
Calcium Dynamics Mediated by the Endoplasmic/Sarcoplasmic Reticulum and Related Diseases

Abstract
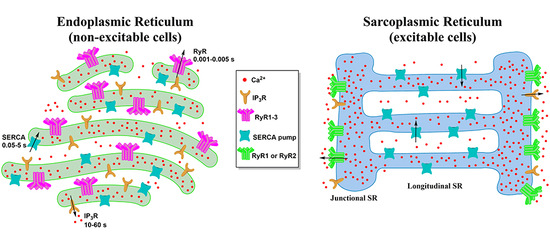
:
1. Introduction
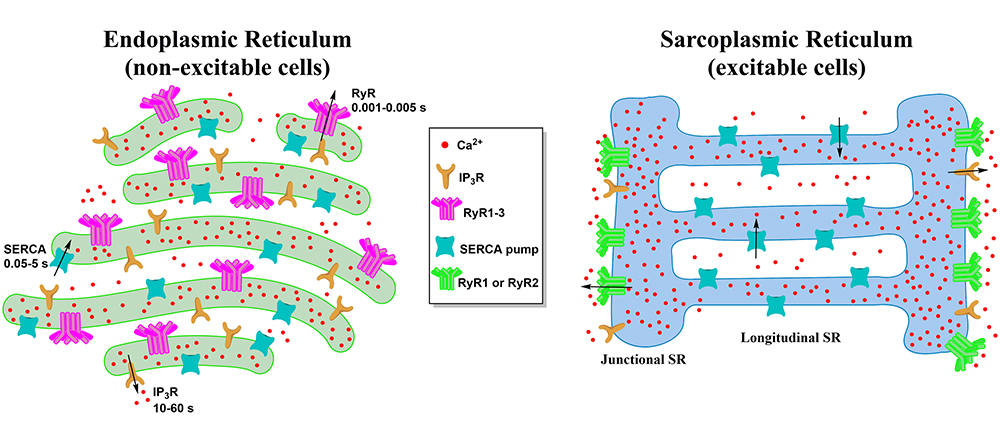
2. Morphology of the ER and the SR
3. ER/SR Mediated Calcium Signaling
3.1. The Ryanodine Receptor
3.2. The Inositol 1,4,5-Trisphosphate Receptor
3.3. The Sarco-Endoplasmic Reticulum Ca2+ ATPase Pump
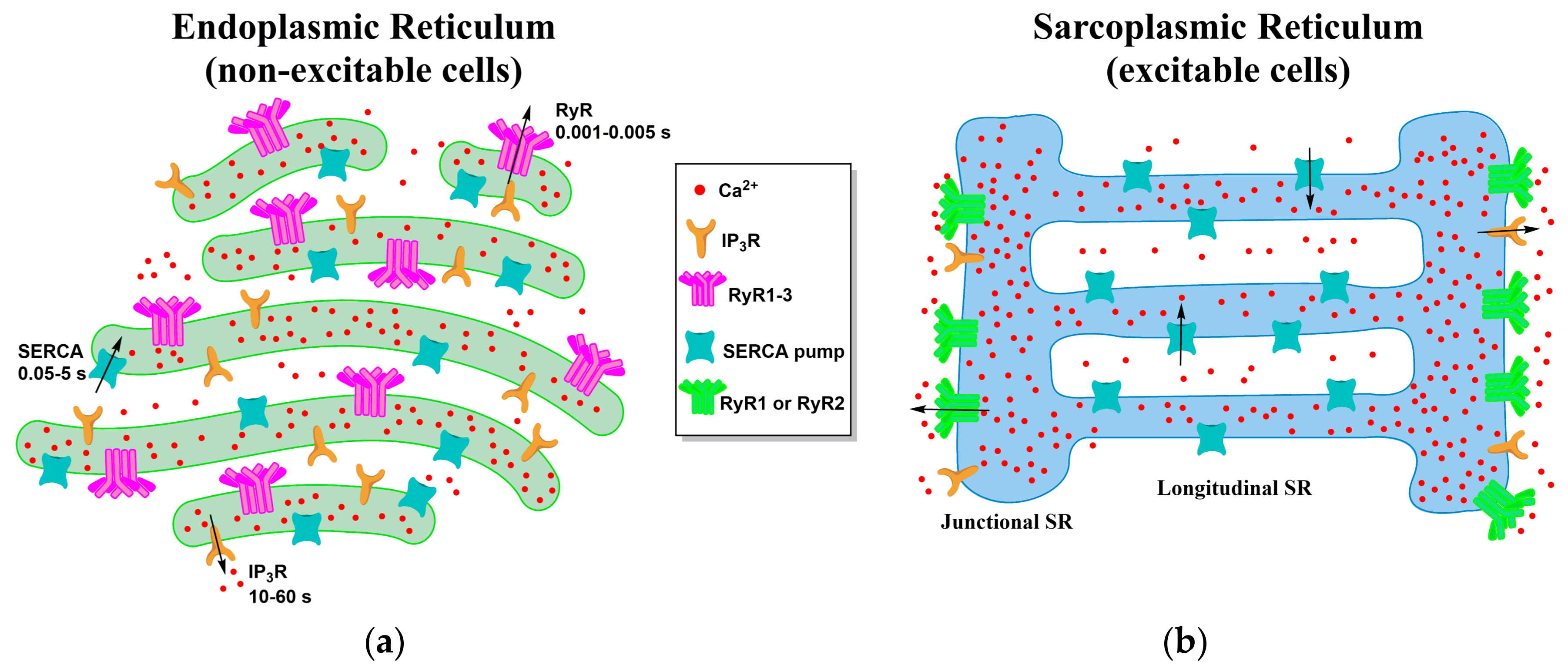
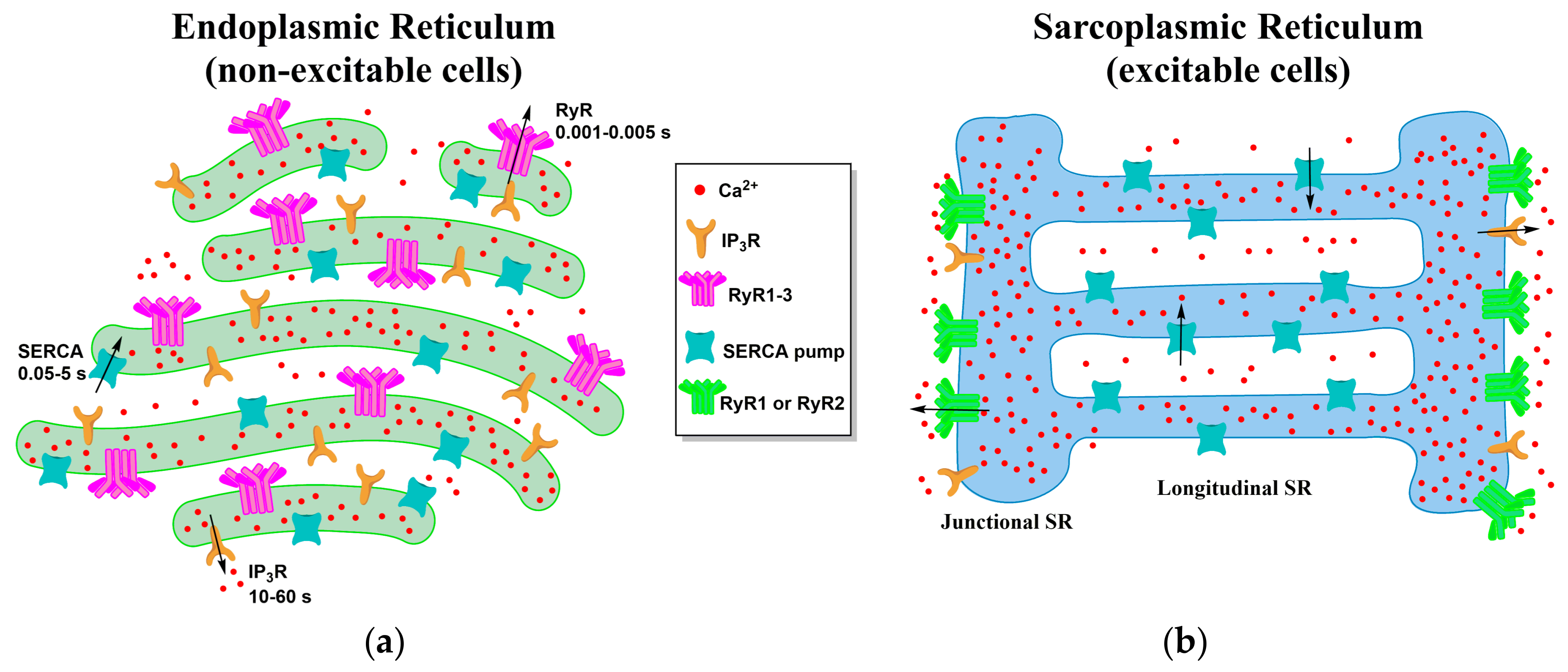
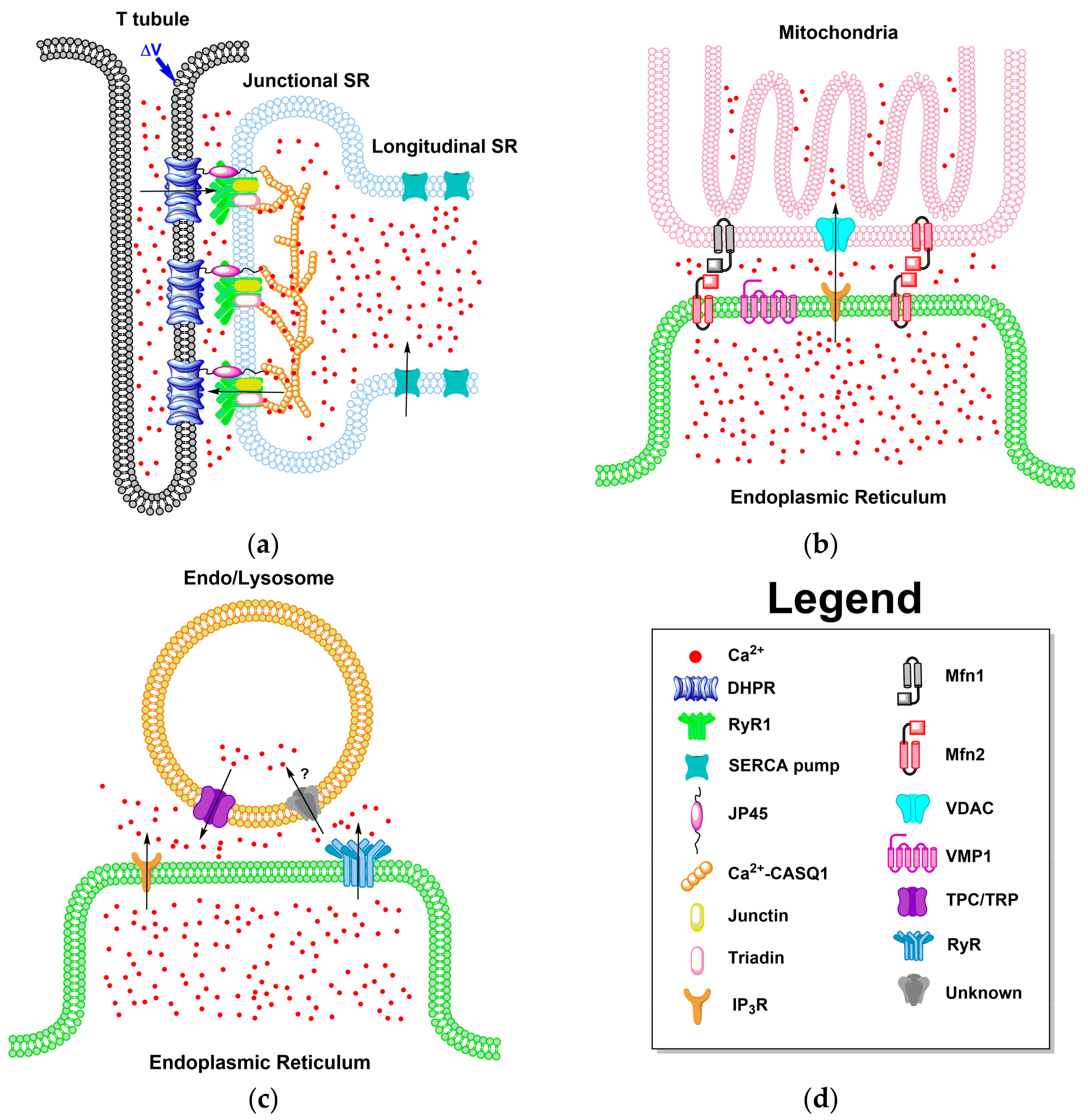
4. Local Domains of ER/SR Calcium Release
4.1. ER-PM Junction of Skeletal Muscle and Cardiac Cells
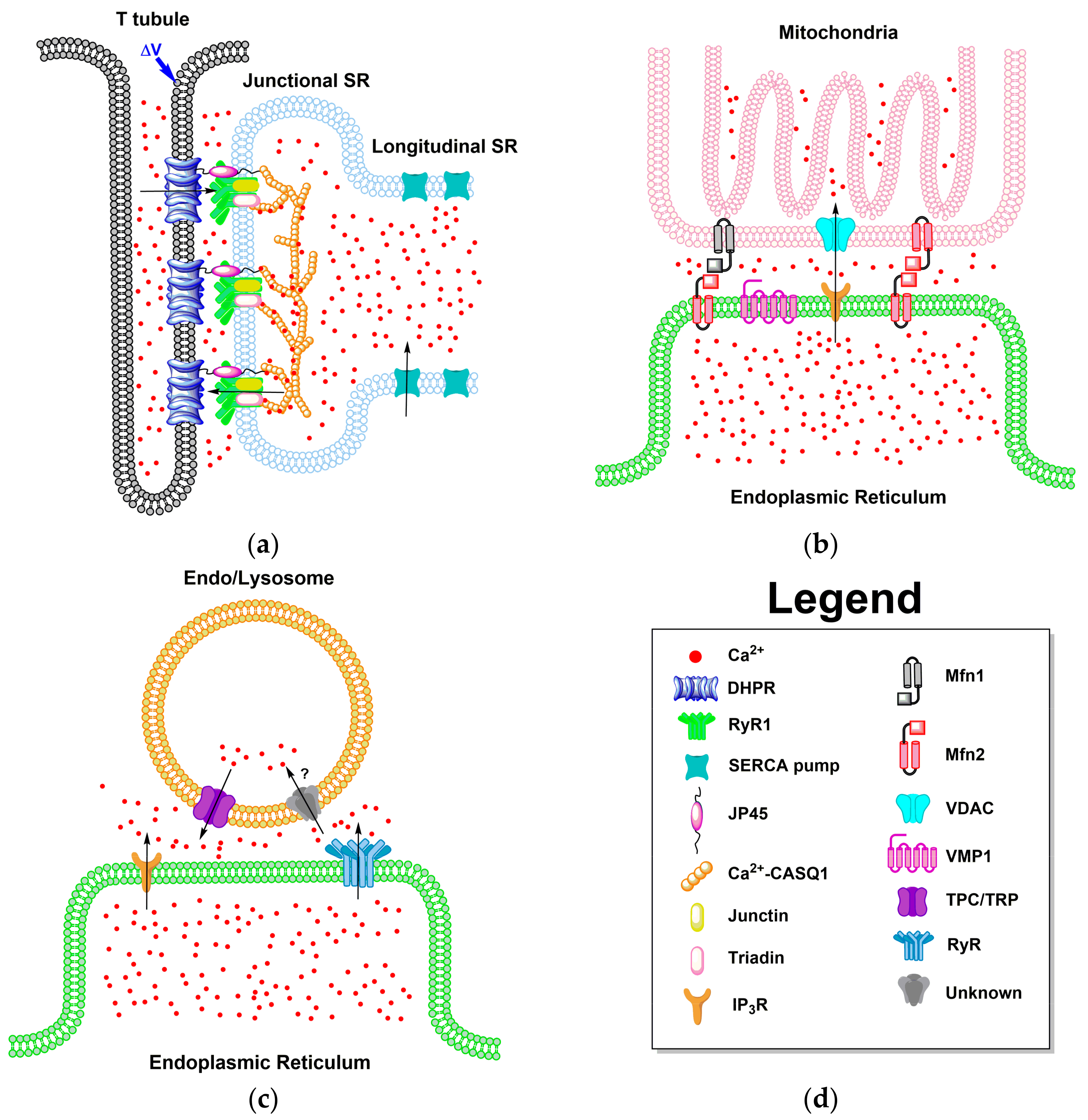
4.2. ER Junctions with Other Organelles
5. Diseases Associated with ER/SR Calcium Signaling
5.1. Brody’s Disease
5.2. Catecholaminergic Polymorphic Ventricular Tachycardia
5.3. Malignant Hyperthermia
5.4. Alzheimer’s Disease
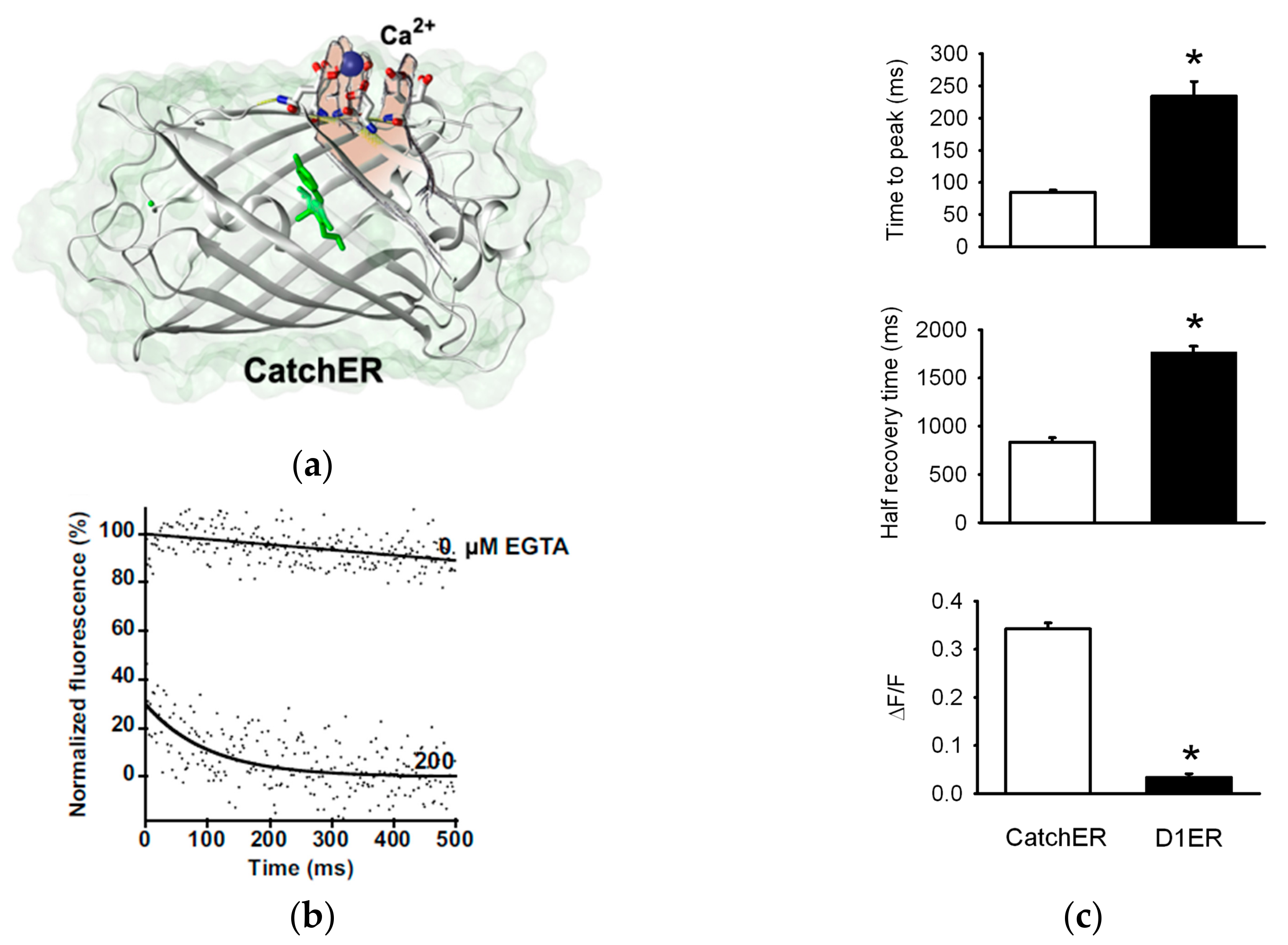
6. ER/SR-Targeted GECIs
7. Summary and Perspective
Acknowledgments
Conflicts of Interest
Abbreviations
| Ca2+ | Calcium |
| ER | Endoplasmic reticulum |
| SR | Sarcoplasmic reticulum |
| RER | Rough ER |
| SER | Smooth ER |
| CaBP | Calcium-binding proteins |
| GECI | Genetically encoded calcium indicators |
| IP3 | Inositol 1,4,5-triphosphate |
| IP3R | Inositol 1,4,5-triphosphate receptor |
| RyR | Ryanodine receptor |
| cADPR | Cyclic ADP-ribose |
| CICR | Calcium induced calcium release |
| SERCA | Sarco-endoplasmic reticulum Ca2+ ATPase |
| UPR | Unfolded protein response |
| MCS | Membrane contact sites |
| PM | Plasma membrane |
| MAM | Mitochondria-associated ER membrane |
| NE | Nuclear envelope |
| TC | Terminal cisternae |
| CaM | Calmodulin |
| 4-CmC | 4-Chloro-m-cresol |
| E-C | Excitation-contraction |
| DHPR | Dihydropyridine receptor |
| PIP2 | Phosphoinositol-4 5-bisphosphate |
| Mg2+ | Magnesium |
| PLC | Phospholipase C |
| GPCR | G-protein coupled receptors |
| PKA | Protein kinase A |
| PKB | Protein kinase B |
| CDK1 | Cycline-dependent kinase 1 |
| CPA | Cyclopiazonic acid |
| JP45 | Junctional protein 45 |
| CASQ1 | Calsequestrin (skeletal muscle) |
| VDAC | Voltage-dependent anion channel |
| MCU | Mitochondrial Ca2+ uniporter |
| VMP1 | Vacuole membrane protein 1 |
| TRP | Transient receptor channels |
| Mfn | Mitofusion proteins |
| TPC | Two-pore channels |
| NAADP | Nicotinic acid adenine dinucleotide phosphate |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| MH | Malignant hyperthermia |
| ATP | Adenosine triphosphate |
| AD | Alzheimer’s disease |
| SAD | Sporadic AD |
| FAD | Familial AD |
| Aβ | Amyloid-beta |
| τ | Tau |
| APP | Amyloid precursor protein |
| PS1 | Presenilins 1 |
| PS2 | Presenilins 2 |
| SNR | Signal to noise ratio |
| TnC | Troponin C |
| FP | Fluorescent protein |
| Ƭrt | Dissociation time constant |
| cpEGFP | Single circularly permuted enhanced green fluorescent protein |
| FDB | Flexor digitorum brevis |
| FLIM | Fluorescence lifetime imaging microscopy |
| FRET | Förster resonance energy transfer |
| CFP | Cyan fluorescent protein |
| YFP | Yellow fluorescent protein |
References
- Al-Shanti, N.; Stewart, C.E. Ca2+/calmodulin-dependent transcriptional pathways: Potential mediators of skeletal muscle growth and development. Biol. Rev. 2009, 84, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Lipskaia, L.; Hulot, J.S.; Lompré, A.M. Role of sarco/endoplasmic reticulum calcium content and calcium ATPase activity in the control of cell growth and proliferation. Pflüg. Arch. Eur. J. Physiol. 2009, 457, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Baylor, S.; Hollingworth, S. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. J. Physiol. 2003, 551, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Dolmetsch, R.E.; Xu, K.; Lewis, R.S. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 1998, 392, 933–936. [Google Scholar] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef]
- Chen, S.; Novick, P.; Ferro-Novick, S. ER structure and function. Curr. Opin. Cell Biol. 2013, 25, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 2009, 1793, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Marrion, N.; Adams, P. Release of intracellular calcium and modulation of membrane currents by caffeine in bull-frog sympathetic neurones. J. Physiol. 1992, 445, 515. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.Y.; Tokimasa, T.; Takasawa, S.; Furuya, Y.; Nohmi, M.; Okamoto, H.; Kuba, K. Cyclic ADP-ribose modulates Ca2+ release channels for activation by physiological Ca2+ entry in bullfrog sympathetic neurons. Neuron 1994, 12, 1073–1079. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Shmigol, A. Calcium-induced calcium release in neurones. Cell Calcium 1996, 19, 1–14. [Google Scholar] [CrossRef]
