
Analysis of Different Approaches for the Selection of Reference Genes in RT-qPCR Experiments: A Case Study in Skeletal Muscle of Growing Mice

Abstract
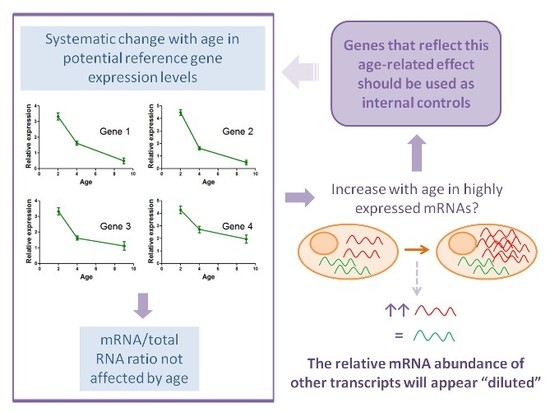
:
1. Introduction
2. Results
2.1. Primer Specificity and Amplification Efficiency
2.2. Selection of Potential Reference Genes for Studies in Skeletal Muscle of Normal and GH-Overexpressing Growing Mice
2.3. Evaluation of Potential Reference Gene Expression Stability in Skeletal Muscle of Normal and GH-Overexpressing Growing Mice
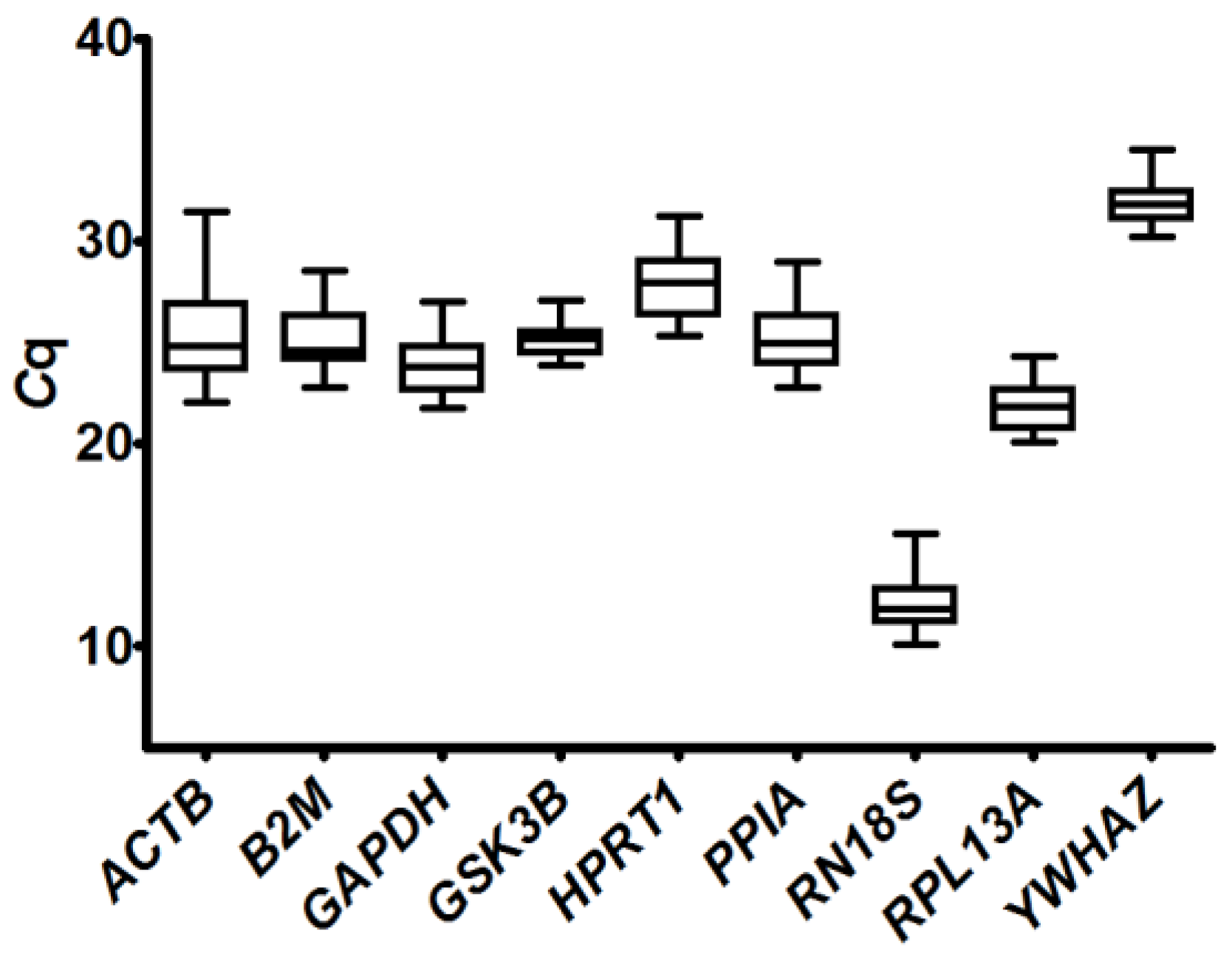
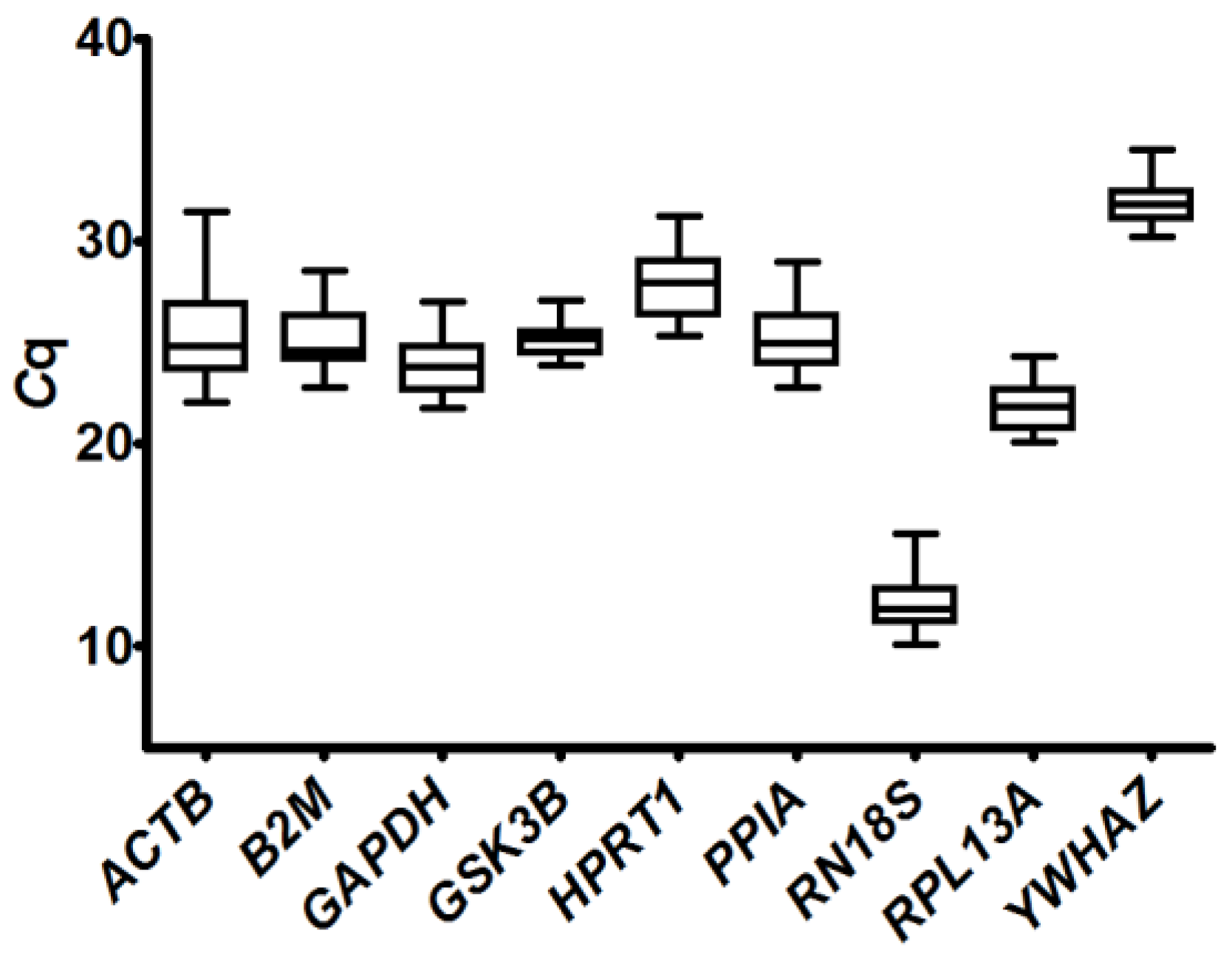
2.3.1. Analysis of Quantification Cycle (Cq) Values Dispersion and Determination of Expression Stability by Algorithms
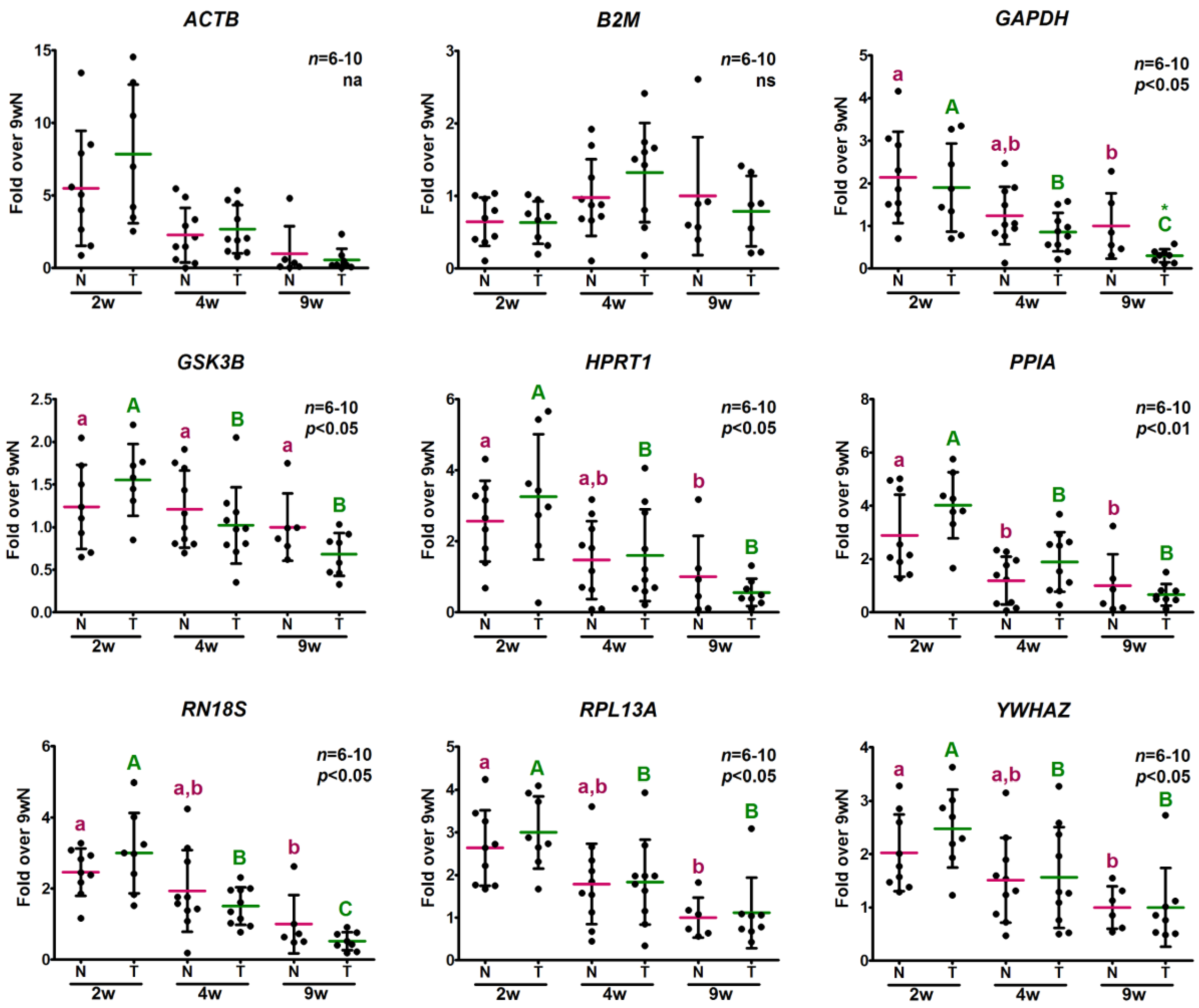
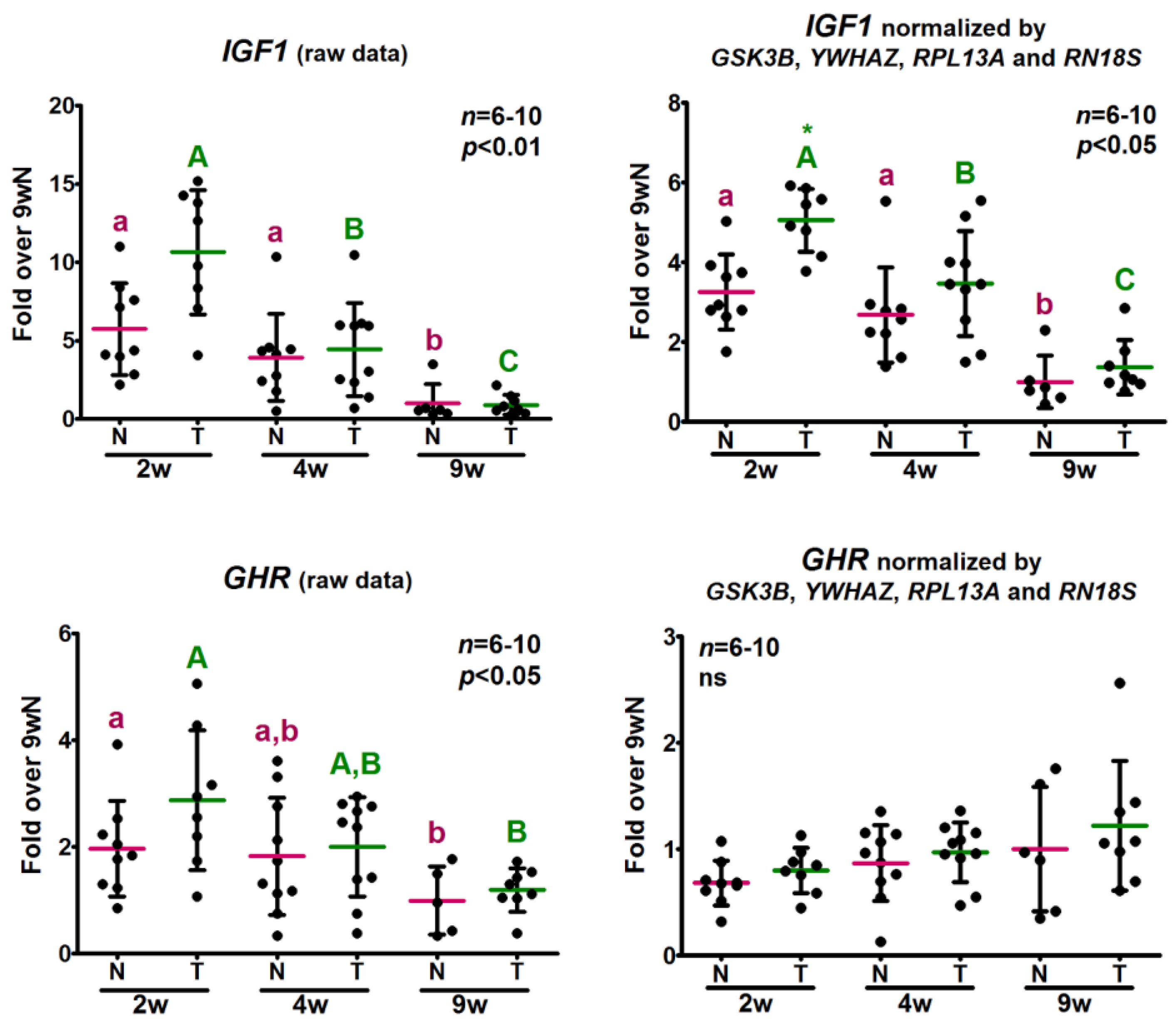
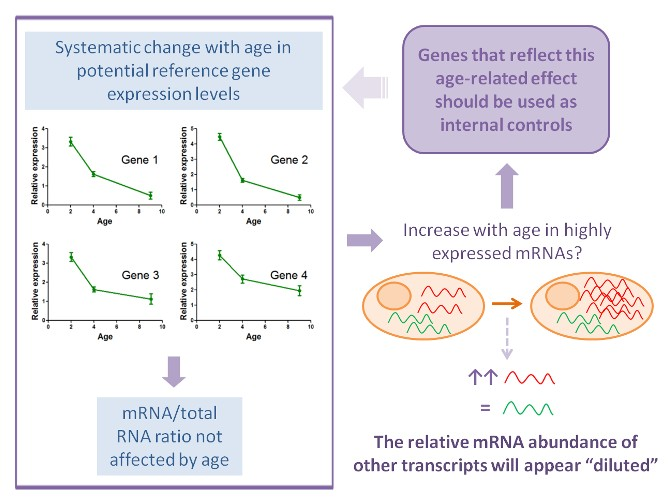
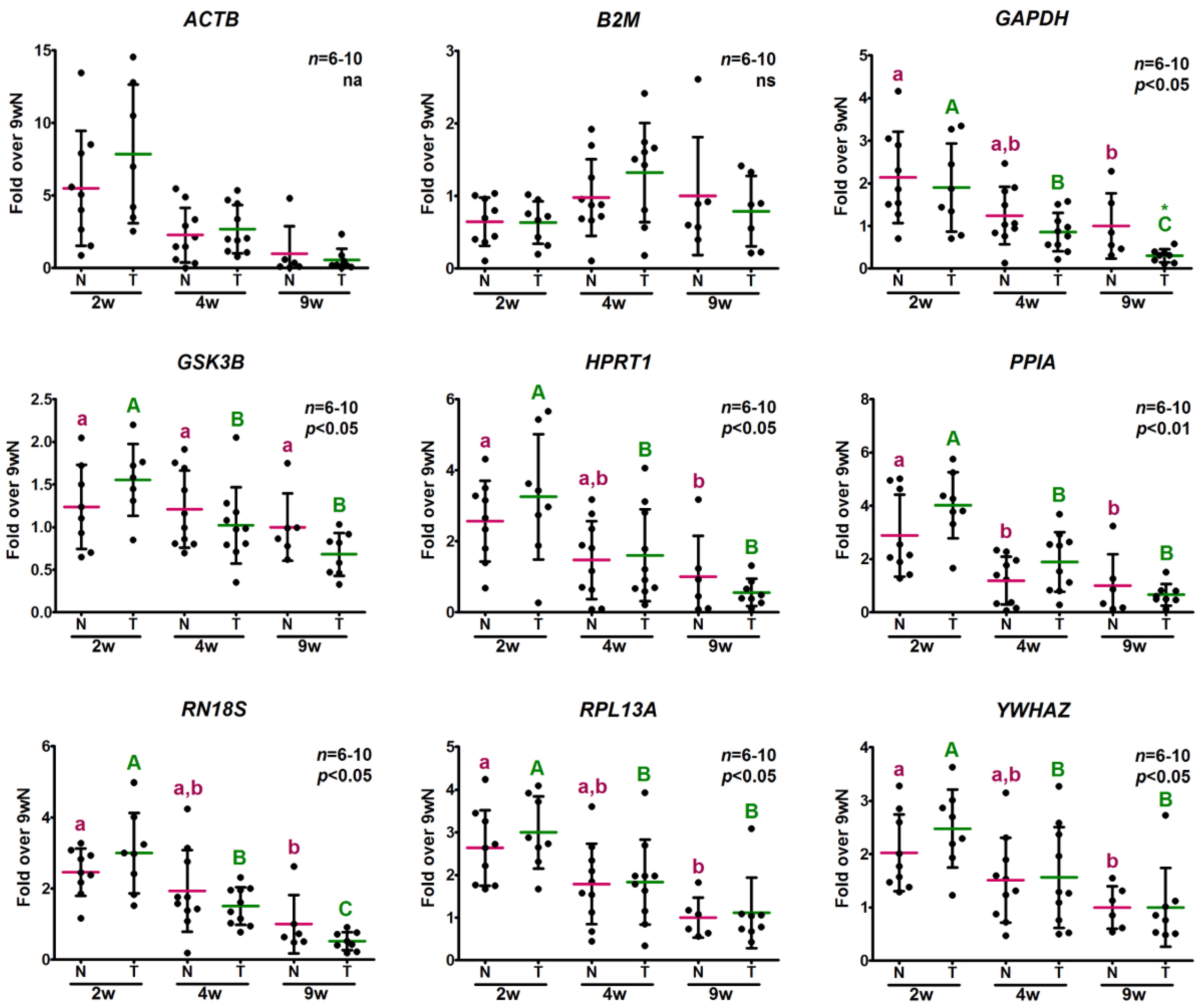
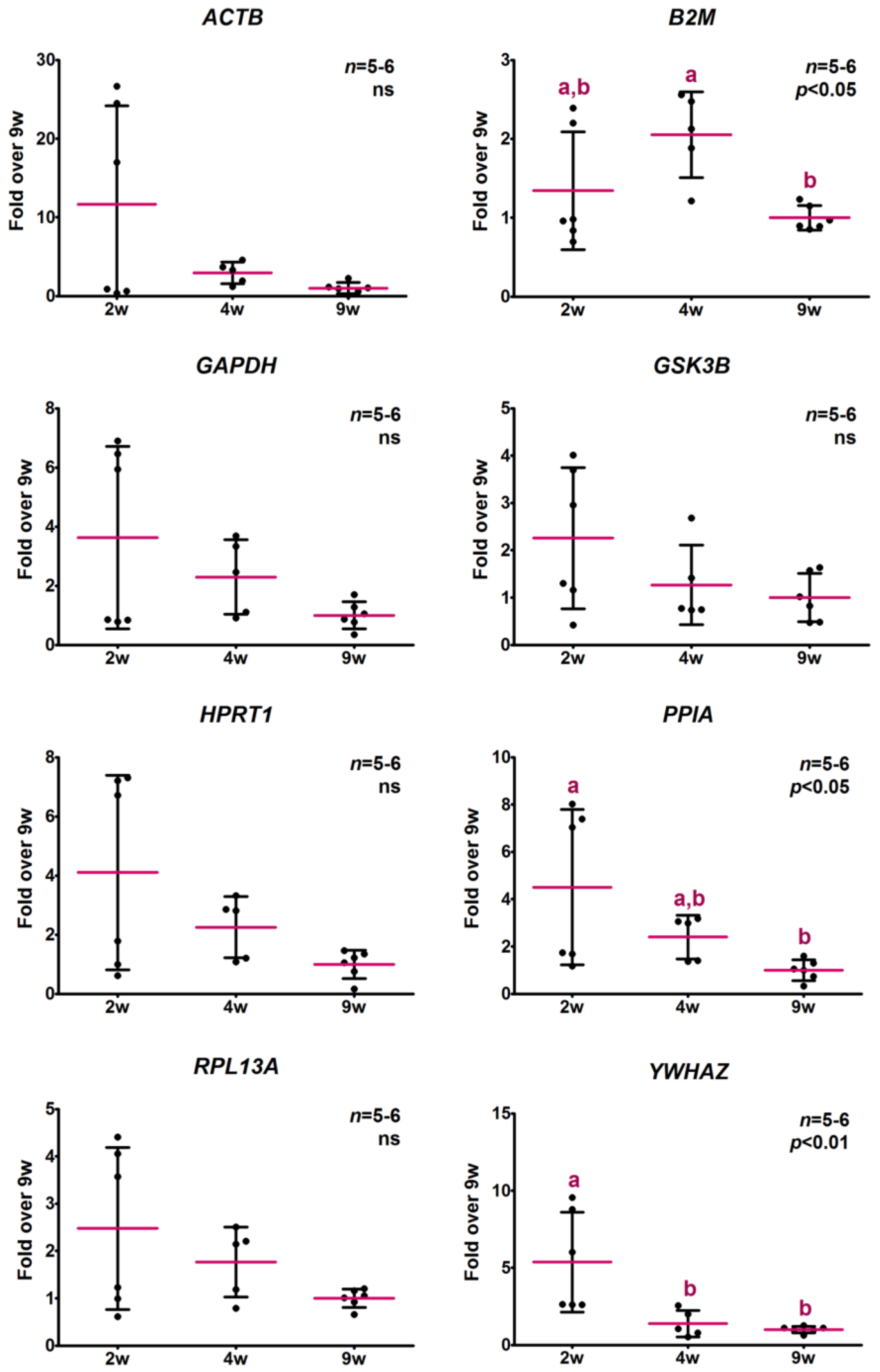
2.3.2. Analysis of Relative Expression Levels
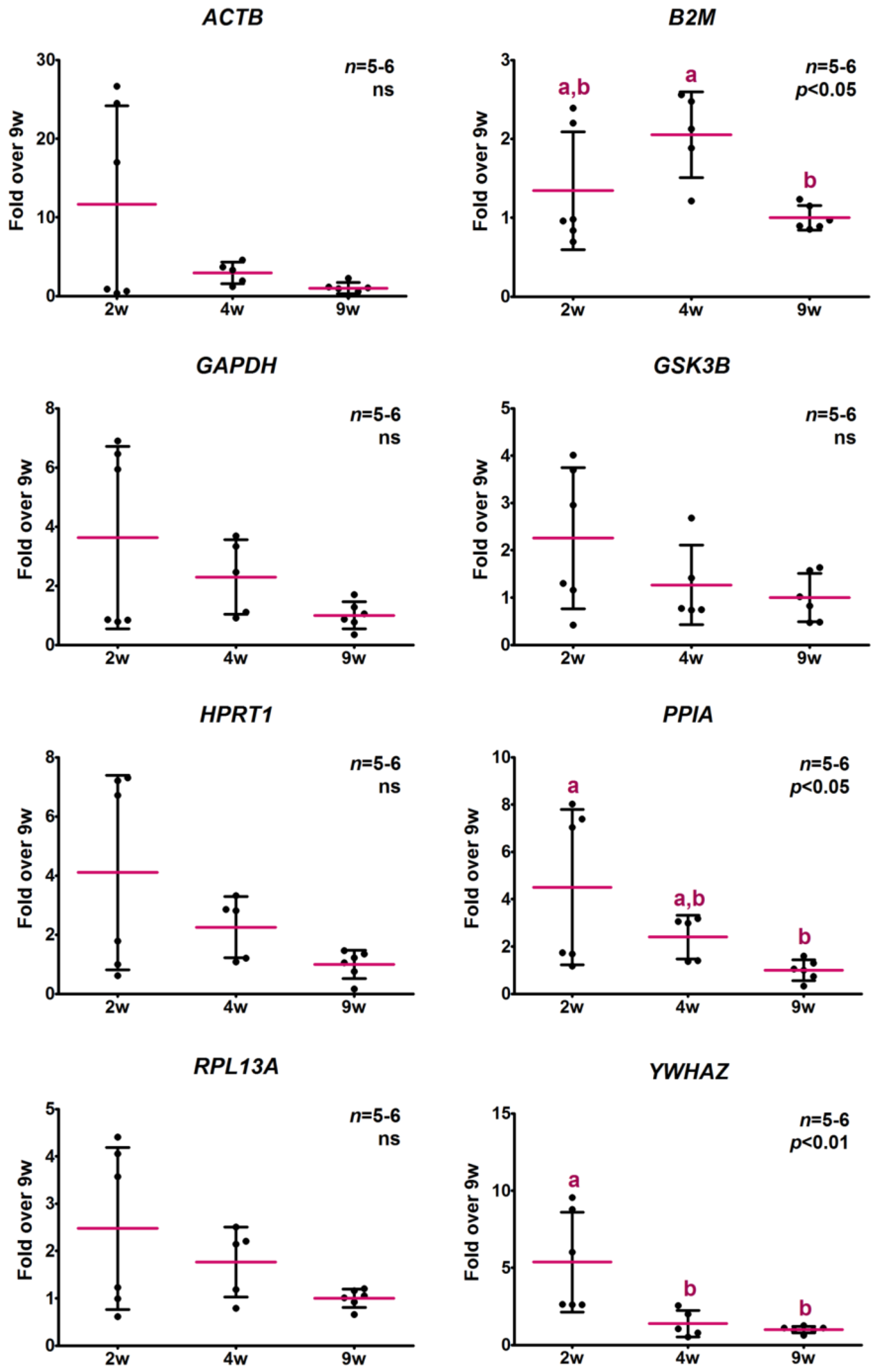
2.4. Determination of Potential Reference Gene Expression Levels in Isolated Skeletal Muscle mRNA from Normal Mice
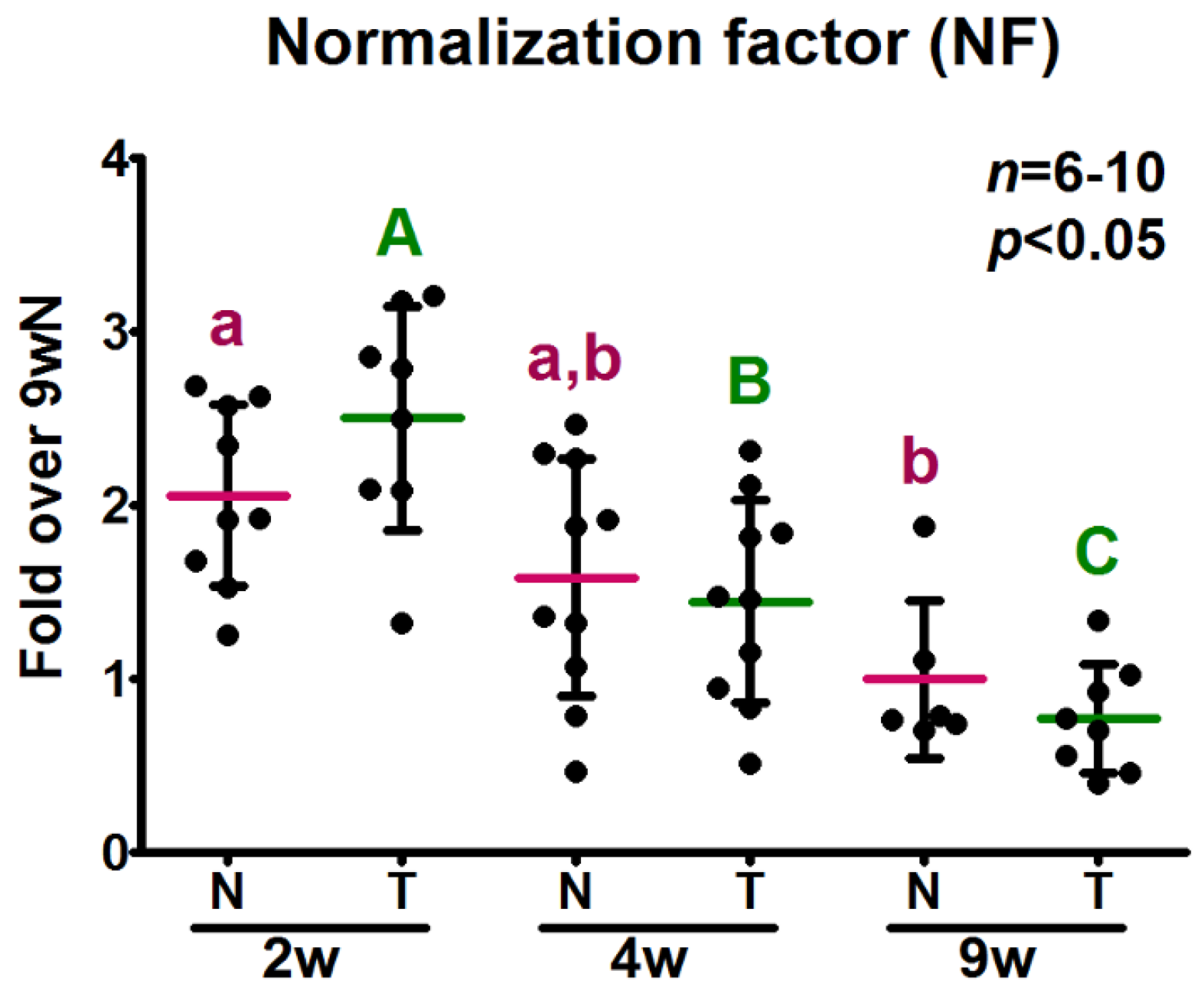
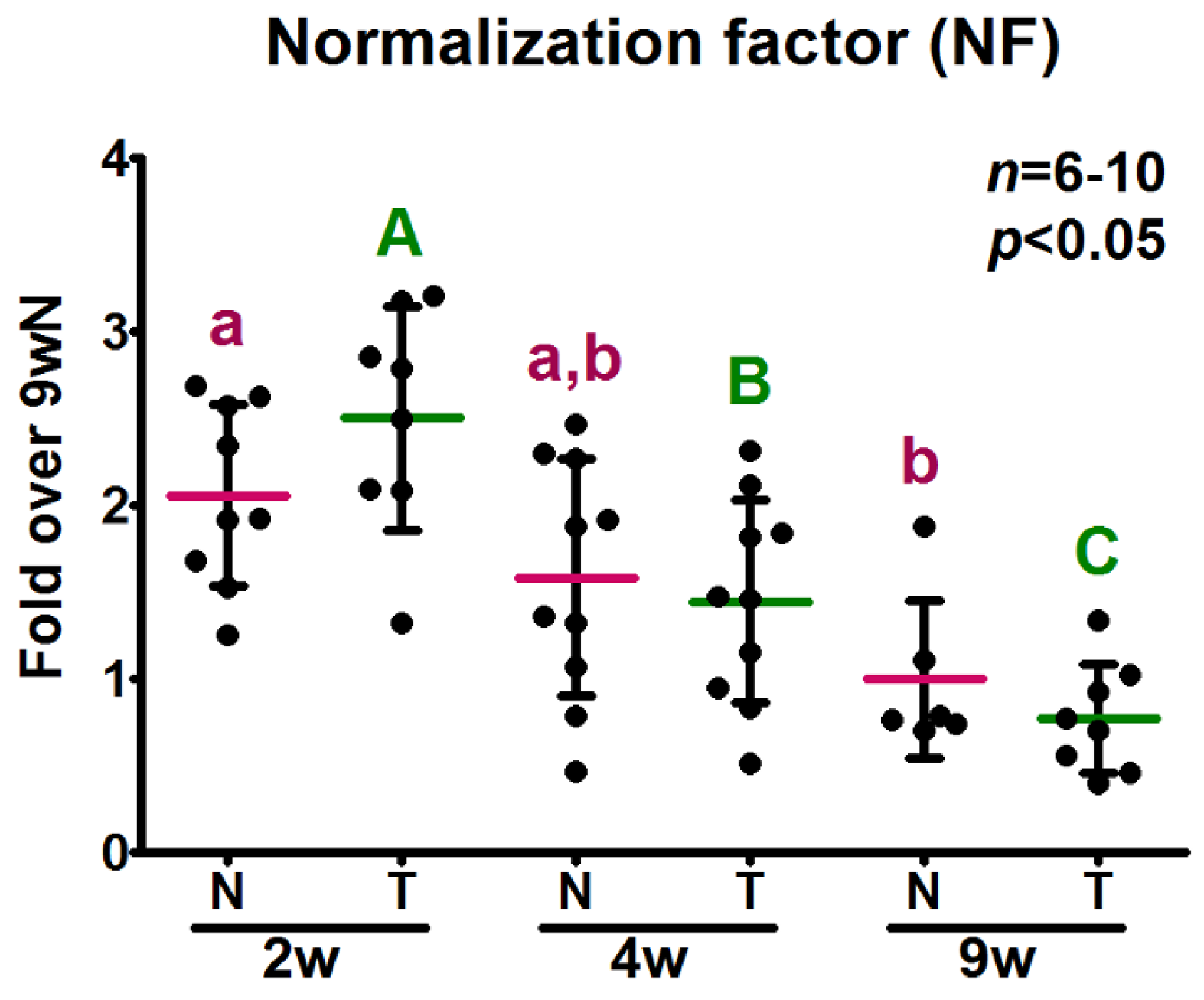
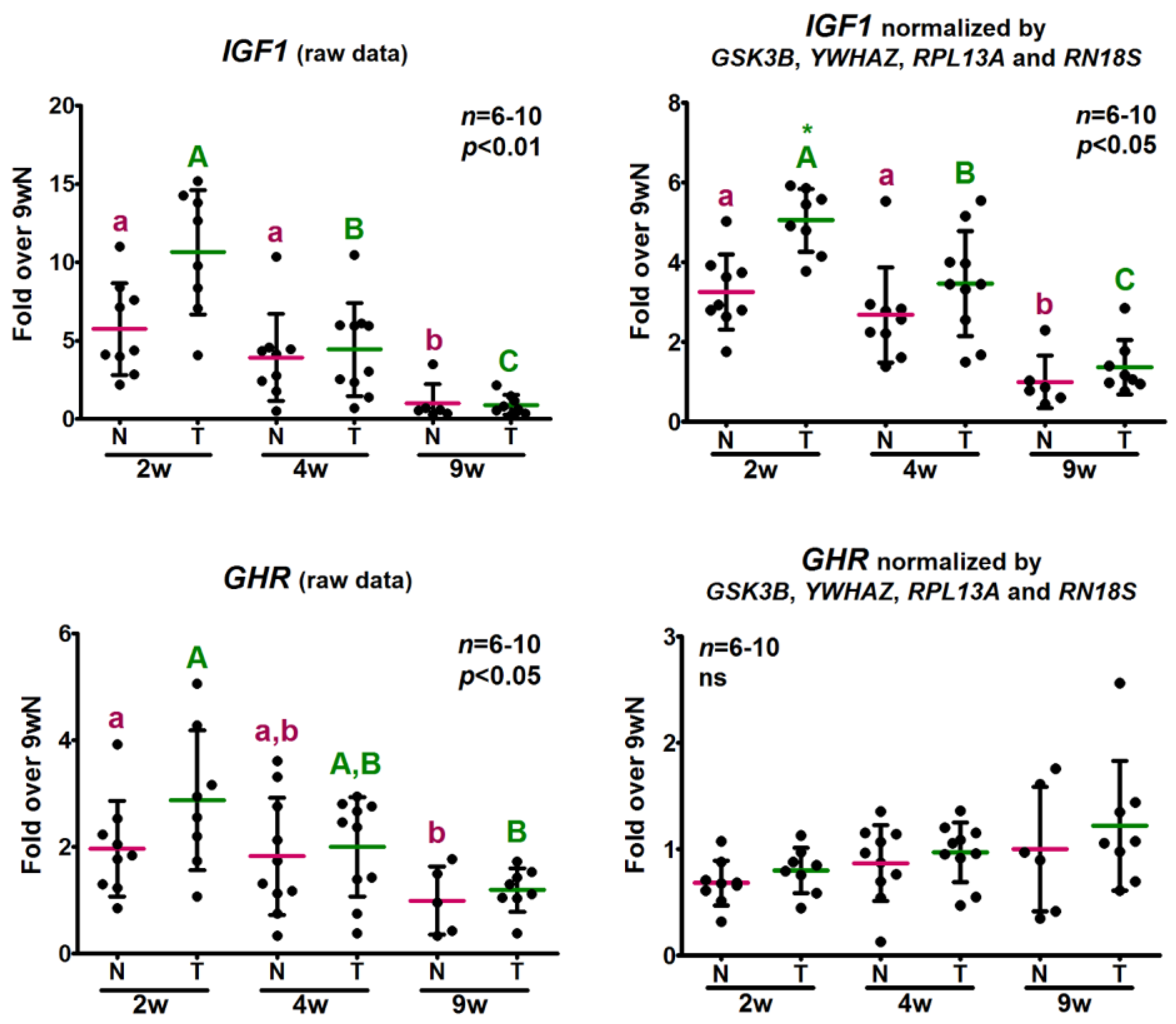
2.5. Analysis of the Relative Expression Levels of Target Genes Normalized against the Selected Reference Genes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Design
4.3. RNA Extraction and Reverse Transcription (RT)
4.4. Messenger RNA (mRNA) Isolation
4.5. Primer Design
4.6. Quantitative PCR (qPCR)
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bustin, S.A.; Nolan, T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech. 2004, 15, 155–166. [Google Scholar] [PubMed]
