
The Effect of VPA on Increasing Radiosensitivity in Osteosarcoma Cells and Primary-Culture Cells from Chemical Carcinogen-Induced Breast Cancer in Rats

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
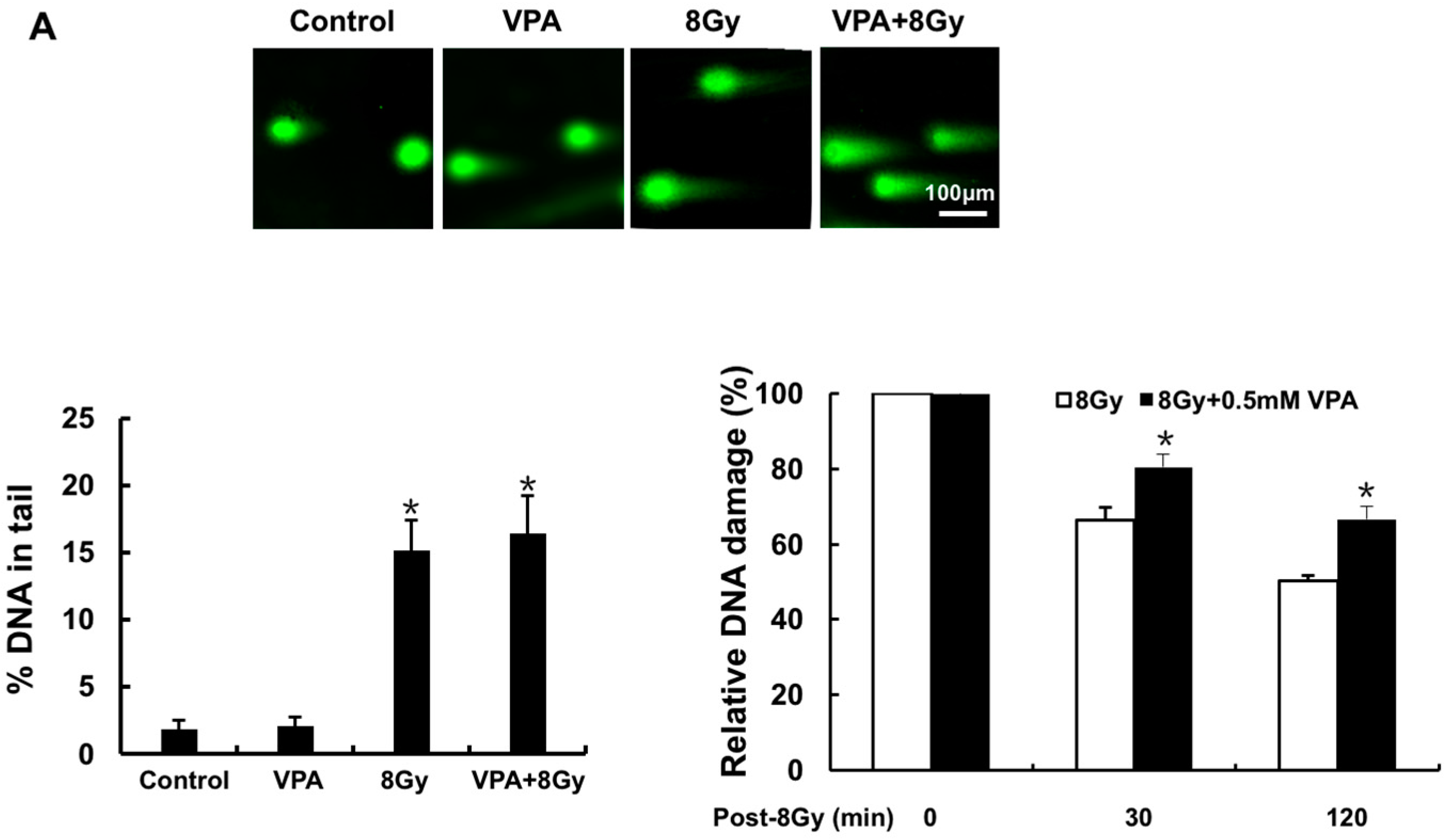
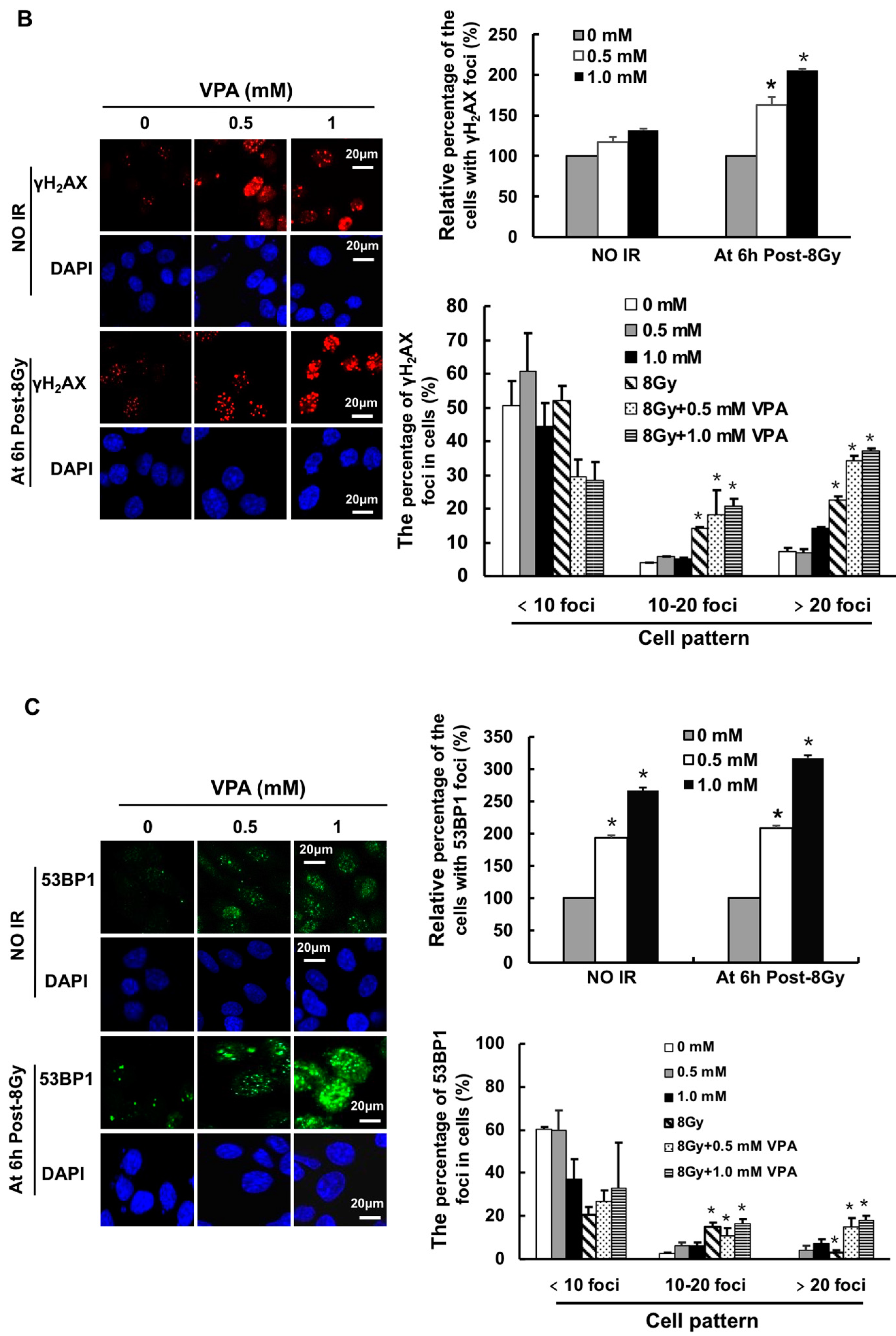
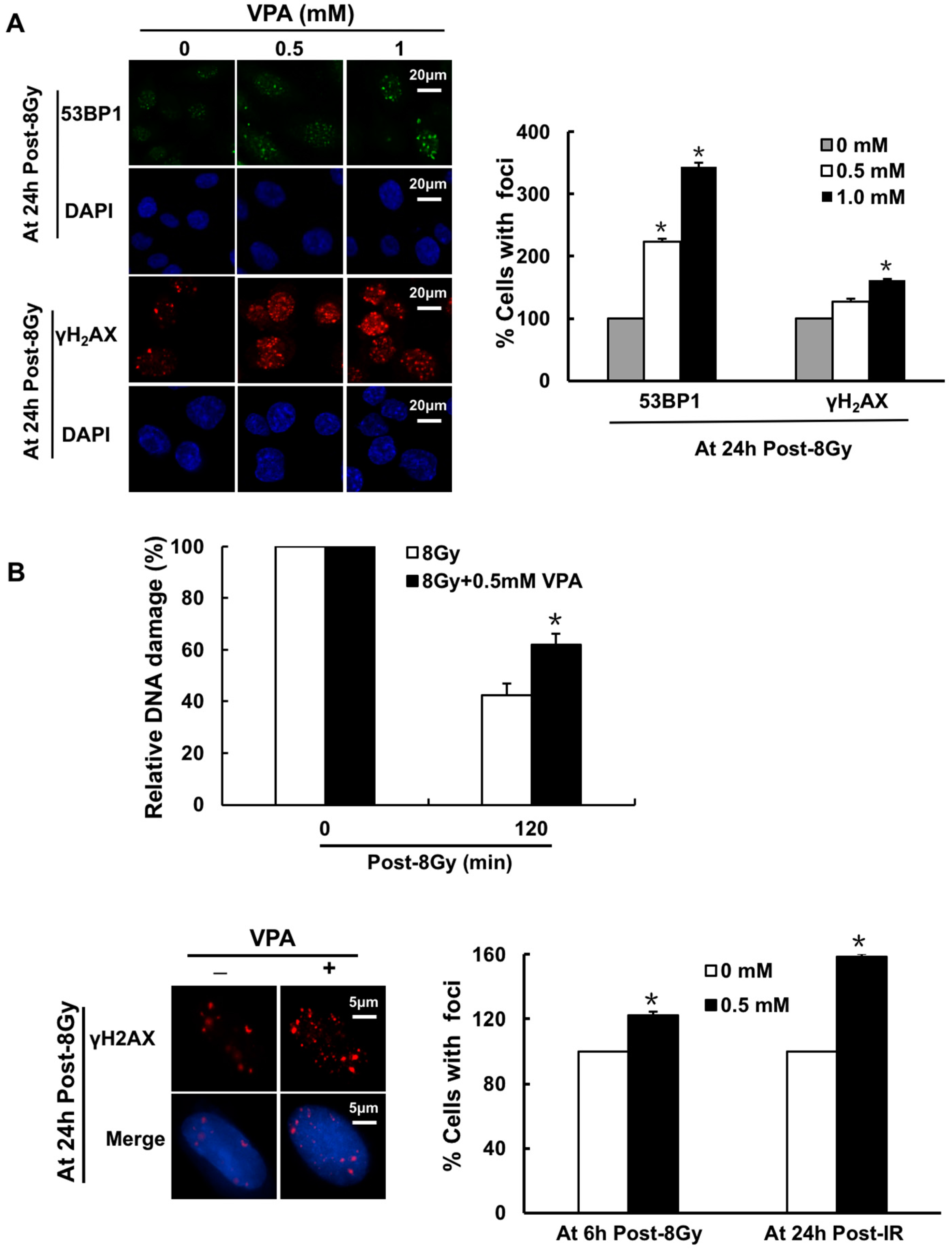
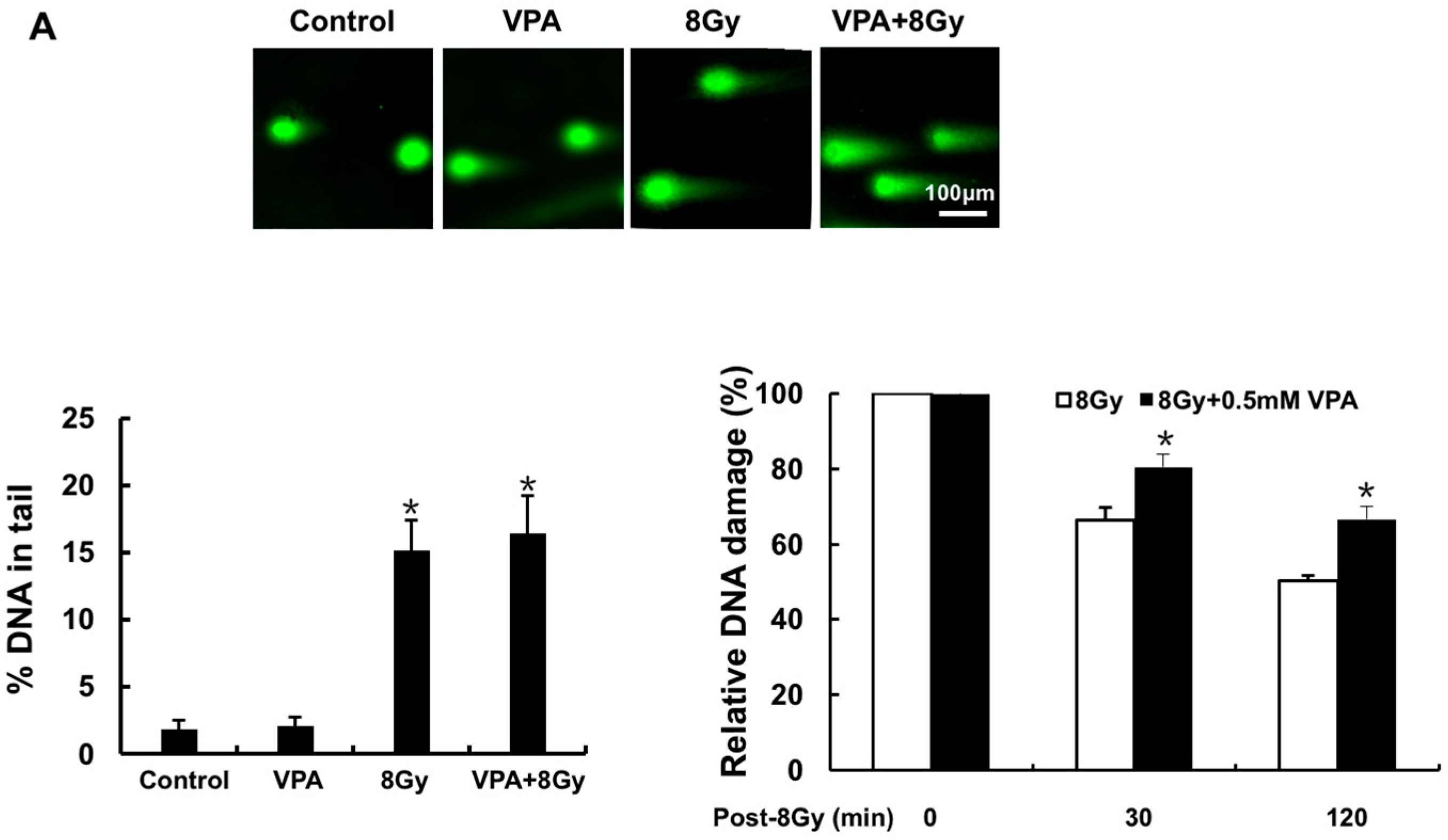
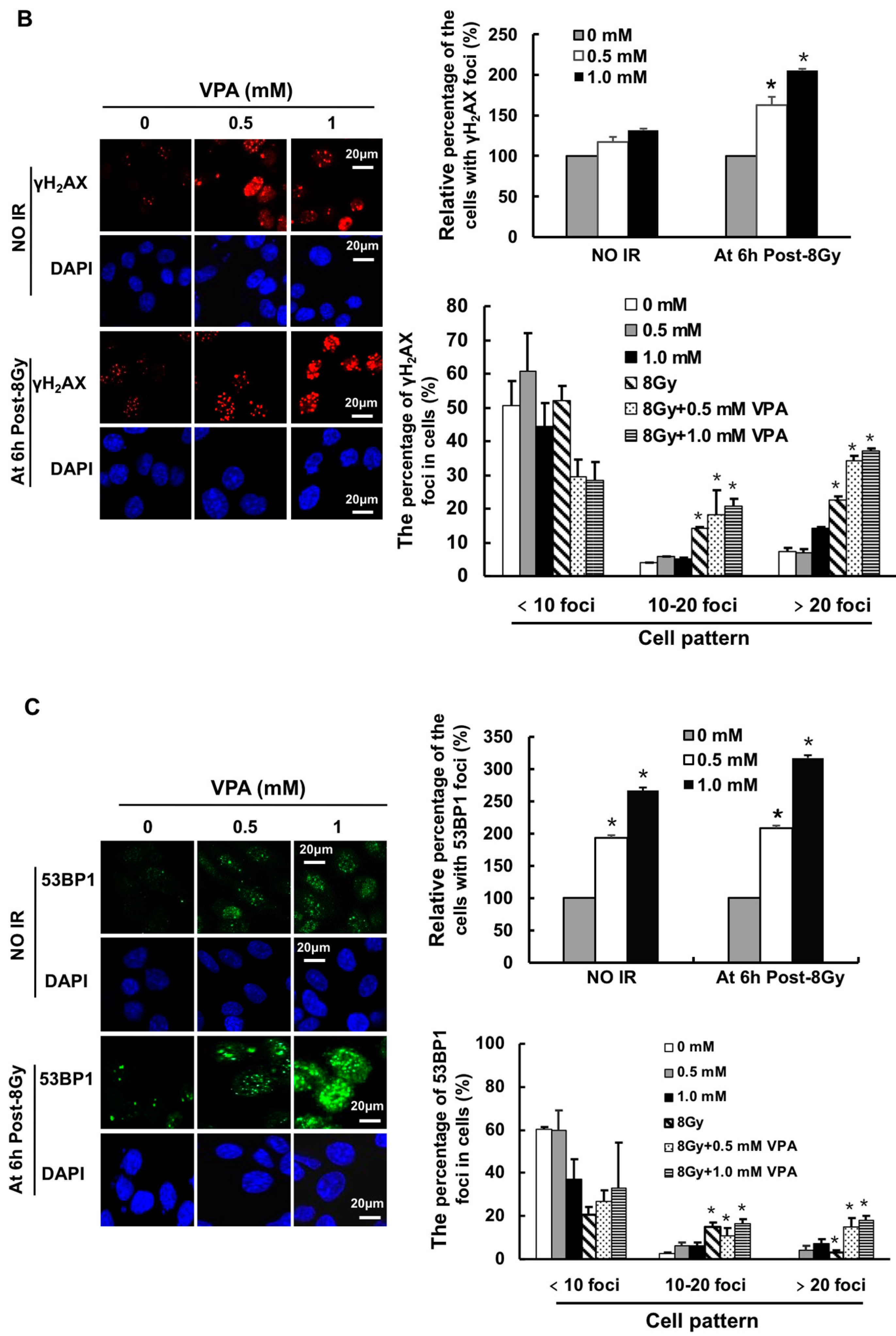
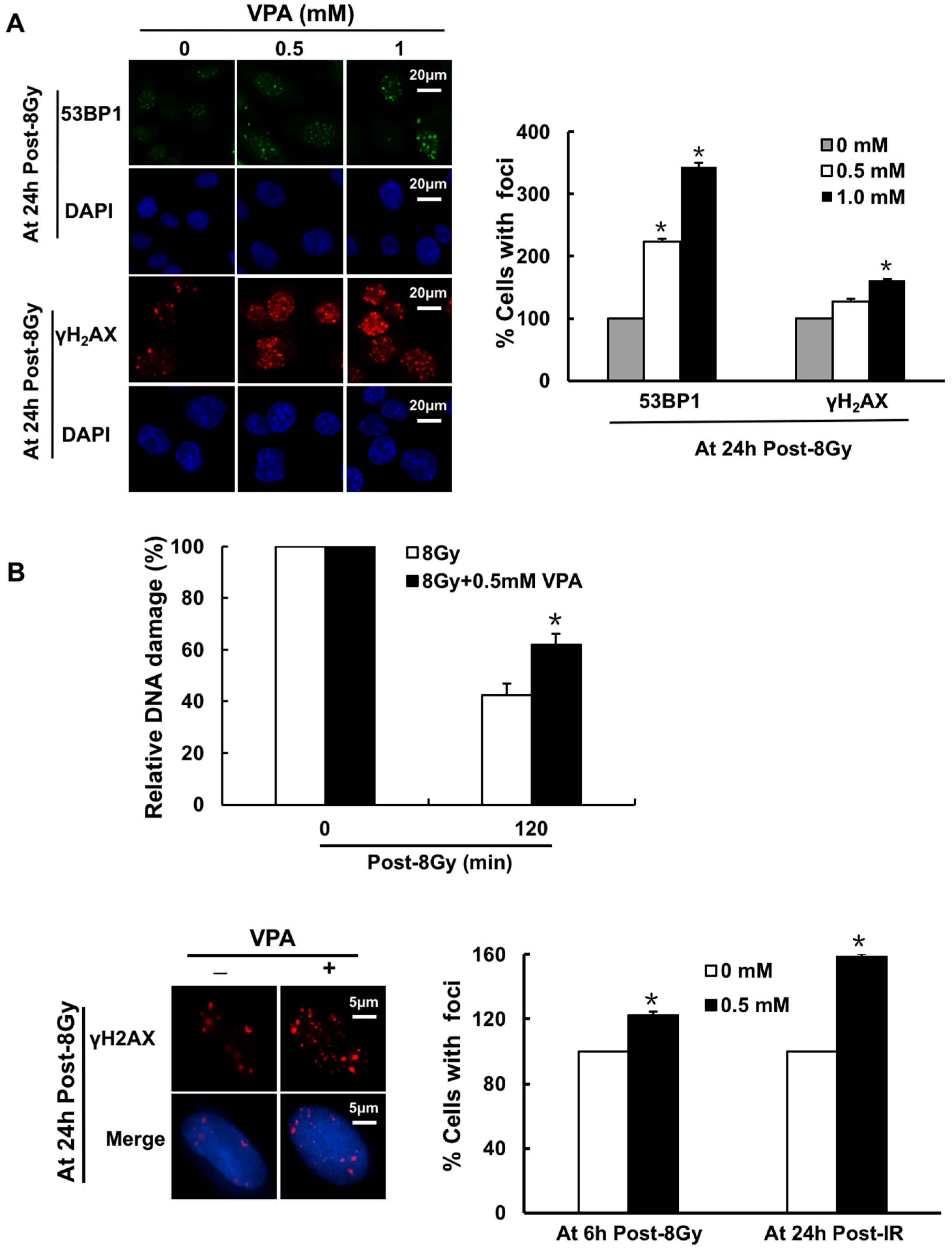
2.1. Effects of VPA on DNA-Double Strand Breaks (DSB) in Osteosarcoma Cells
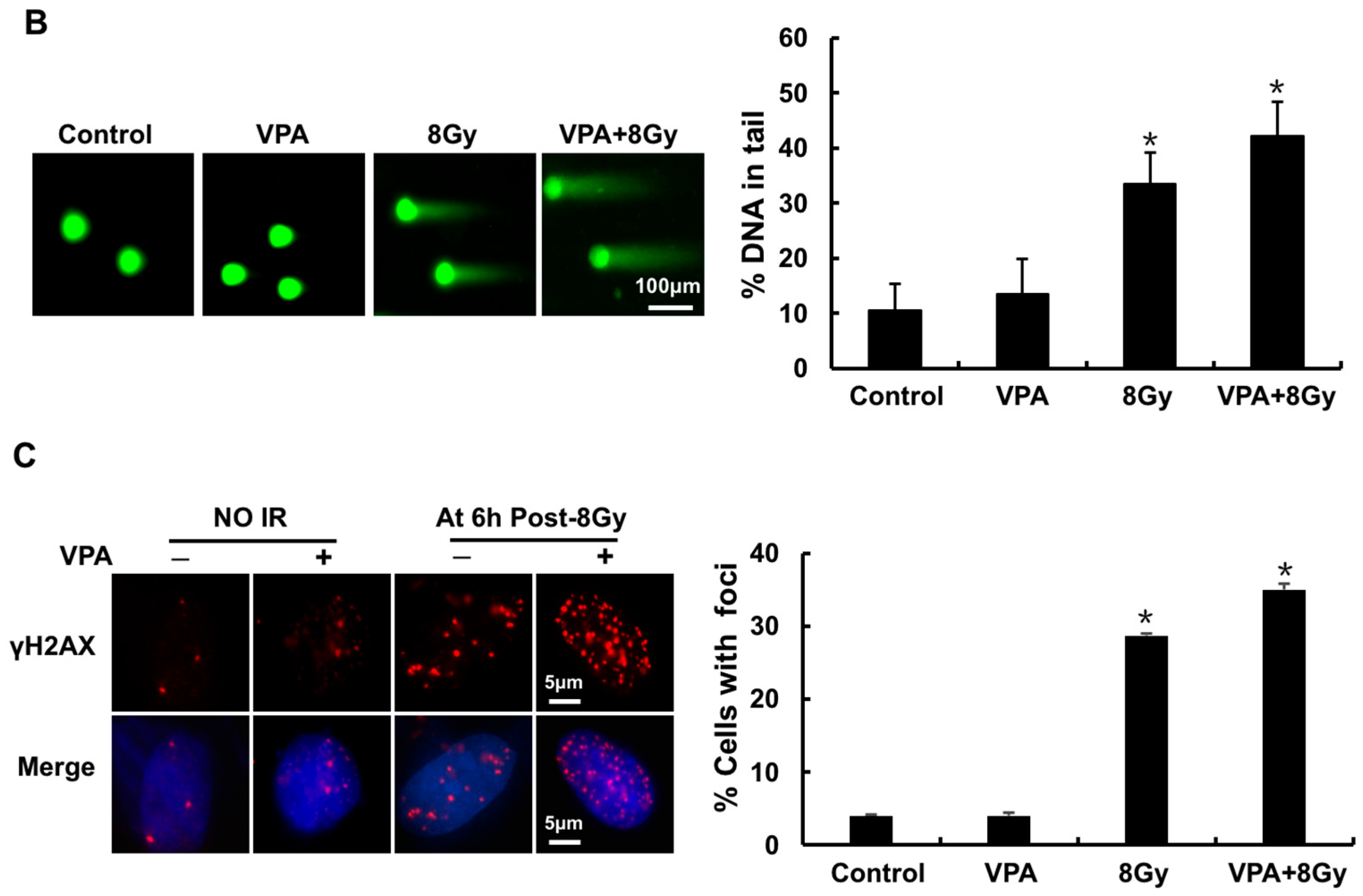
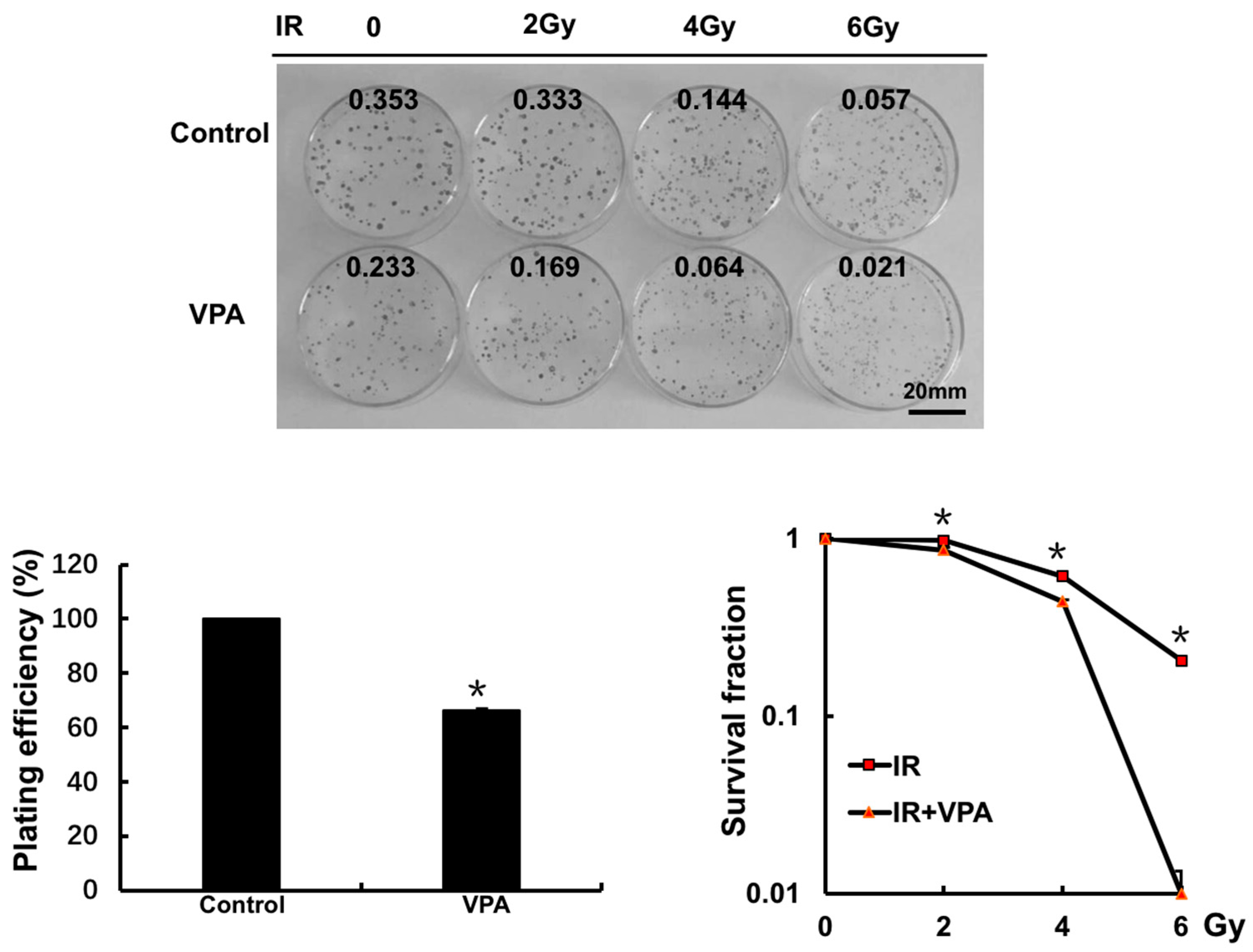
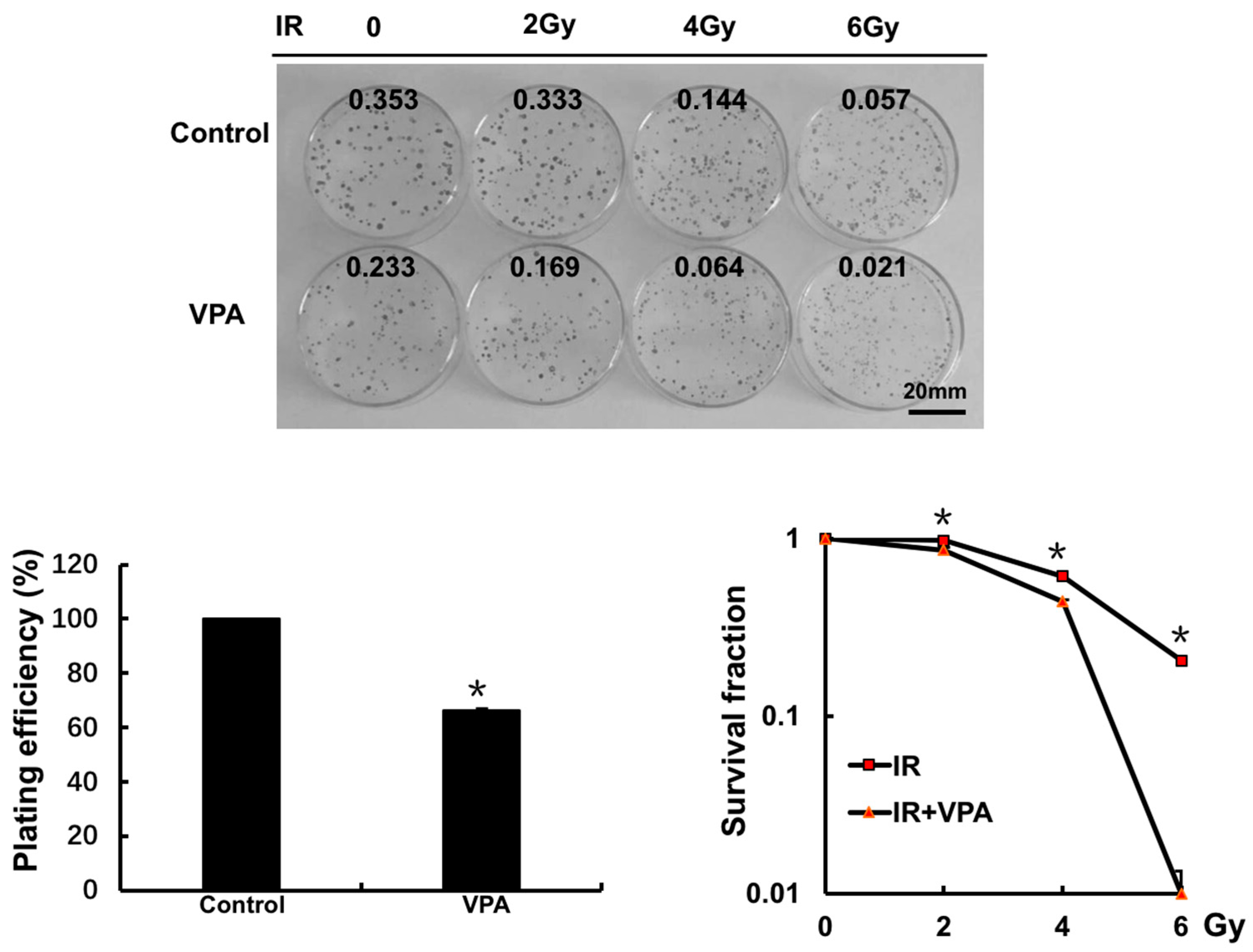
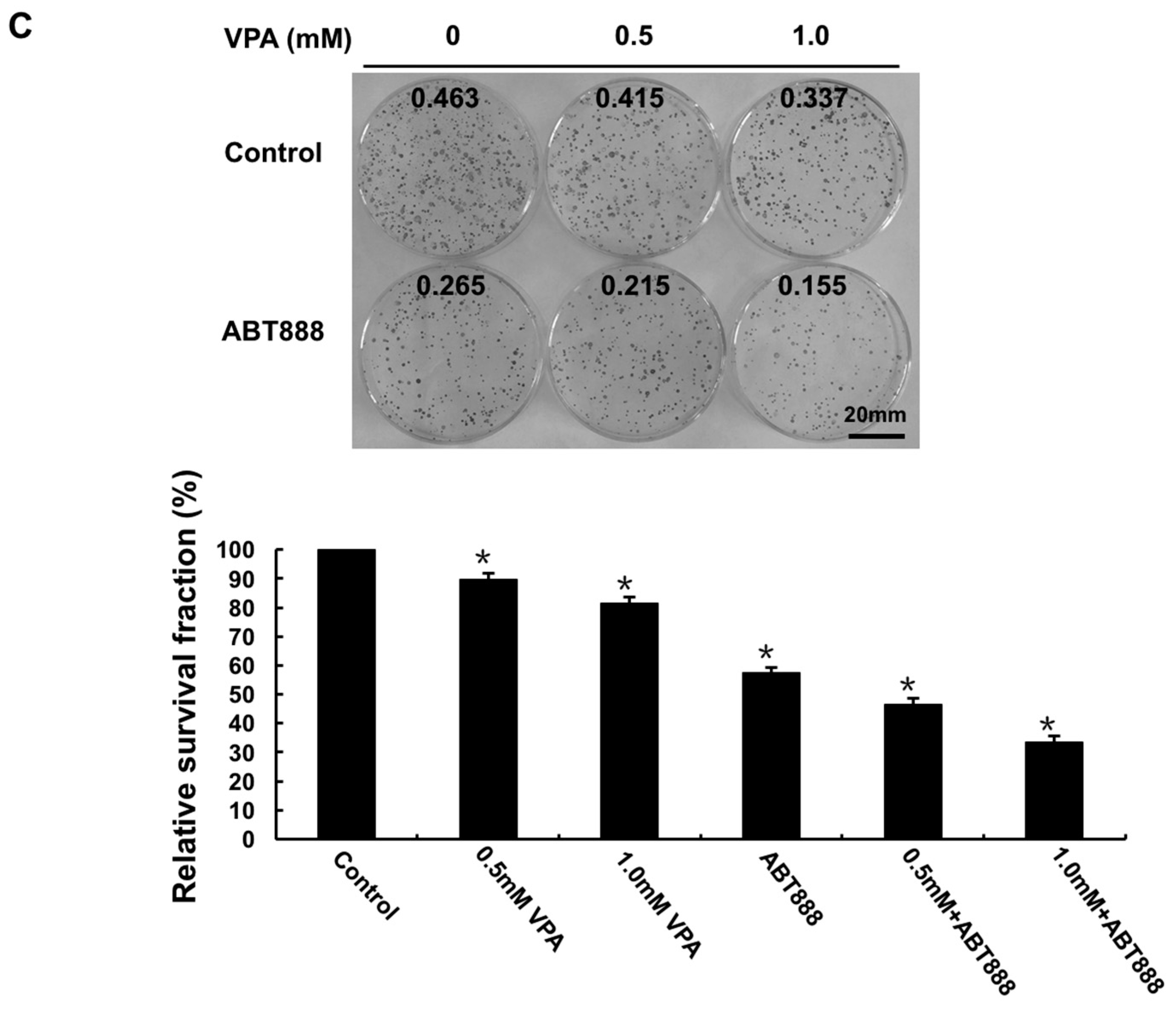
2.2. Effects of VPA on Radiosensitivity of Tumor Cells
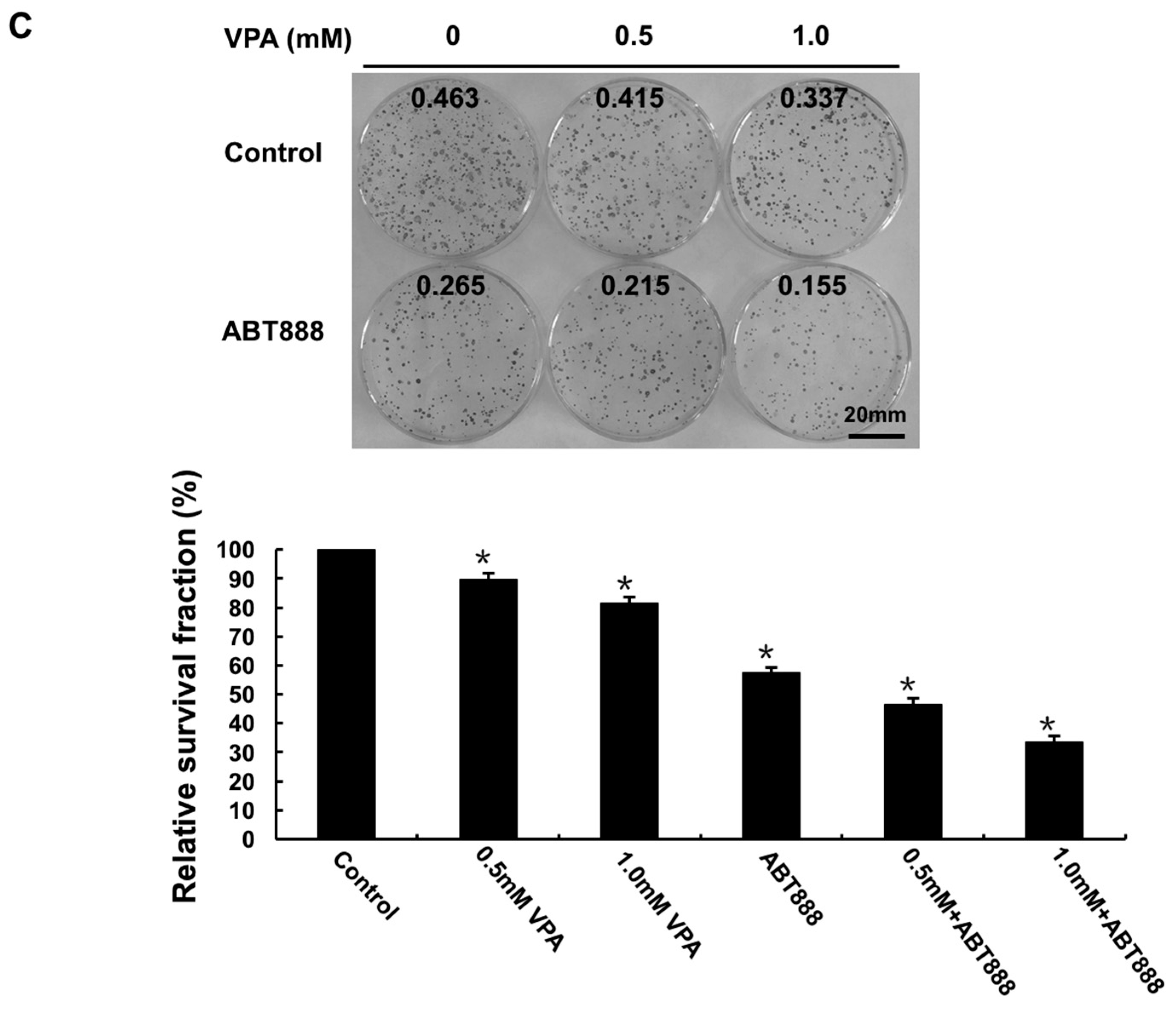
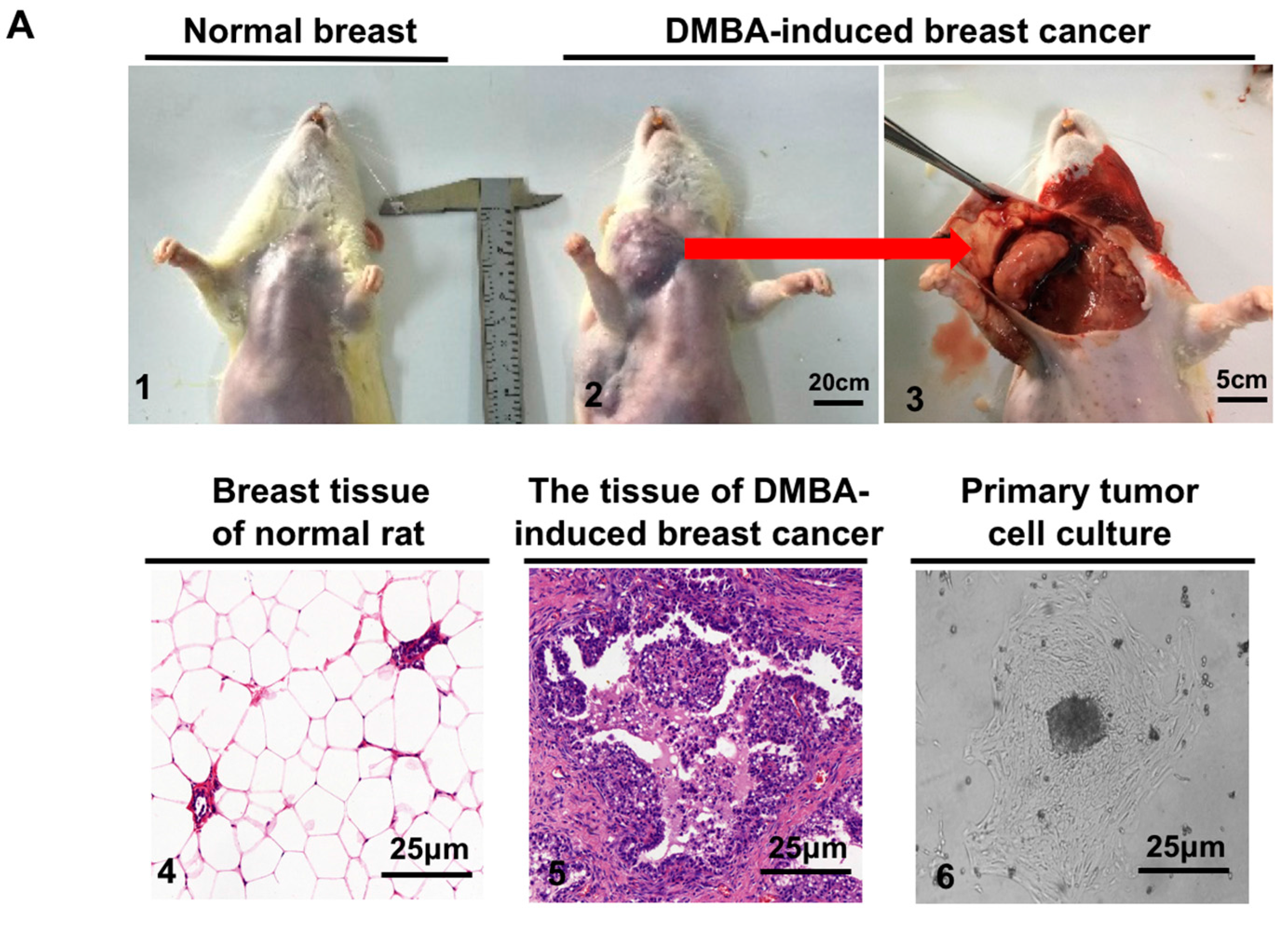
2.3. VPA at Safe Dose Can Arise the Dysfunction of DNA Repair
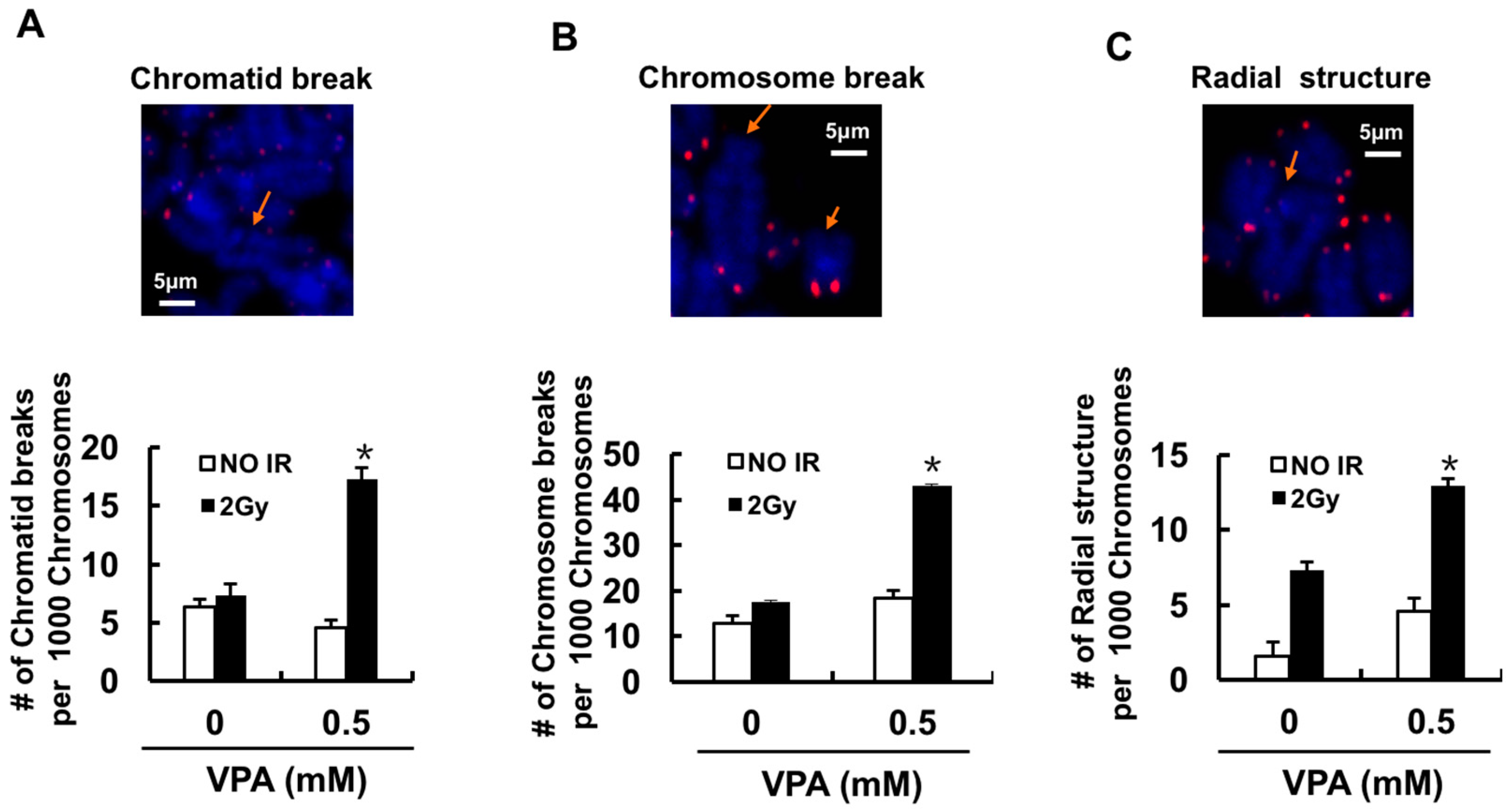
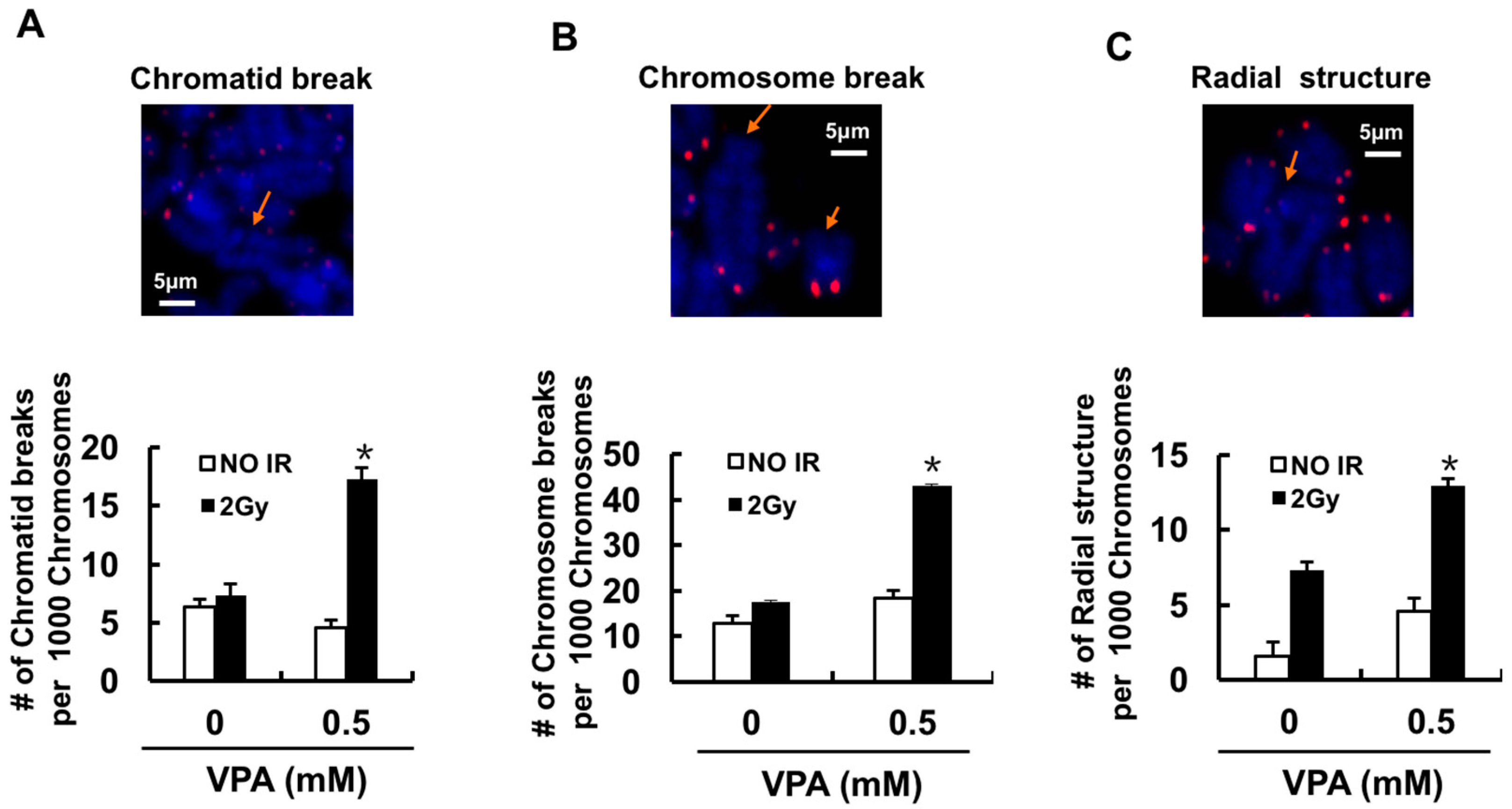
2.4. Effects of VPA on Chromosome Aberrations
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Line
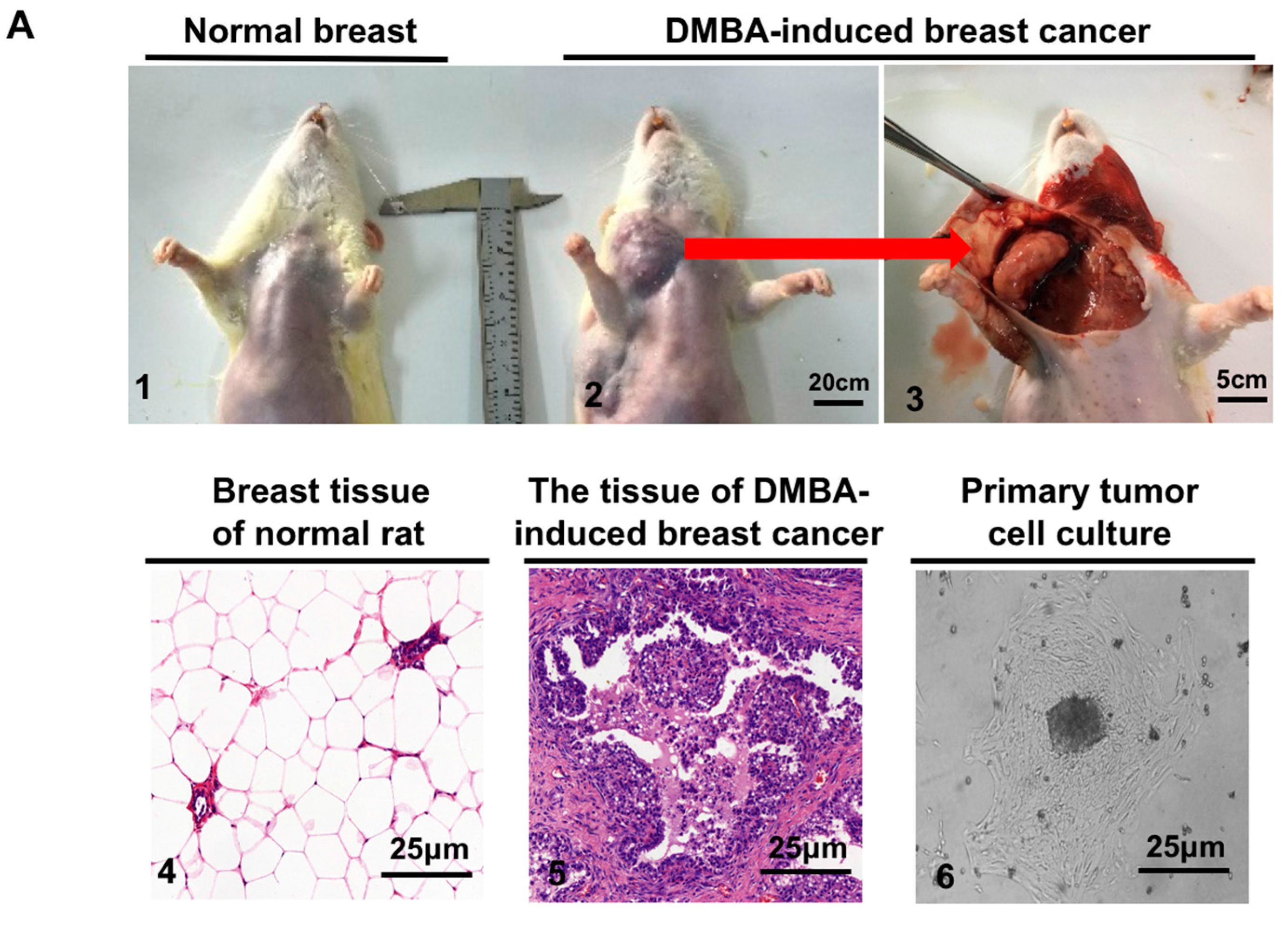
4.3. Tissue Culture and Animal Husbandry
4.4. Clonogenic Survival Assay
4.5. Quantitative Fluorescence In Situ Hybridization (Q-FISH) for Chromosomal Aberration Analysis