- Parsons, R.L.; Barstow, K.L.; Scornik, F.S. Spontaneous miniature hyperpolarizations affect threshold for action potential generation in mudpuppy cardiac neurons. J. Neurophysiol. 2002, 88, 1119–1127. [Google Scholar] [PubMed]
- Seo, M.D.; Enomoto, M.; Ishiyama, N.; Stathopulos, P.B.; Ikura, M. Structural insights into endoplasmic reticulum stored calcium regulation by inositol 1,4,5-trisphosphate and ryanodine receptors. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Endo, M. Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 2009, 89, 1153–1176. [Google Scholar] [CrossRef] [PubMed]
- Vandecaetsbeek, I.; Vangheluwe, P.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. The Ca(2+) pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a004184. [Google Scholar] [CrossRef] [PubMed]
- Burdakov, D.; Petersen, O.H.; Verkhratsky, A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium 2005, 38, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef] [PubMed]
- Pozzan, T.; Rizzuto, R.; Volpe, P.; Meldolesi, J. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 1994, 74, 595–636. [Google Scholar] [PubMed]
- Zhang, H.; Hu, J. Shaping the endoplasmic reticulum into a social network. Trends Cell Biol. 2016, 26, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Navarro, N.; Miller, E. Protein sorting at the ER–Golgi interface. J. Cell Biol. 2016, 215, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, S.; Meyer, T. Stim proteins and the endoplasmic reticulum-plasma membrane junctions. Annu. Rev. Biochem. 2011, 80, 973–1000. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluth, J. Subsurface cisterns and their relationship to the neuronal plasma membrane. J. Cell Biol. 1962, 13, 405–421. [Google Scholar] [CrossRef] [PubMed]
- West, M.; Zurek, N.; Hoenger, A.; Voeltz, G.K. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J. Cell Biol. 2011, 193, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.E.; Dirksen, R.T. Sarcoplasmic reticulum: The dynamic calcium governor of muscle. Muscle Nerve 2006, 33, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Stefan, C.J.; Manford, A.G.; Emr, S.D. ER–PM connections: Sites of information transfer and inter-organelle communication. Curr. Opin. Cell Biol. 2013, 25, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.K.M.; Galione, A. The endoplasmic reticulum and junctional membrane communication during calcium signaling. Biochim. Biophys. Acta 2013, 1833, 2542–2559. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [PubMed]
- Sorrentino, V. Molecular determinants of the structural and functional organization of the sarcoplasmic reticulum. Biochim. Biophys. Acta 2004, 1742, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Barone, V.; Giacomello, E.; Cusimano, V.; Sorrentino, V. The sarcoplasmic reticulum: An organized patchwork of specialized domains. Traffic 2008, 9, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Franzini-Armstrong, C.; Protasi, F.; Ramesh, V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 1999, 77, 1528–1539. [Google Scholar] [CrossRef]
- Sorrentino, V. Sarcoplasmic reticulum: Structural determinants and protein dynamics. Int. J. Biochem. Cell Biol. 2011, 43, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.R.; Palade, G.E. Studies on the endoplasmic reticulum III. Its form and distribution in striated muscle cells. J. Biophys. Biochem. Cytol. 1957, 3, 269–300. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium signalling remodelling and disease. Biochem. Soc. Trans. 2012, 40, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Tsugorka, A.; Rios, E.; Blatter, L.A. Imaging elementary events of calcium release in skeletal muscle cells. Science 1995, 269, 1723. [Google Scholar] [CrossRef] [PubMed]
- Shirokova, N.; García, J.; Ríos, E. Local calcium release in mammalian skeletal muscle. J. Physiol. 1998, 512, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- FranziniArmstrong, C.; Protasi, F. Ryanodine receptors of striated muscles: A complex channel capable of multiple interactions. Physiol. Rev. 1997, 77, 699–729. [Google Scholar]