- Huggett, J.; Dheda, K.; Bustin, S.A.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Kozera, B.; Rapacz, M. Reference genes in real-time PCR. J. Appl. Genet. 2013, 54, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Thellin, O.; Zorzi, W.; Lakaye, B.; de Borman, B.; Coumans, B.; Hennen, G.; Grisar, T.; Igout, A.; Heinen, E. Housekeeping genes as internal standards: Use and limits. J. Biotechnol. 1999, 75, 291–295. [Google Scholar] [CrossRef]
- Tricarico, C.; Pinzani, P.; Bianchi, S.; Paglierani, M.; Distante, V.; Pazzagli, M.; Bustin, S.A.; Orlando, C. Quantitative real-time reverse transcription polymerase chain reaction: Normalization to rRNA or single housekeeping genes is inappropriate for human tissue biopsies. Anal. Biochem. 2002, 309, 293–300. [Google Scholar] [CrossRef]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3. [Google Scholar] [CrossRef]
- Dheda, K.; Huggett, J.; Bustin, S.A.; Johnson, M.A.; Rook, G.; Zumla, A. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 2004, 37, 112–119. [Google Scholar] [PubMed]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaffl, M.W. Quantitative real-time RT-PCR—A perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.; Mauriat, M.; Guénin, S.; Pelloux, J.; Lefebvre, J.F.; Louvet, R.; Rusterucci, C.; Moritz, T.; Guerineau, F.; Bellini, C.; et al. The lack of a systematic validation of reference genes: A serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol. J. 2008, 6, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Dheda, K.; Huggett, J.; Chang, J.S.; Kim, L.U.; Bustin, S.A.; Johnson, M.A.; Rook, G.; Zumla, A. The implications of using an inappropriate reference gene for real-time reverse transcription PCR data normalization. Anal. Biochem. 2005, 344, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: Bestkeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Le Roith, D. Insulin-like growth factor I is essential for postnatal growth in response to growth hormone. Endocrinology 1999, 140, 5178–5184. [Google Scholar] [CrossRef] [PubMed]
