.H. Spontaneous Wheel Running Exercise Induces Brain Recovery via Neurotrophin-3 Expression Following Experimental Traumatic Brain Injury in Rats. J. Phys. Ther. Sci. 2013, 25, 1103–1107. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yan, Z.J.; Zhang, P.; Hu, Y.Q.; Zhang, H.T.; Hong, S.Q.; Zhou, H.L.; Zhang, M.Y.; Xu, R.X. Neural stem-like cells derived from human amnion tissue are effective in treating traumatic brain injury in rat. Neurochem. Res. 2013, 38, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Scheff, S.W. Endogenous neuroprotection factors and traumatic brain injury: Mechanisms of action and implications for therapy. J. Neurotrauma 1994, 11, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Alexi, T.; Hefti, F. Neurotrophin-4/5 selectively protects nigral calbindin-containing neurons in rats with medial forebrain bundle transections. Neuroscience 1996, 72, 911–921. [Google Scholar] [CrossRef]
- Blesch, A.; Yang, H.; Weidner, N.; Hoang, A.; Otero, D. Axonal responses to cellularly delivered NT-4/5 after spinal cord injury. Mol. Cell. Neurosci. 2004, 27, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Haque, N.S.; Hlavin, M.L.; Fawcett, J.W.; Dunnett, S.B. The neurotrophin NT4/5, but not NT3, enhances the efficacy of nigral grafts in a rat model of Parkinson’s disease. Brain Res. 1996, 712, 45–52. [Google Scholar] [CrossRef]
- Sawai, H.; Clarke, D.B.; Kittlerova, P.; Bray, G.M.; Aguayo, A.J. Brain-derived neurotrophic factor and neurotrophin-4/5 stimulate growth of axonal branches from regenerating retinal ganglion cells. J. Neurosci. 1996, 16, 3887–3894. [Google Scholar] [PubMed]
- Perez-Navarro, E.; Canudas, A.M.; Akerund, P.; Alberch, J.; Arenas, E. Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 prevent the death of striatal projection neurons in a rodent model of Huntington’s disease. J. Neurochem. 2000, 75, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Royo, N.C.; Conte, V.; Saatman, K.E.; Shimizu, S.; Belfield, C.M.; Soltesz, K.M.; Davis, J.E.; Fujimoto, S.T.; McIntosh, T.K. Hippocampal vulnerability following traumatic brain injury: A potential role for neurotrophin-4/5 in pyramidal cell neuroprotection. Eur. J. Neurosci. 2006, 23, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Kalish, H.; Phillips, T.M. Analysis of neurotrophins in human serum by immunoaffinity capillary electrophoresis (ICE) following traumatic head injury. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, A.; Genovese, O.; Riccardi, R.; Di Rocco, C.; di Giuda, D.; Mariotti, P.; et al. Intraventricular nerve growth factor infusion: A possible treatment for neurological deficits following hypoxic-ischemic brain injury in infants. Neurol. Res. 2005, 27, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Fantacci, C.; Capozzi, D.; Ferrara, P.; Chiaretti, A. Neuroprotective role of nerve growth factor in hypoxic-ischemic brain injury. Brain Sci. 2013, 3, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.; Cusimano, M.D.; Bendena, W.G. Post-Traumatic Brain Injury: Genetic Susceptibility to Outcome. Neuroscientist 2015, 21, 424–441. [Google Scholar] [CrossRef] [PubMed]
- Failla, M.D.; Kumar, R.G.; Peitzman, A.B.; Conley, Y.P.; Ferrell, R.E.; Wagner, A.K. Variation in the BDNF gene interacts with age to predict mortality in a prospective, longitudinal cohort with severe TBI. Neurorehabil. Neural Repair. 2015, 29, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef]
- Krueger, F.; Pardini, M.; Huey, E.D.; Raymont, V.; Solomon, J.; Lipsky, R.H.; Hodgkinson, C.A.; Goldman, D.; Grafman, J. The role of the Met66 brain-derived neurotrophic factor allele in the recovery of executive functioning after combat-related traumatic brain injury. J. Neurosci. 2011, 31, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Rostami, E.; Krueger, F.; Zoubak, S.; Dal Monte, O.; Raymont, V.; Pardini, M.; Hodgkinson, C.A.; Goldman, D.; Risling, M.; Grafman, J. BDNF polymorphism predicts general intelligence after penetrating traumatic brain injury. PLoS ONE 2011, 6, e27389. [Google Scholar] [CrossRef] [PubMed]