- Hakamata, Y.; Nakai, J.; Takeshima, H.; Imoto, K. Primary structure and distribution of a novel ryanodine receptor/calcium release channel from rabbit brain. FEBS Lett. 1992, 312, 229–235. [Google Scholar] [CrossRef]
- Tarroni, P.; Rossi, D.; Conti, A.; Sorrentino, V. Expression of the ryanodine receptor type 3 calcium release channel during development and differentiation of mammalian skeletal muscle cells. J. Biol. Chem. 1997, 272, 19808–19813. [Google Scholar] [CrossRef] [PubMed]
- Zalk, R.; Clarke, O.B.; des Georges, A.; Grassucci, R.A.; Reiken, S.; Mancia, F.; Hendrickson, W.A.; Frank, J.; Marks, A.R. Structure of a mammalian ryanodine receptor. Nature 2015, 517, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Bai, X.C.; Yan, C.Y.; Wu, J.P.; Li, Z.Q.; Xie, T.; Peng, W.; Yin, C.C.; Li, X.M.; Scheres, S.H.W.; et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 2015, 517, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Capes, E.M.; Loaiza, R.; Valdivia, H.H. Ryanodine receptors. Skelet. Muscle 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- Lamb, G.; Stephenson, D. Effect of Mg2+ on the control of Ca2+ release in skeletal muscle fibres of the toad. J. Physiol. 1991, 434, 507. [Google Scholar] [CrossRef] [PubMed]
- Herrmann-Frank, A.; Lüttgau, H.C.; Stephenson, D.G. Caffeine and excitation–contraction coupling in skeletal muscle: A stimulating story. J. Muscle Res. Cell Motil. 1999, 20, 223–236. [Google Scholar] [CrossRef] [PubMed]
- HerrmannFrank, A.; Richter, M.; Sarkozi, S.; Mohr, U.; LehmannHorn, F. 4-Chloro-m-cresol, a potent and specific activator of the skeletal muscle ryanodine receptor. Biochim. Biophys. Acta Gen. Subj. 1996, 1289, 31–40. [Google Scholar] [CrossRef]
- Nakai, J.; Sekiguchi, N.; Rando, T.A.; Allen, P.D.; Beam, K.G. Two regions of the ryanodine receptor involved in coupling withl-type Ca2+ channels. J. Biol. Chem. 1998, 273, 13403–13406. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.F.; Mukherjee, S.; Allen, P.D. Amino acids 1–1,680 of ryanodine receptor type 1 hold critical determinants of skeletal type for excitation-contraction coupling—Role of divergence domain D2. J. Biol. Chem. 2003, 278, 39644–39652. [Google Scholar] [CrossRef] [PubMed]
- Nakai, J.; Dirksen, R.T.; Nguyen, H.T.; Pessah, I.N.; Beam, K.G.; Allen, P.D. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 1996, 380, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Divet, A.; Paesante, S.; Bleunven, C.; Anderson, A.; Treves, S.; Zorzato, F. Novel sarco(endo) plasmic reticulum proteins and calcium homeostasis in striated muscles. J. Muscle Res. Cell Motil. 2005, 26, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M.; Fameli, N.; Ogunbayo, O.A.; Duan, J.; Navarro-Dorado, J. From contraction to gene expression: Nanojunctions of the sarco/endoplasmic reticulum deliver site- and function-specific calcium signals. Sci. China Life Sci. 2016, 59, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.L.; Boehning, D.; Snyder, S.H. Inositol 1,4,5-trisphosphatereceptors as signal integrators. Annu. Rev. Biochem. 2004, 73, 437–465. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.P.; Bhattacharyya, B.J.; Lin, H.; Gomez, C.M. Skeletal muscle IP3R1 receptors amplify physiological and pathological synaptic calcium signals. J. Neurosci. 2011, 31, 15269–15283. [Google Scholar] [CrossRef]
- Powell, J.A.; Molgó, J.; Adams, D.S.; Colasante, C.; Williams, A.; Bohlen, M.; Jaimovich, E. IP3 receptors and associated Ca2+ signals localize to satellite cells and to components of the neuromuscular junction in skeletal muscle. J. Neurosci. 2003, 23, 8185–8192. [Google Scholar] [PubMed]
- Wu, X.; Zhang, T.; Bossuyt, J.; Li, X.; McKinsey, T.A.; Dedman, J.R.; Olson, E.N.; Chen, J.; Brown, J.H.; Bers, D.M. Local InsP 3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J. Clin. Investig. 2006, 116, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Mikoshiba, K. Ip3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef] [PubMed]
- Serysheva, I.I. Toward a high-resolution structure of IP3R channel. Cell Calcium 2014, 56, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I. The inositol 1,4,5-trisphosphate receptors. Cell Calcium 2005, 38, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Ferris, C.D.; Huganir, R.L.; Bredt, D.S.; Cameron, A.M.; Snyder, S.H. Inositol trisphosphate receptor: Phosphorylation by protein kinase c and calcium calmodulin-dependent protein kinases in reconstituted lipid vesicles. Proc. Natl. Acad. Sci. USA 1991, 88, 2232–2235. [Google Scholar] [CrossRef] [PubMed]