- Lupu, F.; Terwilliger, J.D.; Lee, K.; Segre, G.V.; Efstratiadis, A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol. 2001, 229, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Yu, R.; Lu, Y.; Parlow, A.F.; Liu, J.L. Age-dependent onset of liver-specific IGF-I gene deficiency and its persistence in old age: Implications for postnatal growth and insulin resistance in LID mice. Am. J. Physiol. Endocrinol. Metab. 2005, 289, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Mathews, L.S.; Hammer, R.E.; Brinster, R.L.; Palmiter, R.D. Expression of insulin-like growth factor I in transgenic mice with elevated levels of growth hormone is correlated with growth. Endocrinology 1988, 123, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.S.; Piazza, V.G.; Díaz, M.E.; Boparai, R.K.; Arum, O.; Ramírez, M.C.; González, L.; Becú-Villalobos, D.; Bartke, A.; Turyn, D.; et al. GH/STAT5 signaling during the growth period in livers of mice overexpressing GH. J. Mol. Endocrinol. 2015, 54, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.S.; Piazza, V.G.; González, L.; Fang, Y.; Bartke, A.; Turyn, D.; Miquet, J.G.; Sotelo, A.I. Mitogenic signaling pathways in the liver of growth hormone (GH)-overexpressing mice during the growth period. Cell Cycle 2016, 15, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Miquet, J.G.; González, L.; Matos, M.N.; Hansen, C.E.; Louis, A.; Bartke, A.; Turyn, D.; Sotelo, A.I. Transgenic mice overexpressing GH exhibit hepatic upregulation of GH-signaling mediators involved in cell proliferation. J. Endocrinol. 2008, 198, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Miquet, J.G.; Freund, T.; Martinez, C.S.; González, L.; Díaz, M.E.; Micucci, G.P.; Zotta, E.; Boparai, R.K.; Bartke, A.; Turyn, D.; et al. Hepatocellular alterations and dysregulation of oncogenic pathways in the liver of transgenic mice overexpressing growth hormone. Cell Cycle 2013, 12, 1042–1057. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rider, D.A.; Ruan, R. Identification of valid housekeeping genes and antioxidant enzyme gene expression change in the aging rat liver. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Ciccarone, F.; Guastafierro, T.; Bacalini, M.G.; Calabrese, R.; Moreno-Villanueva, M.; Reale, A.; Chevanne, M.; Bürkle, A.; Caiafa, P. Validation of suitable internal control genes for expression studies in aging. Mech. Ageing Dev. 2010, 131, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.J.; Cinar, M.U.; Tesfaye, D.; Looft, C.; Tholen, E.; Schellander, K. Age-related changes in relative expression stability of commonly used housekeeping genes in selected porcine tissues. BMC Res. Notes 2011, 4, 441. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, J.; Mortier, G.; de Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed]
- Derveaux, S.; Vandesompele, J.; Hellemans, J. How to do successful gene expression analysis using real-time PCR. Methods 2010, 50, 227–230. [Google Scholar] [CrossRef] [PubMed]
- qPCR & RT-qPCR Applications. Available online: http://qrt-pcr-applications.info/ (accessed on 30 March 2017).
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Kortner, T.M.; Valen, E.C.; Kortner, H.; Marjara, I.S.; Krogdahl, A.; Bakke, A.M. Candidate reference genes for quantitative real-time PCR (qPCR) assays during development of a diet-related enteropathy in atlantic salmon (Salmo salar L.) and the potential pitfalls of uncritical use of normalization software tools. Aquaculture 2011, 318, 355–363. [Google Scholar] [CrossRef]
- De Santis, C.; Smith-Keune, C.; Jerry, D.R. Normalizing RT-qPCR data: Are we getting the right answers? An appraisal of normalization approaches and internal reference genes from a case study in the finfish Lates calcarifer. Mar. Biotechnol. 2011, 13, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Bionaz, M.; Loor, J.J. Identification of reference genes for quantitative real-time PCR in the bovine mammary gland during the lactation cycle. Physiol. Genom. 