- Barbey, A.K.; Colom, R.; Paul, E.; Forbes, C.; Krueger, F.; Goldman, D.; Grafman, J. Preservation of general intelligence following traumatic brain injury: Contributions of the Met66 brain-derived neurotrophic factor. PLoS ONE 2014, 9, e88733. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, S.; Minafra, L.; Bravata, V.; Boccagni, C.; Sant′angelo, A.; Castiglione, A.; Andriolo, M.; Lucca, L.F.; De Tanti, A.; Pistarini, C.; et al. Brain-derivedneurotrophic factor (Val66Met) polymorphism does not influence recovery from a post-traumatic vegetative state: A blinded retrospective multi-centric study. J. Neurotrauma 2012, 29, 2050–2059. [Google Scholar] [CrossRef] [PubMed]
- Bronfman, F.C.; Fainzilber, M. Multi-tasking by the p75 neurotrophin receptor: Sortilin things out? EMBO Rep. 2004, 5, 867–871. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gentry, J.J.; Barker, P.A.; Carter, B.D. The p75 neurotrophin receptor: Multiple interactors and numerous functions. Prog. Brain Res. 2004, 146, 25–39. [Google Scholar] [PubMed]
- Chen, L.W.; Zhang, J.P.; Kwok-Yan Shum, D.; Chan, Y.S. Localization of nerve growth factor, neurotrophin-3, and glial cell line-derived neurotrophic factor in nestin-expressing reactive astrocytes in the caudate-putamen of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated C57/Bl mice. J. Comp. Neurol. 2006, 497, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, M.; Matute, C. Expression of nerve growth factor in astrocytes of the hippocampal CA1 area following transient forebrain ischemia. Neuroscience 1999, 91, 1027–1034. [Google Scholar] [CrossRef]
- Lee, T.H.; Kato, H.; Chen, S.T.; Kogure, K.; Itoyama, Y. Expression of nerve growth factor and trkA after transient focal cerebral ischemia in rats. Stroke 1998, 29, 1687–1696. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.L.; Escartin, C.; Ayata, C.; Bonvento, G.; Shuttleworth, C.W. Multifaceted roles for astrocytes in spreading depolarization: A target for limiting spreading depolarization in acute brain injury? Glia 2016, 64, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Kurkowska-Jastrzebska, I. Inflammation and gliosis in neurological diseases—Clinical implications. J. Neuroimmunol. 2011, 231, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Kerschensteiner, M.; Gallmeier, E.; Behrens, L.; Leal, V.V.; Misgeld, T.; Klinkert, W.E.; Kolbeck, R.; Hoppe, E.; Oropeza-Wekerle, R.L.; Bartke, I.; et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: A neuroprotective role of inflammation? J. Exp. Med. 1999, 189, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Betsholtz, C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003, 314, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, R.; Yamashita, T. Pericyte function in the physiological central nervous system. Neurosci. Res. 2014, 81, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Edelman, D.A.; Jiang, Y.; Tyburski, J.G.; Wilson, R.F.; Steffes, C.P. Cytokine production in lipopolysaccharide-exposed rat lung pericytes. J. Trauma 2007, 62, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Jansson, D.; Smyth, L.C.; Dragunow, M. Brain Pericytes As Mediators of Neuroinflammation. Trends Pharmacol. Sci. 2016, 38, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Navarro, R.; Compte, M.; Alvarez-Vallina, L.; Sanz, L. Immune Regulation by Pericytes: Modulating Innate and Adaptive Immunity. Front. Immunol. 2016, 7, 480. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lauridsen, H.M.; Amezquita, R.A.; Pierce, R.W.; Jane-Wit, D.; Fang, C.; Pellowe, A.S.; Kirkiles-Smith, N.C.; Gonzalez, A.L.; Pober, J.S. IL-17 Promotes Neutrophil-Mediated Immunity by Activating Microvascular Pericytes and Not Endothelium. J. Immunol. 2016, 197, 2400–2408. [Google Scholar] [CrossRef] [PubMed]