- Dubyak, G.R.; el-Moatassim, C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am. J. Physiol. Cell Physiol. 1993, 265, C577–C606. [Google Scholar] [PubMed]
- Song, Z.; Vijayaraghavan, S.; Sladek, C.D. Atp increases intracellular calcium in supraoptic neurons by activation of both P2X and P2Y purinergic receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R423–R431. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.Z.; Zhao, D.; Khan, S.H.; Yang, L. Regulatory mechanisms of endoplasmic reticulum resident IP3 receptors. J. Mol. Neurosci. MN 2015, 56, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.K.; Khan, I. Calcium-pump isoforms—Diversity, selectivity and plasticity. Cell Calcium 1992, 13, 9–17. [Google Scholar] [CrossRef]
- Periasamy, M.; Kalyanasundaram, A. Serca pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, C.; Nomura, H.; Sugita, Y. Crystal structures of Ca2+-ATPase in various physiological states. In Na,k-Atpase and Related Cation Pumps: Structure, Function, and Regulatory Mechanisms; Jorgensen, P.L., Karlish, S.J.D., Maunsbach, A.B., Eds.; New York Academy of Sciences: New York, NY, USA, 2003; Volume 986, pp. 1–8. [Google Scholar]
- Toyoshima, C.; Nakasako, M.; Nomura, H.; Ogawa, H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 2000, 405, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Michelangeli, F.; East, J.M. A diversity of serca Ca2+ pump inhibitors. Biochem. Soc. Trans. 2011, 39, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Rietdorf, K.; Hardy, H.; Dautova, Y.; Corps, E.; Pierro, C.; Stapleton, E.; Kang, E.; Proudfoot, D. Calcium signalling and regulation of cell function. eLS 2006. [Google Scholar] [CrossRef]
- Bootman, M.D.; Lipp, P.; Berridge, M.J. The organisation and functions of local Ca2+ signals. J. Cell Sci. 2001, 114, 2213–2222. [Google Scholar] [PubMed]
- Sanchez, E.J.; Lewis, K.M.; Danna, B.R.; Kang, C. High-capacity Ca2+ binding of human skeletal calsequestrin. J. Biol. Chem. 2012, 287, 11592–11601. [Google Scholar] [CrossRef] [PubMed]
- Royer, L.; Rios, E. Deconstructing calsequestrin. Complex buffering in the calcium store of skeletal muscle. J. Physiol. Lond. 2009, 587, 3101–3111. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Wu, S.; Dunker, A.K.; Kang, C. Polymerization of calsequestrin: Implications for Ca2+ regulation. J. Biol. Chem. 2003, 278, 16176–16182. [Google Scholar] [CrossRef] [PubMed]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.; Horton, K.D.; Weissman, N.J.; Holinstat, I. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef] [PubMed]
- Beard, N.A.; Laver, D.R.; Dulhunty, A.F. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 2004, 85, 33–69. [Google Scholar] [CrossRef] [PubMed]
- Manno, C.; Figueroa, L.C.; Gillespie, D.; Fitts, R.; Kang, C.; Franzini-Armstrong, C.; Rios, E. Calsequestrin depolymerizes when calcium is depleted in the sarcoplasmic reticulum of working muscle. Proc. Natl. Acad. Sci. USA 2017, 114, E638–E647. [Google Scholar] [CrossRef] [PubMed]
- Zorzato, F.; Anderson, A.A.; Ohlendieck, K.; Froemming, G.; Guerrini, R.; Treves, S. Identification of a novel 45 kDa protein (JP-45) from rabbit sarcoplasmic-reticulum junctional-face membrane. Biochem. J. 2000, 351, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.A.; Treves, S.; Biral, D.; Betto, R.; Sandona, D.; Ronjat, M.; Zorzato, F. The novel skeletal muscle sarcoplasmic reticulum JP-45 protein—Molecular cloning, tissue distribution, developmental expression, and interaction with α1.1 subunit of the voltage-gated calcium channel. J. Biol. Chem. 2003, 278, 39987–39992. [Google Scholar] [CrossRef] [PubMed]
- Delbono, O.; Xia, J.Y.; Treves, S.; Wang, Z.M.; Jimenez-Moreno, R.; Payne, A.M.; Messi, M.L.; Briguet, A.; Schaerer, F.; Nishi, M.; et al. Loss of skeletal muscle strength by ablation of the sarcoplasmic reticulum protein JP45. Proc. Natl. Acad. Sci. USA 2007, 104, 20108–20113. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ochoa, E.O.; Pratt, S.J.P.; Lovering, R.M.; Schneider, M.F. Critical role of intracellular RyR1 calcium release channels in skeletal muscle function and disease. Front. Physiol. 2016, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kelley, J.; Schmeisser, G.; Kobayashi, Y.M.; Jones, L.R. Complex formation between junction, triadin, calsequestrin, and the ryanodine receptor—Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 1997, 272, 23389–23397. [Google Scholar] [CrossRef] [PubMed]