2007, 29, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Tramontana, S.; Bionaz, M.; Sharma, A.; Graugnard, D.E.; Cutler, E.A.; Ajmone-Marsan, P.; Hurley, W.L.; Loor, J.J. Internal controls for quantitative polymerase chain reaction of swine mammary glands during pregnancy and lactation. J. Dairy Sci. 2008, 91, 3057–3066. [Google Scholar] [CrossRef] [PubMed]
- Monaco, E.; Bionaz, M.; Sobreira de Lima, A.; Hurley, W.L.; Loor, J.J.; Wheeler, M.B. Selection and reliability of internal reference genes for quantitative PCR verification of transcriptomics during the differentiation process of porcine adult mesenchymal stem cells. Stem Cell Res. Ther. 2010, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Bionaz, M.; Sloboda, D.M.; Ehrlich, L.; Li, S.; Newnham, J.P.; Dudenhausen, J.W.; Henrich, W.; Plagemann, A.; Challis, J.R.; et al. The dilution effect and the importance of selecting the right internal control genes for RT-qPCR: A paradigmatic approach in fetal sheep. BMC Res. Notes 2015, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Lee, J.N.; Bionaz, M.; Deng, X.Y.; Wang, Y. Evaluation of suitable internal control genes for RT-qPCR in yak mammary tissue during the lactation cycle. PLoS ONE 2016, 11, e0147705. [Google Scholar] [CrossRef] [PubMed]
- Solanas, M.; Moral, R.; Escrich, E. Unsuitability of using ribosomal RNA as loading control for northern blot analyses related to the imbalance between messenger and ribosomal RNA content in rat mammary tumors. Anal. Biochem. 2001, 288, 99–102. [Google Scholar] [CrossRef] [PubMed]
- White, R.J. RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 2008, 24, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.C.; Nielsen, A.K.; Molin, S.; Hammer, K.; Kilstrup, M. Changes in rRNA levels during stress invalidates results from mRNA blotting: Fluorescence in situ rRNA hybridization permits renormalization for estimation of cellular mRNA levels. J. Bacteriol. 