
Impact of Acetazolamide, a Carbonic Anhydrase Inhibitor, on the Development of Intestinal Polyps in Min Mice


Abstract
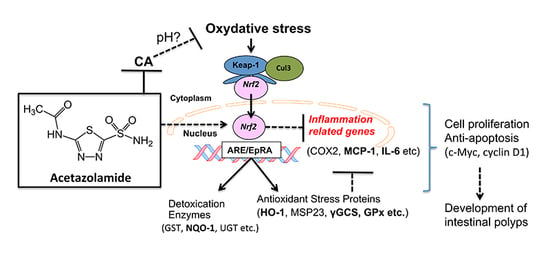
:
1. Introduction
2. Results
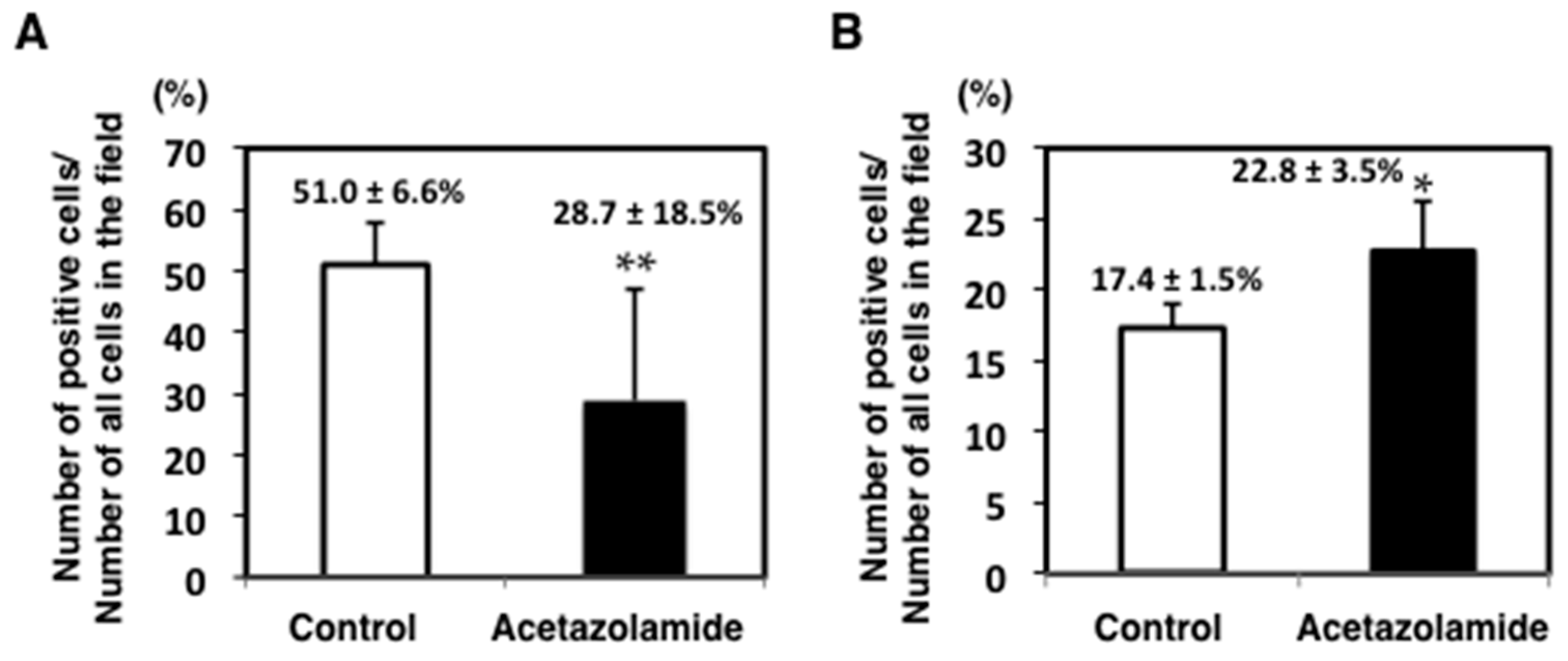
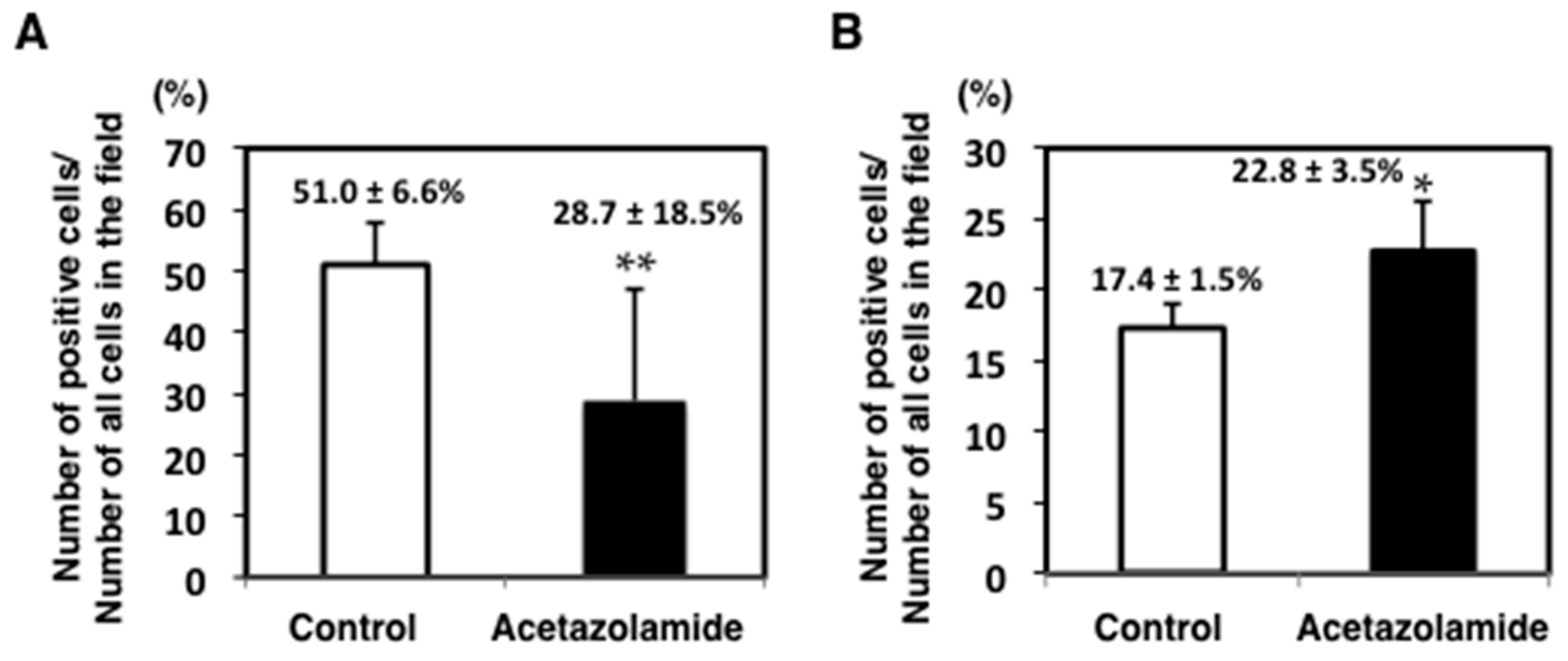