- Lamb, G.D. Excitation–contraction coupling in skeletal muscle: Comparisons with cardiac muscle. Clin. Exp. Pharmacol. Physiol. 2000, 27, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Lu, F.; Sun, T.; Xu, J.; Li, L.L.; Wang, Y.; Wang, G.; Chen, L.; Wang, X.; Cannell, M.B.; et al. Imaging Ca2+ nanosparks in heart with a new targeted biosensor. Circ. Res. 2014, 114, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Shui, B.; Bossuyt, J.; Lang, D.; Kotlikoff, M.I.; Bers, D.M. Junctional cleft Ca2+ (I) measurements using novel cleft-targeted Ca2+ sensors. Circ. Res. 2014, 115, 339. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Li, L.; Shuai, J. Optimal microdomain crosstalk between endoplasmic reticulum and mitochondria for Ca2+ oscillations. Sci. Rep. 2015, 5, 7984. [Google Scholar] [CrossRef] [PubMed]
- Tabara, L.C.; Escalante, R. Vmp1 establishes ER-microdomains that regulate membrane contact sites and autophagy. PLoS ONE 2016, 11, e0166499. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2: A mitochondria-shaping protein with signaling roles beyond fusion. Antioxid. Redox Signal. 2008, 10, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Hassan, S.; Wahl-Schott, C.; Biel, M. Role of trpml and two-pore channels in endolysosomal cation homeostasis. J. Pharmacol. Exp. Ther. 2012, 342, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Ruas, M.; Rietdorf, K.; Arredouani, A.; Davis, L.C.; Lloyd-Evans, E.; Koegel, H.; Funnell, T.M.; Morgan, A.J.; Ward, J.A.; Watanabe, K.; et al. Purified tpc isoforms form naadp receptors with distinct roles for Ca(2+) signaling and endolysosomal trafficking. Curr. Biol. CB 2010, 20, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Dong, X.P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. Tpc proteins are phosphoinositide-activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Puertollano, R. Role of trp channels in the regulation of the endosomal pathway. Physiology 2011, 26, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanjurjo, C.I.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Lysosomes shape ins(1,4,5)P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J. Cell Sci. 2013, 126, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Ríos, E.; Figueroa, L.; Manno, C.; Kraeva, N.; Riazi, S. The couplonopathies: A comparative approach to a class of diseases of skeletal and cardiac muscle. J. Gen. Physiol. 2015, 145, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Mikoshiba, K. Role of IP3 receptor signaling in cell functions and diseases. Adv. Biol. Regul. 2015, 57, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Benders, A.A.; Veerkamp, J.H.; Oosterhof, A.; Jongen, P.J.; Bindels, R.J.; Smit, L.M.; Busch, H.F.; Wevers, R.A. Ca2+ homeostasis in brody’s disease. A study in skeletal muscle and cultured muscle cells and the effects of dantrolene an verapamil. J. Clin. Investig. 1994, 94, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Bublitz, M.; Musgaard, M.; Poulsen, H.; Thøgersen, L.; Olesen, C.; Schiøtt, B.; Morth, J.P.; Møller, J.V.; Nissen, P. Ion pathways in the sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 2013, 288, 10759–10765. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Guo, T. Calcium signaling in cardiac ventricular myocytes. In Communicative Cardiac Cell; Sideman, S., Beyar, R., Landesberg, A., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2005; Volume 1047, pp. 86–98. [Google Scholar]
- Leenhardt, A.; Denjoy, I.; Guicheney, P. Catecholaminergic polymorphic ventricular tachycardia. Circ. Arrhythm. Electrophysiol. 2012, 5, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Faggioni, M.; Kryshtal, D.O.; Knollmann, B.C. Calsequestrin mutations and catecholaminergic polymorphic ventricular tachycardia. Pediatr. Cardiol. 2012, 33, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Yamamoto, T.; Kobayashi, S.; Matsuzaki, M. Role of ryanodine receptor as a Ca2+ regulatory center in normal and failing hearts. J. Cardiol. 2009, 53, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Shannon, T.R. Calcium movements inside the sarcoplasmic reticulum of cardiac myocytes. J. Mol. Cell. Cardiol. 2013, 58, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.; Carpenter, D.; Shaw, M.A.; Halsall, J.; Hopkins, P. Mutations in RyR1 in malignant hyperthermia and central core disease. Hum. Mutat. 2006, 27, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Mickelson, J.R.; Louis, C.F. Malignant hyperthermia: Excitation-contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol. Rev. 1996, 76, 537–592. [Google Scholar] [PubMed]