2001, 183, 4747–4751. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Igoa, M.; Manzano, R.; Oliván, S.; Calvo, A.C.; Toivonen, J.M.; Osta, R. Effects of gene therapy on muscle 18S rRNA expression in mouse model of ALS. BMC Res. Notes 2010, 3, 275. [Google Scholar] [CrossRef] [PubMed]
- Parent, R.; Beretta, L. Translational control plays a prominent role in the hepatocytic differentiation of HepaRG liver progenitor cells. Genome Biol. 2008, 9, R19. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, 2002–2007. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Efficiency (%) | R2 |
|---|---|---|
| ACTB | 101 | 0.982 |
| B2M | 98 | 0.998 |
| GAPDH | 95 | 0.999 |
| GHR | 93 | 0.995 |
| GSK3B | 91 | 0.995 |
| HPRT1 | 105 | 0.991 |
| IGF1 | 108 | 0.996 |
| PPIA | 101 | 0.998 |
| RN18S | 101 | 0.998 |
| RPL13A | 100 | 0.998 |
| YWHAZ | 99 | 0.980 |
| Ranking | Bestkeeper | geNorm | Comparative ΔCq | NormFinder | ||||
|---|---|---|---|---|---|---|---|---|
| Gene Symbol | Stability Value | Gene Symbol | Stability Value | Gene Symbol | Stability Value | Gene Symbol | Stability Value | |
| 1 | GSK3B | 0.587 | RPL13A/YWHAZ | 0.458 | RPL13A | 1.073 | RPL13A | 0.202 |
| 2 | YWHAZ | 0.762 | GSK3B | 0.549 | YWHAZ | 1.092 | RN18S | 0.279 |
| 3 | RPL13A | 0.916 | RN18S | 0.698 | GSK3B | 1.146 | YWHAZ | 0.291 |
| 4 | RN18S | 0.962 | GAPDH | 0.757 | RN18S | 1.159 | PPIA | 0.398 |
| 5 | GAPDH | 1.194 | PPIA | 0.871 | PPIA | 1.279 | GSK3B | 0.409 |
| 6 | B2M | 1.263 | HPRTA | 0.989 | HPRT1 | 1.427 | HPRT1 | 0.420 |
| 7 | PPIA | 1.317 | B2M | 1.104 | GAPDH | 1.479 | GAPDH | 0.421 |
| 8 | HPRT1 | 1.399 | ACTB | 1.279 | B2M | 1.780 | B2M | 0.762 |
| 9 | ACTB | 1.958 | - | - | ACTB | 1.891 | ACTB | 0.822 |
| Ranking | Bestkeeper | geNorm | Comparative ΔCq | NormFinder | ||||
|---|---|---|---|---|---|---|---|---|
| Gene Symbol | Stability Value | Gene Symbol | Stability Value | Gene Symbol | Stability Value | Gene Symbol | Stability Value | |