4.6. Comet Assay for DNA DSBs
4.7. Immunofluorescence Assay of γH2AX and 53BP1
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Groselj, B.; Sharma, N.L.; Hamdy, F.C.; Kerr, M.; Kiltie, A.E. Histone deacetylase inhibitors as radiosensitisers: Effects on DNA damage signalling and repair. Br. J. Cancer 2013, 108, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Mawatari, T.; Ninomiya, I.; Inokuchi, M.; Harada, S.; Hayashi, H.; Oyama, K.; Makino, I.; Nakagawara, H.; Miyashita, T.; Tajima, H.; et al. Valproic acid inhibits proliferation of HER2-expressing breast cancer cells by inducing cell cycle arrest and apoptosis through Hsp70 acetylation. Int. J. Oncol. 2015, 47, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Elbadawi, M.A.A.; Awadalla, M.K.A.; Hamid, M.M.A.; Mohamed, M.A.; Awad, T.A. Valproic Acid as a Potential Inhibitor of Plasmodium falciparum Histone Deacetylase 1 (PfHDAC1): An in Silico Approach. Int. J. Mol. Sci. 2015, 16, 3915–3931. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.; Marchion, D.; Bicaku, E.; Schmitt, M.; Lee, J.H.; DeConti, R.; Simon, G.; Fishman, M.; Minton, S.; Garrett, C.; et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: A clinical and translational study. J. Clin. Oncol. 