2.1. Suppression of Intestinal Polyp Formation in Min Mice by Acetazolamide Treatment
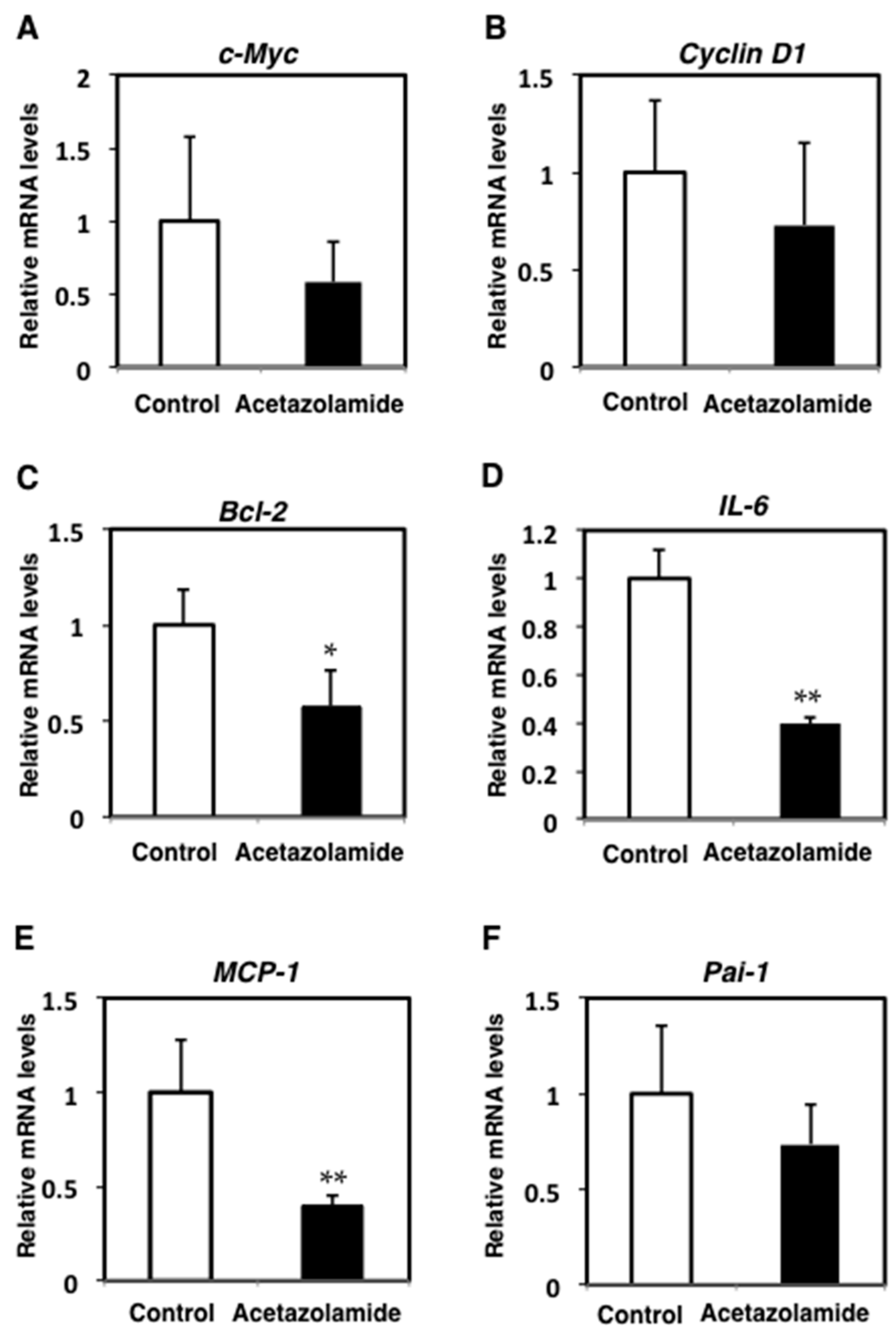
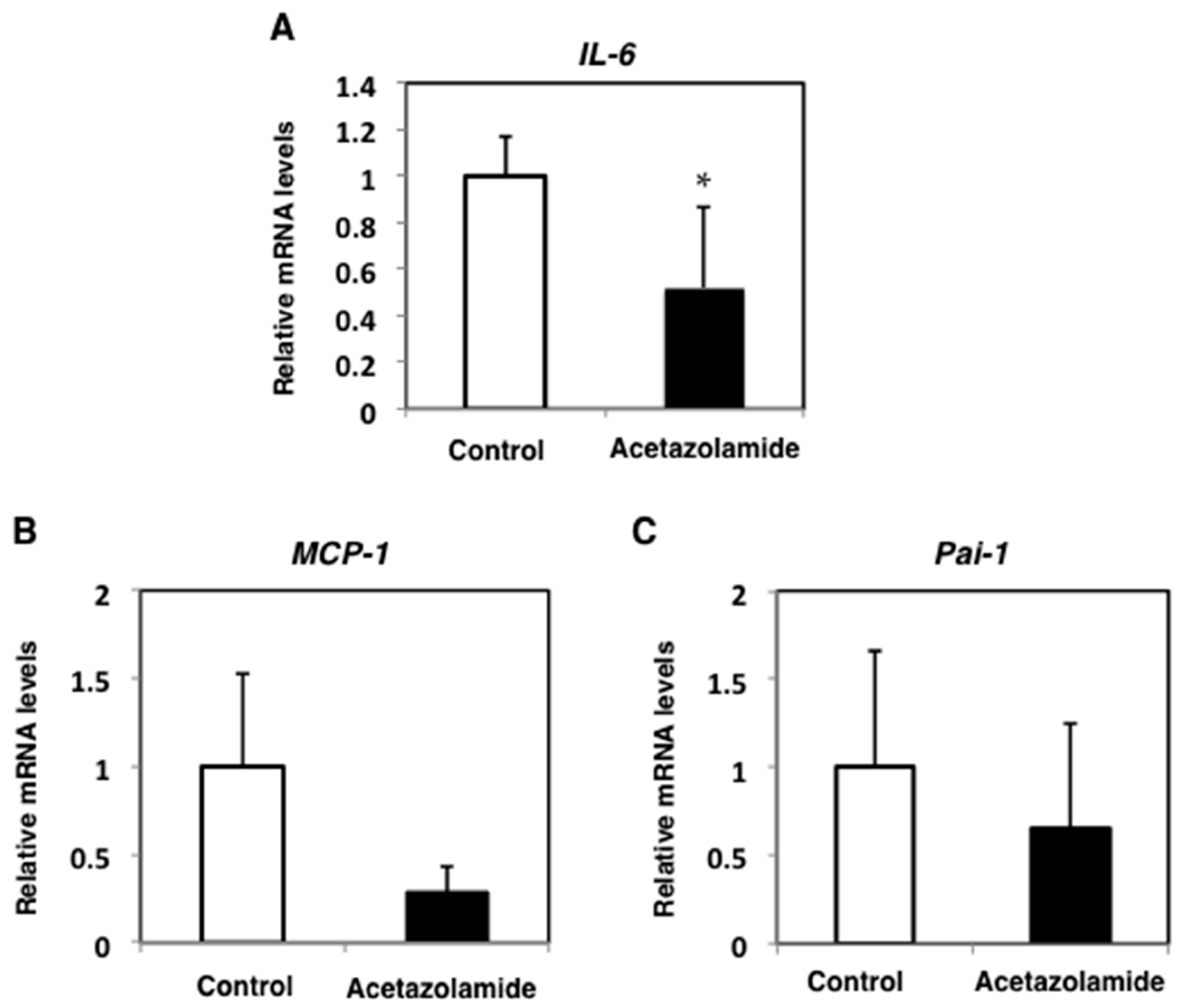