| 1 | GSK3B | 0.646 | RPL13A/YWHAZ | 0.518 | YWHAZ | 1.173 | RPL13A | 0.169 |
| 2 | YWHAZ | 0.796 | GSK3B | 0.593 | RPL13A | 1.188 | RN18S | 0.227 |
| 3 | RN18S | 0.952 | RN18S | 0.701 | RN18S | 1.197 | GAPDH | 0.233 |
| 4 | RPL13A | 1.010 | GAPDH | 0.722 | GSK3B | 1.240 | YWHAZ | 0.268 |
| 5 | GAPDH | 1.207 | PPIA | 0.900 | PPIA | 1.492 | GSK3B | 0.449 |
| 6 | B2M | 1.378 | B2M | 1.031 | HPRT1 | 1.558 | PPIA | 0.486 |
| 7 | HPRT1 | 1.488 | HPRT1 | 1.152 | GAPDH | 1.611 | HPRT1 | 0.498 |
| 8 | PPIA | 1.516 | ACTB | 1.344 | B2M | 1.996 | B2M | 0.718 |
| 9 | ACTB | 2.273 | - | - | ACTB | 2.029 | ACTB | 0.813 |
| Gene Symbol | Gene Name | GenBank Accession Number | Primer Sequence (5′–3′) a |
|---|---|---|---|
| ACTB | Actin, β | NM_007393.5 | F: GTGCCCATCTACGAGGGCTATGCT |
| R: TACCCAAGAAGGAAGGCTGGAAAA | |||
| B2M | β-2 microglobulin | NM_009735.3 | F: AAGTATACTCACGCCACCCA |
| R: AAGACCAGTCCTTGCTGAAG | |||
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | NM_008084.3 | F: AGTGCCAGCCTCGTCCCGTAG |
| R: GTGCCGTTGAATTTGCCGTGAGTG | |||
| GHR | Growth hormone receptor | NM_001286370.1 | F: CCAACTCGCCTCTACACCG |
| R: GGGAAAGGACTACACCACCTG | |||
| GSK3B | Glycogen synthase kinase 3β | NM_019827.6 | F: CCACCATCCTTATCCCTCCAC |
| R: GTATCTGAGGCTGCTGTGGC | |||
| HPRT1 | Hypoxanthine guanine phosphoribosyl transferase | NM_013556.2 | F: CAGTCCCAGCGTCGTGATTA |
| R: TCGAGCAAGTCTTTCAGTCCT | |||
| IGF1 | Insulin-like growth factor 1 | NM_010512.4 | F: CCAAACACAATTCTCCTTCC |
| R: GCTACAGCAACCTGTGATTG | |||
| PPIA | Peptidylprolyl isomerase A | NM_008907.1 | F: GCGTCTCCTTGAGCTGTT |
| R: AAGTCACCACCCTGGCAC | |||
| RN18S | 18S ribosomal RNA | NR_003278.3 | F: ACGGACAGGATTGACAGATT |
| R: GCCAGAGTCTCGTTCGTTAT | |||
| RPL13A | Ribosomal protein L13A | NM_009438.5 | F: TGACAAGAAAAAGCGGATGGTG |
| R: GCTGTCACTGCCTGGTACTT | |||
| YWHAZ | Tyr 3-monooxygenase/Trp 5-monooxygenase activation protein, Z polypeptide | NM_011740.3 | F: CCAGGACCTAAAAGGGTCGG |