- Rosenberg, H.; Pollock, N.; Schiemann, A.; Bulger, T.; Stowell, K. Malignant hyperthermia: A review. Orphanet J. Rare Dis. 2015, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Betzenhauser, M.J.; Marks, A.R. Ryanodine receptor channelopathies. Pflug. Arch. 2010, 460, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Monnier, N.; Procaccio, V.; Stieglitz, P.; Lunardi, J. Malignant-hyperthermia susceptibility is associated with a mutation of the α1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am. J. Hum. Genet. 1997, 60, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, R.T.; Avila, G. Distinct effects on Ca2+ handling caused by malignant hyperthermia and central core disease mutations in RyR1. Biophys. J. 2004, 87, 3193–3204. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H.; Chen, S. Store overload-induced Ca2+ release as a triggering mechanism for cpvt and mh episodes caused by mutations in RyR and Casq genes. J. Physiol. 2009, 587, 3113–3115. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H. Ca2+ signalling and muscle disease. Eur. J. Biochem. 2000, 267, 5291–5297. [Google Scholar] [CrossRef] [PubMed]
- Krause, T.; Gerbershagen, M.U.; Fiege, M.; Weisshorn, R.; Wappler, F. Dantrolene—A review of its pharmacology, therapeutic use and new developments. Anaesthesia 2004, 59, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Bannister, R.A. Dantrolene-induced inhibition of skeletal L-type Ca2+ current requires RyR1 expression. BioMed Res. Int. 2013, 2013, 390493. [Google Scholar] [CrossRef] [PubMed]
- Oo, Y.W.; Gomez-Hurtado, N.; Walweel, K.; van Helden, D.F.; Imtiaz, M.S.; Knollmann, B.C.; Laver, D.R. Essential role of calmodulin in ryr inhibition by dantrolene. Mol. Pharmacol. 2015, 88, 57–63. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Supnet, C.; Bezprozvanny, I. The dysregulation of intracellular calcium in alzheimer disease. Cell Calcium 2010, 47, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Chan, S.L. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 2003, 34, 385–397. [Google Scholar] [CrossRef]
- Liang, J.Y.; Kulasiri, D.; Samarasinghe, S. Ca2+ dysregulation in the endoplasmic reticulum related to Alzheimer’s disease: A review on experimental progress and computational modeling. Biosystems 2015, 134, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.R.; Lyckman, A.; Oddo, S.; LaFerla, F.M.; Querfurth, H.W.; Shtifman, A. Increased intraneuronal resting Ca2+ in adult Alzheimer’s disease mice. J. Neurochem. 2008, 105, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.H.; Shineman, D.; Muller, M.; Cardenas, C.; Mei, L.J.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.Y.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP(3) receptor channel gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Parker, I. Cytotoxicity of intracellular a β42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 2013, 33, 3824–3833. [Google Scholar] [CrossRef] [PubMed]
- Shilling, D.; Muller, M.; Takano, H.; Mak, D.O.D.; Abel, T.; Coulter, D.A.; Foskett, J.K. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer’s disease pathogenesis. J. Neurosci. 2014, 34, 6910–6923. [Google Scholar] [CrossRef] [PubMed]
- Oules, B.; Del Prete, D.; Greco, B.; Zhang, X.X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Brechot, P.; Trebak, M.; Checler, F.; et al. Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of alzheimer disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef] [PubMed]
- Gerasimenko, J.V.; Petersen, O.H.; Gerasimenko, O.V. Monitoring of intra-ER free Ca2+. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2014, 3, 63–71. [Google Scholar] [CrossRef]
- Hofer, A.M.; Schulz, I. Quantification of intraluminal free ca in the agonist-sensitive internal calcium store using compartmentalized fluorescent indicators: Some considerations. Cell Calcium 1996, 20, 235–242. [Google Scholar] [CrossRef]
- Ziman, A.P.; Ward, C.W.; Rodney, G.G.; Lederer, W.J.; Bloch, R.J. Quantitative measurement of Ca2+ in the sarcoplasmic reticulum lumen of mammalian skeletal muscle. Biophys. J. 2010, 99, 2705–2714. [Google Scholar] [CrossRef] [PubMed]