- da Silva Meirelles, L.; Malta, T.M.; de Deus Wagatsuma, V.M.; Palma, P.V.; Araujo, A.G.; Ribeiro Malmegrim, K.C.; et al. Cultured Human Adipose Tissue Pericytes and Mesenchymal Stromal Cells Display a Very Similar Gene Expression Profile. Stem Cells Dev. 2015, 24, 2822–2840. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Li, Y.; Smith, D.S.; Sheibani, N.; Huang, S.; Kern, T.; et al. Retinal pericytes inhibit activated T cell proliferation. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9005–9010. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Meirelles, L.; Caplan, A.I.; Nardi, N.B. In search of the in vivo identity of mesenchymal stem cells. Stem Cells 2008, 26, 2287–2299. [Google Scholar] [CrossRef] [PubMed]
- da Silva Meirelles, L.; Bellagamba, B.C.; Camassola, M.; Nardi, N.B. Mesenchymal stem cells and their relationship to pericytes. Front. Biosci. 2016, 21, 130–156. [Google Scholar] [CrossRef]
- Caplan, A.I. What‘s in a name? Tissue Eng. Part A 2010, 16, 2415–2417. [Google Scholar] [CrossRef] [PubMed]
- Dore-Duffy, P.; Wang, S.; Mehedi, A.; Katyshev, V.; Cleary, K.; Tapper, A.; Reynolds, C.; Ding, Y.; Zhan, P.; Rafols, J.; et al. Pericyte-mediated vasoconstriction underlies TBI-induced hypoperfusion. Neurol Res. 2011, 33, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Dore-Duffy, P.; Owen, C.; Balabanov, R.; Murphy, S.; Beaumont, T.; Rafols, J.A. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc. Res. 2000, 60, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Zehendner, C.M.; Sebastiani, A.; Hugonnet, A.; Bischoff, F.; Luhmann, H.J.; Thal, S.C. Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Sci. Rep. 2015, 5, 13497. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Sano, Y.; Abe, M.A.; Maeda, T.; Ohtsuki, S.; Terasaki, T.; Kanda, T. Peripheral nerve pericytes modify the blood-nerve barrier function and tight junctional molecules through the secretion of various soluble factors. J. Cell. Physiol. 2011, 226, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, K.; Ago, T.; Arimura, K.; Nakamura, K.; Tokami, H.; Makihara, N.; Kuroda, J.; Kamouchi, M.; Kitazono, T. Neurotrophin production in brain pericytes during hypoxia: A role of pericytes for neuroprotection. Microvasc. Res. 2012, 83, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Meirelles, L.; de Deus Wagatsuma, V.M.; Malta, T.M.; Bonini Palma, P.V.; Araujo, A.G.; Panepucci, R.A.; Silva, W.A.; Kashima, S.; Covas, D.T. The gene expression profile of non-cultured, highly purified human adipose tissue pericytes: Transcriptomic evidence that pericytes are stem cells in human adipose tissue. Exp. Cell Res. 2016, 349, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Chen, H.; Zhang, K.; Yang, H.; Liu, J.; Huang, Q. Protective effect of nerve growth factor on neurons after traumatic brain injury. J. Basic Clin. Physiol. Pharmacol. 2003, 14, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Guo, R.; Yue, X.; Lv, Q.; Ye, X.; Wang, Z.; Chen, Z.; Wu, B.; Xu, G.; Liu, X. Intranasal administration of nerve growth factor ameliorate β-amyloid deposition after traumatic brain injury in rats. Brain Res. 2012, 1440, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Longo, F.M.; Massa, S.M. A small molecule p75NTR ligand protects neurogenesis after traumatic brain injury. Stem Cells 2013, 31, 2561–2574. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, A.; Golz, C.; Werner, C.; Schafer, M.K.; Engelhard, K.; Thal, S.C. Proneurotrophin Binding to P75 Neurotrophin Receptor P75NTR Is Essential for Brain Lesion Formation and Functional Impairment after Experimental Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Delbary-Gossart, S.; Lee, S.; Baroni, M.; Lamarche, I.; Arnone, M.; Canolle, B.; Lin, A.; Sacramento, J.; Salegio, E.A.; Castel, M.N.; et al. A novel inhibitor of p75-neurotrophin receptor improves functional outcomes in two models of traumatic brain injury. Brain 2016, 139, 1762–1782. [Google Scholar] [CrossRef] [PubMed]