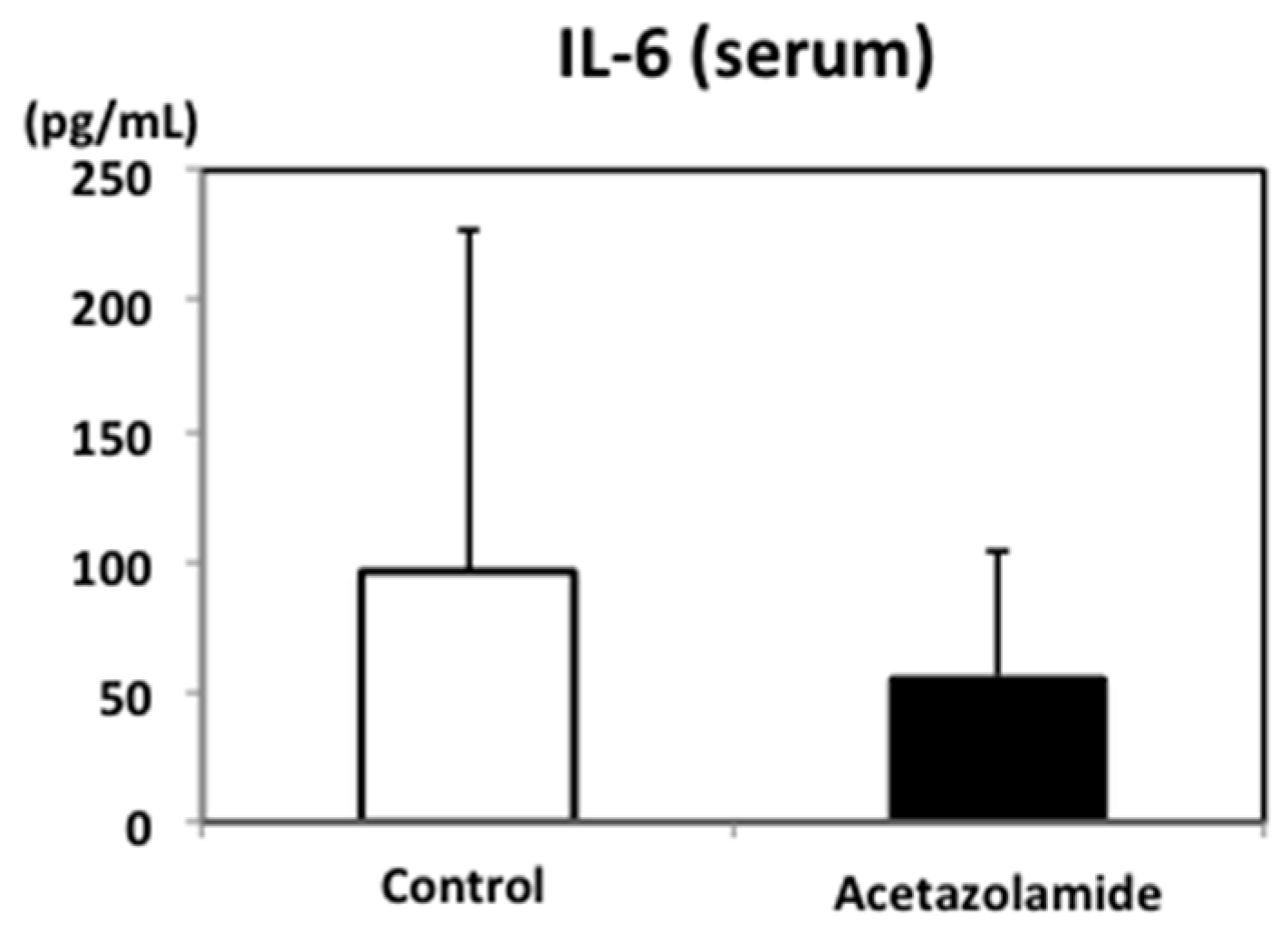
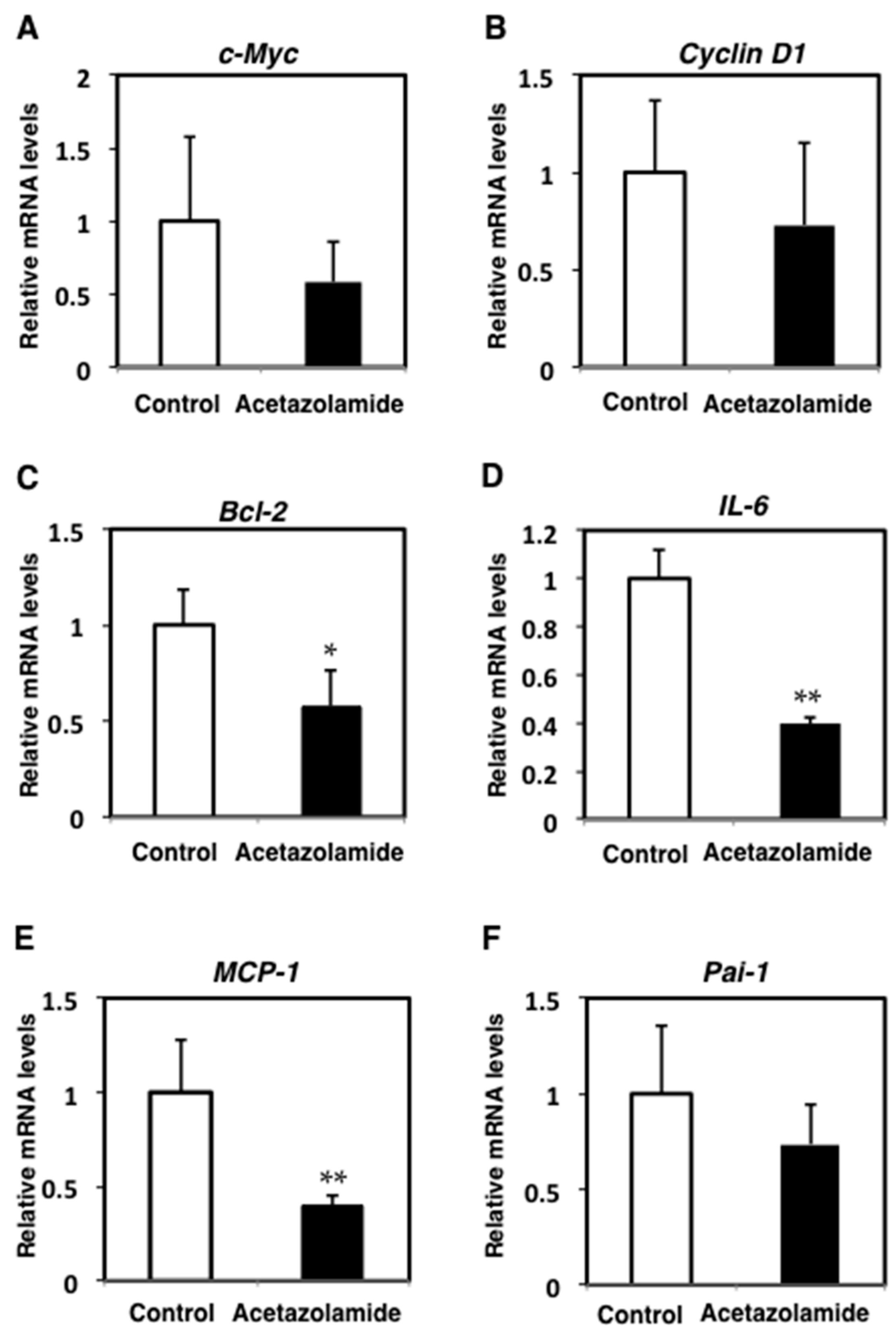
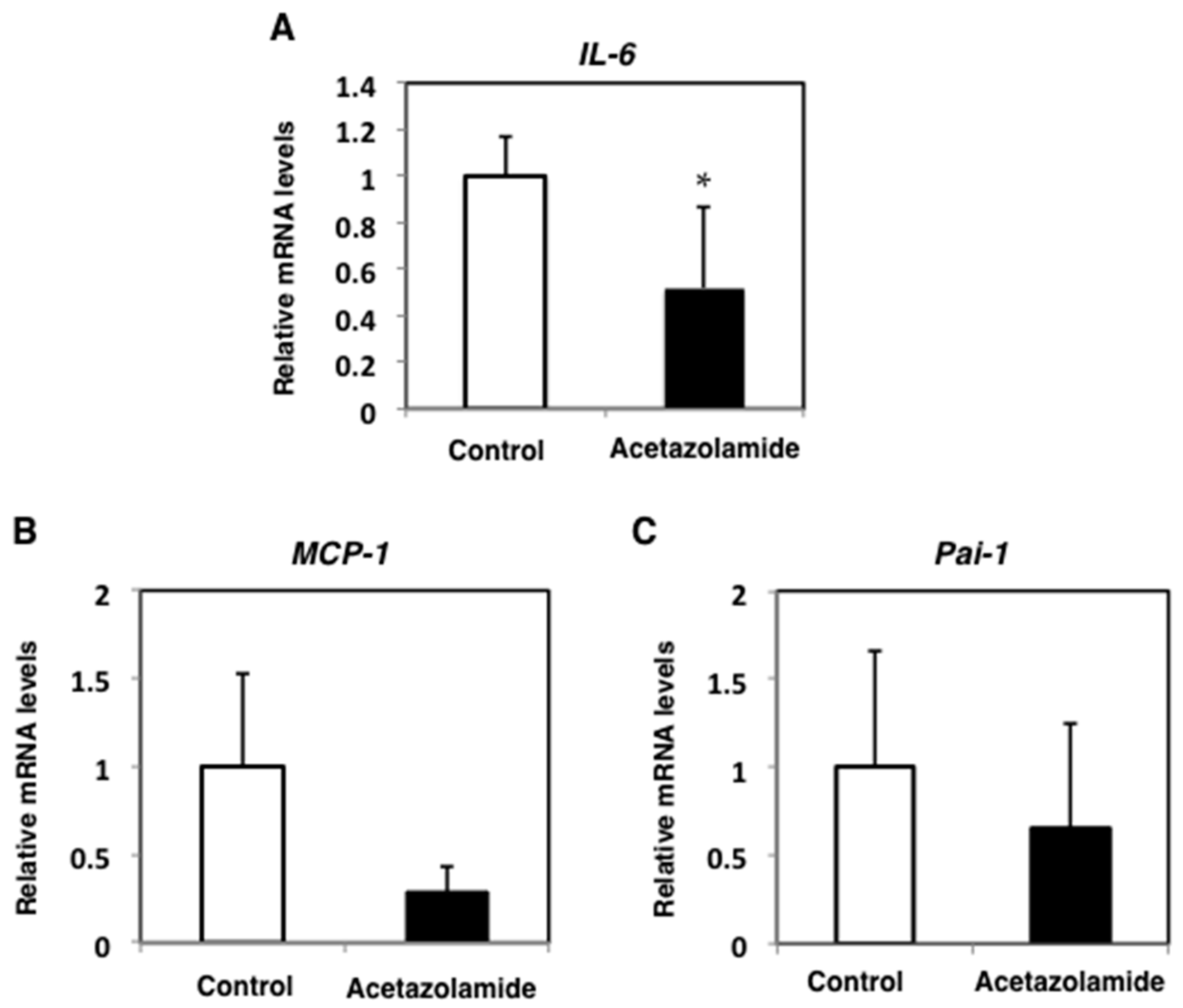