| R: ACACACCGAACTGTTGTCGT |
| Gene Symbol | Exon–Exon Junction a,b | Tm (°C) | Product Length (bp) |
|---|---|---|---|
| ACTB | F: no | 63 | 319 |
| R: yes | 58 | ||
| B2M | F: no | 56 | 162 |
| R: no | 55 | ||
| GAPDH | F: yes | 64 | 171 |
| R: no | 61 | ||
| GHR | F: no | 58 | 104 |
| R: no | 57 | ||
| GSK3B | F: no | 57 | 79 |
| R: yes | 58 | ||
| HPRT1 | F: yes | 62 | 142 |
| R: yes | 59 | ||
| IGF1 | F: no | 57 | 97 |
| R: no | 57 | ||
| PPIA | F: no | 55 | 145 |
| R: yes | 58 | ||
| RN18S | F: no | 57 | 118 |
| R: no | 57 | ||
| RPL13A | F: yes | 64 | 126 |
| R: no | 58 | ||
| YWHAZ | F: no | 63 | 115 |
| R: no | 59 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piazza, V.G.; Bartke, A.; Miquet, J.G.; Sotelo, A.I. Analysis of Different Approaches for the Selection of Reference Genes in RT-qPCR Experiments: A Case Study in Skeletal Muscle of Growing Mice. Int. J. Mol. Sci. 2017, 18, 1060. https://doi.org/10.3390/ijms18051060
Piazza VG, Bartke A, Miquet JG, Sotelo AI. Analysis of Different Approaches for the Selection of Reference Genes in RT-qPCR Experiments: A Case Study in Skeletal Muscle of Growing Mice. International Journal of Molecular Sciences. 2017; 18(5):1060. https://doi.org/10.3390/ijms18051060
Chicago/Turabian StylePiazza, Verónica G., Andrzej Bartke, Johanna G. Miquet, and Ana I. Sotelo. 2017. "Analysis of Different Approaches for the Selection of Reference Genes in RT-qPCR Experiments: A Case Study in Skeletal Muscle of Growing Mice" International Journal of Molecular Sciences 18, no. 5: 1060. https://doi.org/10.3390/ijms18051060
APA StylePiazza, V. G., Bartke, A., Miquet, J. G., & Sotelo, A. I. (2017). Analysis of Different Approaches for the Selection of Reference Genes in RT-qPCR Experiments: A Case Study in Skeletal Muscle of Growing Mice. International Journal of Molecular Sciences, 18(5), 1060. https://doi.org/10.3390/ijms18051060