- Siao, C.J.; Lorentz, C.U.; Kermani, P.; Marinic, T.; Carter, J.; McGrath, K.; Padow, V.A.; Mark, W.; Falcone, D.J.; Cohen-Gould, L.; et al. ProNGF, a cytokine induced after myocardial infarction in humans, targets pericytes to promote microvascular damage and activation. J. Exp. Med. 2012, 209, 2291–2305. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Evaldt, J.; Nabinger, D.D.; Fontana, M.F.; Klein, M.G.; do Amaral Gomes, J.; Regner, A. Plasma matrix metalloproteinase-9 levels predict intensive care unit mortality early after severe traumatic brain injury. Brain Inj. 2017, 31, 390–395. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Probe Name | Gene Symbol | Description | ncPCs | cPCs (PC Medium) | cPCs (MSC Medium) | ATMSCs | HUVECs | PBWBCs |
|---|---|---|---|---|---|---|---|---|
| A_23_P115190 | NGF | nerve growth factor (β polypeptide) | 7.721 | 4.458 | 6.394 | 7.998 | 3.967 | 4.790 |
| A_23_P127891 | BDNF | brain-derived neurotrophic factor, transcript variant 1 | 6.635 | 7.903 | 11.047 | 8.457 | 6.650 | 2.812 |
| A_32_P7316 | BDNF | brain-derived neurotrophic factor, transcript variant 1 | 2.739 | 2.975 | 8.517 | 5.321 | 4.026 | 2.572 |
| A_23_P127891 | BDNF | brain-derived neurotrophic factor, transcript variant 1 | 6.635 | 7.903 | 11.047 | 8.457 | 6.650 | 2.812 |
| A_23_P360797 | NTF3 | neurotrophin 3, transcript variant 2 | 8.034 | 3.479 | 3.916 | 8.211 | 4.513 | 3.945 |
| A_23_P4899 | NTF4 | neurotrophin 4 (NTF4) | 2.052 | 2.046 | 5.979 | 2.293 | 2.266 | 2.352 |
| A_24_P25544 | GDNF | glial cell derived neurotrophic factor, transcript variant 1 | 2.277 | 2.294 | 4.050 | 2.979 | 1.758 | 1.948 |
| A_23_P167683 | GDNF | glial cell derived neurotrophic factor, transcript variant 1 | 4.375 | 3.359 | 4.955 | 2.883 | 1.868 | 1.852 |
| A_32_P377880 | GDNF | glial cell derived neurotrophic factor, precursor | 7.660 | 7.685 | 9.894 | 6.382 | 1.790 | 2.378 |
| A_23_P90359 | NRTN | Neurturin | 2.839 | 2.461 | 4.375 | 2.853 | 2.636 | 3.578 |
| A_23_P410507 | PSPN | Persephin | 8.082 | 7.490 | 6.954 | 6.349 | 6.620 | 8.475 |
| A_23_P389897 | NGFR | nerve growth factor receptor (p75) | 10.493 | 2.299 | 3.673 | 5.042 | 2.448 | 3.644 |
 | ||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Silva Meirelles, L.; Simon, D.; Regner, A. Neurotrauma: The Crosstalk between Neurotrophins and Inflammation in the Acutely Injured Brain. Int. J. Mol. Sci. 2017, 18, 1082. https://doi.org/10.3390/ijms18051082
Da Silva Meirelles L, Simon D, Regner A. Neurotrauma: The Crosstalk between Neurotrophins and Inflammation in the Acutely Injured Brain. International Journal of Molecular Sciences. 2017; 18(5):1082. https://doi.org/10.3390/ijms18051082
Chicago/Turabian StyleDa Silva Meirelles, Lindolfo, Daniel Simon, and Andrea Regner. 2017. "Neurotrauma: The Crosstalk between Neurotrophins and Inflammation in the Acutely Injured Brain" International Journal of Molecular Sciences 18, no. 5: 1082. https://doi.org/10.3390/ijms18051082
APA StyleDa Silva Meirelles, L., Simon, D., & Regner, A. (2017). Neurotrauma: The Crosstalk between Neurotrophins and Inflammation in the Acutely Injured Brain. International Journal of Molecular Sciences, 18(5), 1082. https://doi.org/10.3390/ijms18051082