2007, 25, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Marchion, D.C.; Bicaku, E.; Daud, A.I.; Sullivan, D.M.; Munster, P.N. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res. 2005, 65, 3815–3822. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Ninomiya, I.; Tsukada, T.; Okamoto, K.; Harada, S.; Nakanuma, S.; Sakai, S.; Makino, I.; Kinoshita, J.; Hayashi, H.; et al. Inhibitory effects of valproic acid in DNA double-strand break repair after irradiation in esophageal squamous carcinoma cells. Oncol. Rep. 2015, 34, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, H.; Zhao, X.P.; Dong, C.; Zhang, F.M.; Guo, G.; Guo, G.S.; Wang, X.W.; Powell, S.N.; Feng, Z.H. Valproic acid causes radiosensitivity of breast cancer cells via disrupting the DNA repair pathway. Toxicol. Res. UK 2016, 5, 859–870. [Google Scholar] [CrossRef]
- Van Oorschot, B.; Granata, G.; Di Franco, S.; ten Cate, R.; Rodermond, H.M.; Todaro, M.; Medema, J.P.; Franken, N.A.P. Targeting DNA double strand break repair with hyperthermia and DNA-PKCS inhibition to enhance the effect of radiation treatment. Oncotarget 2016, 7, 65504–65513. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, P.; Cerna, D.; Burgan, W.E.; Beam, K.; Williams, E.S.; Camphausen, K.; Tofilon, P.J. Postradiation sensitization of the histone deacetylase inhibitor valproic acid. Clin. Cancer Res. 2008, 14, 5410–5415. [Google Scholar] [CrossRef] [PubMed]
- Mamo, T.; Mladek, A.C.; Shogren, K.L.; Gustafson, C.; Gupta, S.K.; Riester, S.M.; Maran, A.; Galindo, M.; van Wijnen, A.J.; Sarkaria, J.N.; et al. Inhibiting DNA-PKCS radiosensitizes human osteosarcoma cells. Biochem. Biophys. Res. Commun. 2017, 486, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Zuch, D.; Giang, A.H.; Shapovalov, Y.; Schwarz, E.; Rosier, R.; O’Keefe, R.; Eliseev, R.A. Targeting radioresistant osteosarcoma cells with parthenolide. J. Cell. Biochem. 2012, 113, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Wang, X.; Pan, Y.; Lee, D.H.; Chowdhury, D.; Kimmelman, A.C. Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PLoS ONE 2012, 7, e39588. [Google Scholar] [CrossRef] [PubMed]
- Lobrich, M.; Shibata, A.; Beucher, A.; Fisher, A.; Ensminger, M.; Goodarzi, A.A.; Barton, O.; Jeggo, P.A. gamma H2AX foci analysis for monitoring DNA double-strand break repair Strengths, limitations and optimization. Cell Cycle 2010, 9, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Malewicz, M. The role of 53BP1 protein in homology-directed DNA repair: Things get a bit complicated. Cell Death Differ. 2016, 23, 1902–1903. [Google Scholar] [CrossRef] [PubMed]