- Paredes, R.M.; Etzler, J.C.; Watts, L.T.; Zheng, W.; Lechleiter, J.D. Chemical calcium indicators. Methods 2008, 46, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Raju, B.; Murphy, E.; Levy, L.A.; Hall, R.D.; London, R.E. A fluorescent indicator for measuring cytosolic free magnesium. Am. J. Physiol. Cell Physiol. 1989, 256, C540–C548. [Google Scholar]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Koldenkova, V.P.; Nagai, T. Genetically encoded Ca2+ indicators: Properties and evaluation. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Kanemaru, K.; Iino, M. Genetically encoded fluorescent indicators for organellar calcium imaging. Biophys. J. 2016, 111, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- McCombs, J.E.; Palmer, A.E. Measuring calcium dynamics in living cells with genetically encodable calcium indicators. Methods 2008, 46, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.H.; Fosbrink, M.D.; Zhang, J. Genetically encodable fluorescent biosensors for tracking signaling dynamics in living cells. Chem. Rev. 2011, 111, 3614–3666. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [PubMed]
- Palmer, A.E.; Jin, C.; Reed, J.C.; Tsien, R.Y. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. USA 2004, 101, 17404–17409. [Google Scholar] [CrossRef] [PubMed]
- Sztretye, M.; Yi, J.X.; Figueroa, L.; Zhou, J.S.; Royer, L.; Rios, E. D4cpv-calsequestrin: A sensitive ratiometric biosensor accurately targeted to the calcium store of skeletal muscle. J. Gen. Physiol. 2011, 138, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Waldeck-Weiermair, M.; Bischof, H.; Blass, S.; Deak, A.; Klec, C.; Graier, T.; Roller, C.; Rost, R.; Eroglu, E.; Gottschalk, B.; et al. Generation of red-shifted cameleons for imaging Ca2+ dynamics of the endoplasmic reticulum. Sensors 2015, 15, 13052–13068. [Google Scholar] [CrossRef] [PubMed]
- Nakai, J.; Ohkura, M.; Imoto, K. A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001, 19, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Akerboom, J.; Rivera, J.D.V.; Guilbe, M.M.R.; Malave, E.C.A.; Hernandez, H.H.; Tian, L.; Hires, S.A.; Marvin, J.S.; Looger, L.L.; Schreiter, E.R. Crystal structures of the gcamp calcium sensor reveal the mechanism of fluorescence signal change and aid rational design. J. Biol. Chem. 2009, 284, 6455–6464. [Google Scholar] [CrossRef] [PubMed]
- Tallini, Y.N.; Ohkura, M.; Choi, B.R.; Ji, G.J.; Imoto, K.; Doran, R.; Lee, J.; Plan, P.; Wilson, J.; Xin, H.B.; et al. Imaging cellular signals in the heart in vivo: Cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc. Natl. Acad. Sci. USA 2006, 103, 4753–4758. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Hires, S.A.; Mao, T.; Huber, D.; Chiappe, M.E.; Chalasani, S.H.; Petreanu, L.; Akerboom, J.; McKinney, S.A.; Schreiter, E.R.; et al. Imaging neural activity in worms, flies and mice with improved gcamp calcium indicators. Nat. Methods 2009, 6, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Akerboom, J.; Chen, T.W.; Wardill, T.J.; Tian, L.; Marvin, J.S.; Mutlu, S.; Calderon, N.C.; Esposti, F.; Borghuis, B.G.; Sun, X.R.; et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 2012, 32, 13819–13840. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Mikoshiba, K. Quantitative comparison of novel GCaMP-type genetically encoded Ca2+ indicators in mammalian neurons. Front. Cell. Neurosci. 2012, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Prole, D.L.; Shen, Y.; Lin, Z.H.; Gnanasekaran, A.; Liu, Y.J.; Chen, L.D.; Zhou, H.; Cheng, S.R.W.; Usachev, Y.M.; et al. Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum. Biochem. J. 2014, 464, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Reddish, F.; Zhuo, Y.; Yang, J.J. Fast kinetics of calcium signaling and sensor design. Curr. Opin. Chem. Biol. 2015, 27, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wilkins, A.L.; Ye, Y.M.; Liu, Z.R.; Li, S.Y.; Urbauer, J.L.; Hellinga, H.W.; Kearney, A.; van der Merwe, P.A.; Yang, J.J. Design of a calcium-binding protein with desired structure in a cell adhesion molecule. J. Am. Chem. Soc. 2005, 127, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Jones, L.M.; Isley, L.; Ye, Y.M.; Lee, H.W.; Wilkins, A.; Liu, Z.R.; Hellinga, H.W.; Malchow, R.; Ghazi, M.; et al. Rational design of a calcium-binding protein. J. Am. Chem. Soc. 2003, 125, 6165–6171. [Google Scholar] [CrossRef] [PubMed]
- Maniccia, A.W.; Yang, W.; Li, S.Y.; Johnson, J.A.; Yang, J.J. Using protein design to dissect the effect of charged residues on metal binding and protein stability. Biochemistry 2006, 45, 5848–5856. [Google Scholar] [CrossRef] [PubMed]