- Maes, K.; De Smedt, E.; Lemaire, M.; De Raeve, H.; Menu, E.; Van Valckenborgh, E.; McClue, S.; Vanderkerken, K.; De Bruyne, E. The role of DNA damage and repair in decitabine-mediated apoptosis in multiple myeloma. Oncotarget 2014, 5, 3115–3129. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, M.; Hanscheid, H.; Gassen, D.; Biko, J.; Meineke, V.; Reiners, C.; Scherthan, H. In Vivo Formation of gamma-H2AX and 53BP1 DNA Repair Foci in Blood Cells After Radioiodine Therapy of Differentiated Thyroid Cancer. J. Nucl. Med. 2010, 51, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Croco, E.; Marchionni, S.; Bocchini, M.; Angeloni, C.; Stamato, T.; Stefanelli, C.; Hrelia, S.; Sell, C.; Lorenzini, A. DNA Damage Detection by 53BP1: Relationship to Species Longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.B. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Brunen, D.; Bernards, R. Drug therapy: Exploiting synthetic lethality to improve cancer therapy. Nat. Rev. Clin. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.S.; Zhang, F.M.; Gao, R.J.; Delsite, R.; Feng, Z.H.; Powell, S.N. DNA repair and synthetic lethality. Int. J. Oral Sci. 2011, 3, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Ninomiya, I.; Makino, I.; Kinoshita, J.; Nakamura, K.; Oyama, K.; Nakagawara, H.; Fujita, H.; Tajima, H.; Takamura, H.; et al. Valproic acid, a histone deacetylase inhibitor, enhances radiosensitivity in esophageal squamous cell carcinoma. Int. J. Oncol. 2012, 40, 2140–2146. [Google Scholar] [CrossRef] [PubMed]
- Defoort, E.N.; Kim, P.M.; Winn, L.M. Valproic acid increases conservative homologous recombination frequency and reactive oxygen species formation: A potential mechanism for valproic acid-induced neural tube defects. Mol. Pharmacol. 2006, 69, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wong, P.; Radany, E.; Wong, J.Y. HDAC inhibitor, valproic acid, induces p53-dependent radiosensitization of colon cancer cells. Cancer Biother. Radiopharm. 2009, 24, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Adimoolam, S.; Sirisawad, M.; Chen, J.; Thiemann, P.; Ford, J.M.; Buggy, J.J. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA 2007, 104, 19482–19487. [Google Scholar] [CrossRef] [PubMed]
- Koprinarova, M.; Botev, P.; Russev, G. Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair 2011, 10, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Kachhap, S.K.; Rosmus, N.; Collis, S.J.; Kortenhorst, M.S.; Wissing, M.D.; Hedayati, M.; Shabbeer, S.; Mendonca, J.; Deangelis, J.; Marchionni, L.; et al. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS ONE 2010, 5, e11208. [Google Scholar] [CrossRef] [PubMed]
- Blattmann, C.; Oertel, S.; Ehemann, V.; Thiemann, M.; Huber, P.E.; Bischof, M.; Witt, O.; Deubzer, H.E.; Kulozik, A.E.; Debus, J.; et al. Enhancement of radiation response in osteosarcoma and rhabdomyosarcoma cell lines by histone deacetylase inhibition. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Zhang, F.; Luo, Y.; Wang, H.; Zhao, X.; Guo, G.; Powell, S.N.; Feng, Z. p53 suppresses hyper-recombination by modulating BRCA1 function. DNA Repair 2015, 33, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.H.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.S.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.H.; Zhang, J.R. A dual role of BRCA1 in two distinct homologous recombination mediated repair in response to replication arrest. Nucleic Acids Res. 2012, 40, 726–738. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Wang, H.; Zhang, F.; Tian, Y.; Tian, Z.; Cai, Z.; Lim, D.; Feng, Z. The Effect of VPA on Increasing Radiosensitivity in Osteosarcoma Cells and Primary-Culture Cells from Chemical Carcinogen-Induced Breast Cancer in Rats. Int. J. Mol. Sci. 2017, 18, 1027. https://doi.org/10.3390/ijms18051027
Liu G, Wang H, Zhang F, Tian Y, Tian Z, Cai Z, Lim D, Feng Z. The Effect of VPA on Increasing Radiosensitivity in Osteosarcoma Cells and Primary-Culture Cells from Chemical Carcinogen-Induced Breast Cancer in Rats. International Journal of Molecular Sciences. 2017; 18(5):1027. https://doi.org/10.3390/ijms18051027
Chicago/Turabian StyleLiu, Guochao, Hui Wang, Fengmei Zhang, Youjia Tian, Zhujun Tian, Zuchao Cai, David Lim, and Zhihui Feng. 2017. "The Effect of VPA on Increasing Radiosensitivity in Osteosarcoma Cells and Primary-Culture Cells from Chemical Carcinogen-Induced Breast Cancer in Rats" International Journal of Molecular Sciences 18, no. 5: 1027. https://doi.org/10.3390/ijms18051027
APA StyleLiu, G., Wang, H., Zhang, F., Tian, Y., Tian, Z., Cai, Z., Lim, D., & Feng, Z. (2017). The Effect of VPA on Increasing Radiosensitivity in Osteosarcoma Cells and Primary-Culture Cells from Chemical Carcinogen-Induced Breast Cancer in Rats. International Journal of Molecular Sciences, 18(5), 1027. https://doi.org/10.3390/ijms18051027