- Maniccia, A.L.W.; Yang, W.; Johnson, J.; Li, S.; Harianto, T.; Zhou, H.X.; Shaket, L.A.; Yang, J.J. Inverse tuning of metal binding affinity and protein stability by altering charged coordination residues in designed calcium binding proteins. PMC Biophys. 2009, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Kirberger, M.; Wang, X.; Deng, H.; Yang, W.; Chen, G.T.; Yang, J.J. Statistical analysis of structural characteristics of protein Ca2+-binding sites. J. Biol. Inorg. Chem. 2008, 13, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Kirberger, M.; Wang, X.; Zhao, K.; Tang, S.; Chen, G.T.; Yang, J.J. Integration of diverse research methods to analyze and engineer Ca2+-binding proteins: From prediction to production. Curr. Bioinform. 2010, 5, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wong, H.C.; Wang, Z.M.; Huang, Y.; Zou, J.; Zhuo, Y.; Pennati, A.; Gadda, G.; Delbono, O.; Yang, J.J. Design and application of a class of sensors to monitor Ca2+ dynamics in high Ca2+ concentration cellular compartments. Proc. Natl. Acad. Sci. USA 2011, 108, 16265–16270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Reddish, F.; Tang, S.; Zhuo, Y.; Wang, Y.F.; Yang, J.J.; Weber, I.T. Structural basis for a hand-like site in the calcium sensor catcher with fast kinetics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2309–2319. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Solntsev, K.M.; Reddish, F.; Tang, S.; Yang, J.J. Effect of Ca2+ on the steady-state and time-resolved emission properties of the genetically encoded fluorescent sensor CatchER. J. Phys. Chem. B 2014, 119, 2103. [Google Scholar] [CrossRef] [PubMed]
- Gadella, T.W.J.; Jovin, T.M.; Clegg, R.M. Fluorescence lifetime imaging microscopy (FLIM)—Spatial-resolution of microstructures on the nanosecond time-scale. Biophys. Chem. 1993, 48, 221–239. [Google Scholar] [CrossRef]
- Griesbeck, O.; Baird, G.S.; Campbell, R.E.; Zacharias, D.A.; Tsien, R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001, 276, 29188–29194. [Google Scholar] [CrossRef] [PubMed]
- Kabbara, A.A.; Allen, D.G. The use of the indicator fluo-5N to measure sarcoplasmic reticulum calcium in single muscle fibres of the cane toad. J. Physiol. 2001, 534, 87–97. [Google Scholar] [CrossRef] [PubMed]
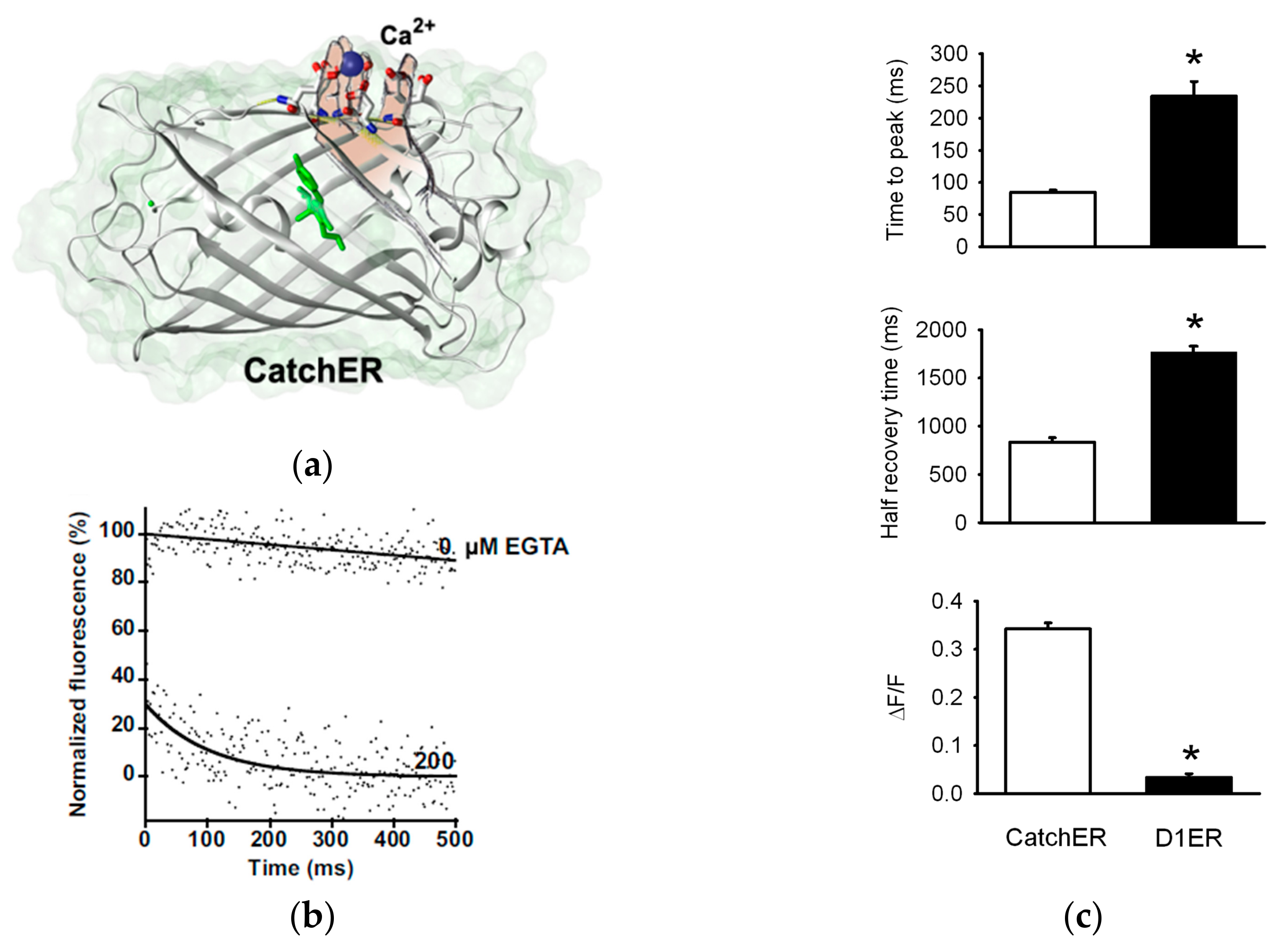

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sensor Name | kon (M−1·s−1) | koff (s−1) | Kd (µM) | λEx (nm) | λEm (nm) | Reference |
|---|---|---|---|---|---|---|
| CatchER | 3.89 × 106 | 700 | 120–180 | 395, 488 | 510 | [160] |
| D1ER | 3.86 × 106 | 256 | 0.8–60 | 436 (CFP) | 465 (CFP) | [164] |
| 500 (YFP) | 535 (YFP) | |||||
| D1ERCmR2 | - | - | 200 | 490 | 510,560 | [144] |
| Fluo-5N | - | - | 90 | 494 | 516 | [165] |
| G-CEPIA1er | - | - | 672 | (−Ca2+) 402,498 | (−Ca2+) 499 | [152] |
| (+Ca2+) 401,497 | (+Ca2+) 498 | |||||
| GEM-CEPIA1er | - | - | 558 | (−Ca2+) 401 | (−Ca2+) 381,395 | [152] |
| (+Ca2+) 391 | (+Ca2+) 381,394 | |||||
| LAR-GECO1 | - | - | 24 | (−Ca2+) 574 | 598 | [151] |
| (+Ca2+) 561 | ||||||
| LAR-GECO1.2 | - | - | 12 | (−Ca2+) 570 | (−Ca2+) 594 | [151] |
| (+Ca2+) 557 | (+Ca2+) 584 | |||||
| Mag-Fura-2 | 7.5 × 108 | 600, 26, 760 | 50 | 345 | 490 | [135] |
| R-CEPIA1er | - | - | 565 | (−Ca2+) 445,576 | (−Ca2+) 570 | [152] |
| (+Ca2+) 448,562 | (+Ca2+) 561 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddish, F.N.; Miller, C.L.; Gorkhali, R.; Yang, J.J. Calcium Dynamics Mediated by the Endoplasmic/Sarcoplasmic Reticulum and Related Diseases. Int. J. Mol. Sci. 2017, 18, 1024. https://doi.org/10.3390/ijms18051024
Reddish FN, Miller CL, Gorkhali R, Yang JJ. Calcium Dynamics Mediated by the Endoplasmic/Sarcoplasmic Reticulum and Related Diseases. International Journal of Molecular Sciences. 2017; 18(5):1024. https://doi.org/10.3390/ijms18051024
Chicago/Turabian StyleReddish, Florence N., Cassandra L. Miller, Rakshya Gorkhali, and Jenny J. Yang. 2017. "Calcium Dynamics Mediated by the Endoplasmic/Sarcoplasmic Reticulum and Related Diseases" International Journal of Molecular Sciences 18, no. 5: 1024. https://doi.org/10.3390/ijms18051024
APA StyleReddish, F. N., Miller, C. L., Gorkhali, R., & Yang, J. J. (2017). Calcium Dynamics Mediated by the Endoplasmic/Sarcoplasmic Reticulum and Related Diseases. International Journal of Molecular Sciences, 18(5), 1024. https://doi.org/10.3390/ijms18051024