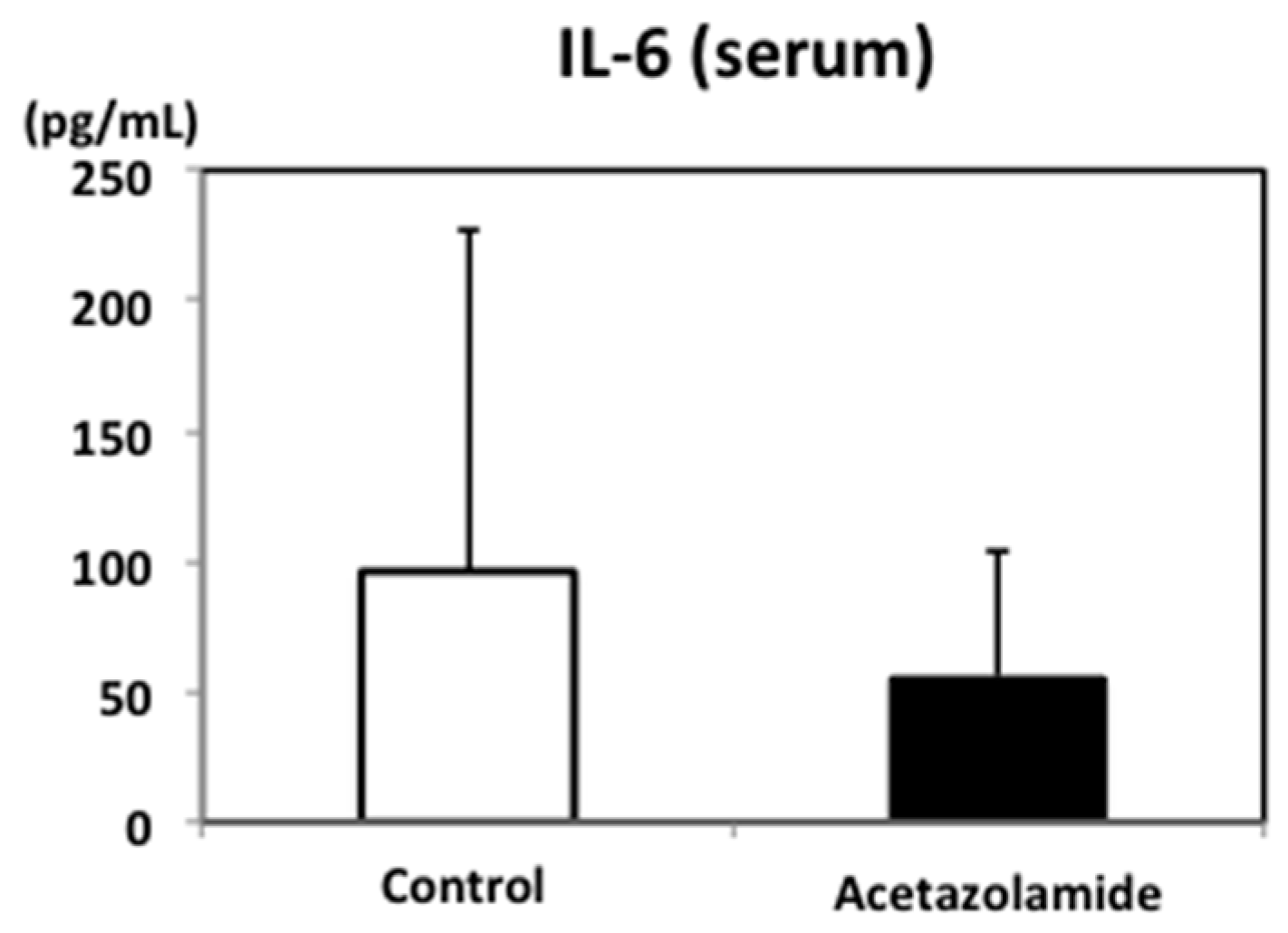
2.2. Suppression of Inflammatory Cytokine mRNA Levels in the Intestinal Polyps and Liver in Min Mice Treated with Acetazolamide
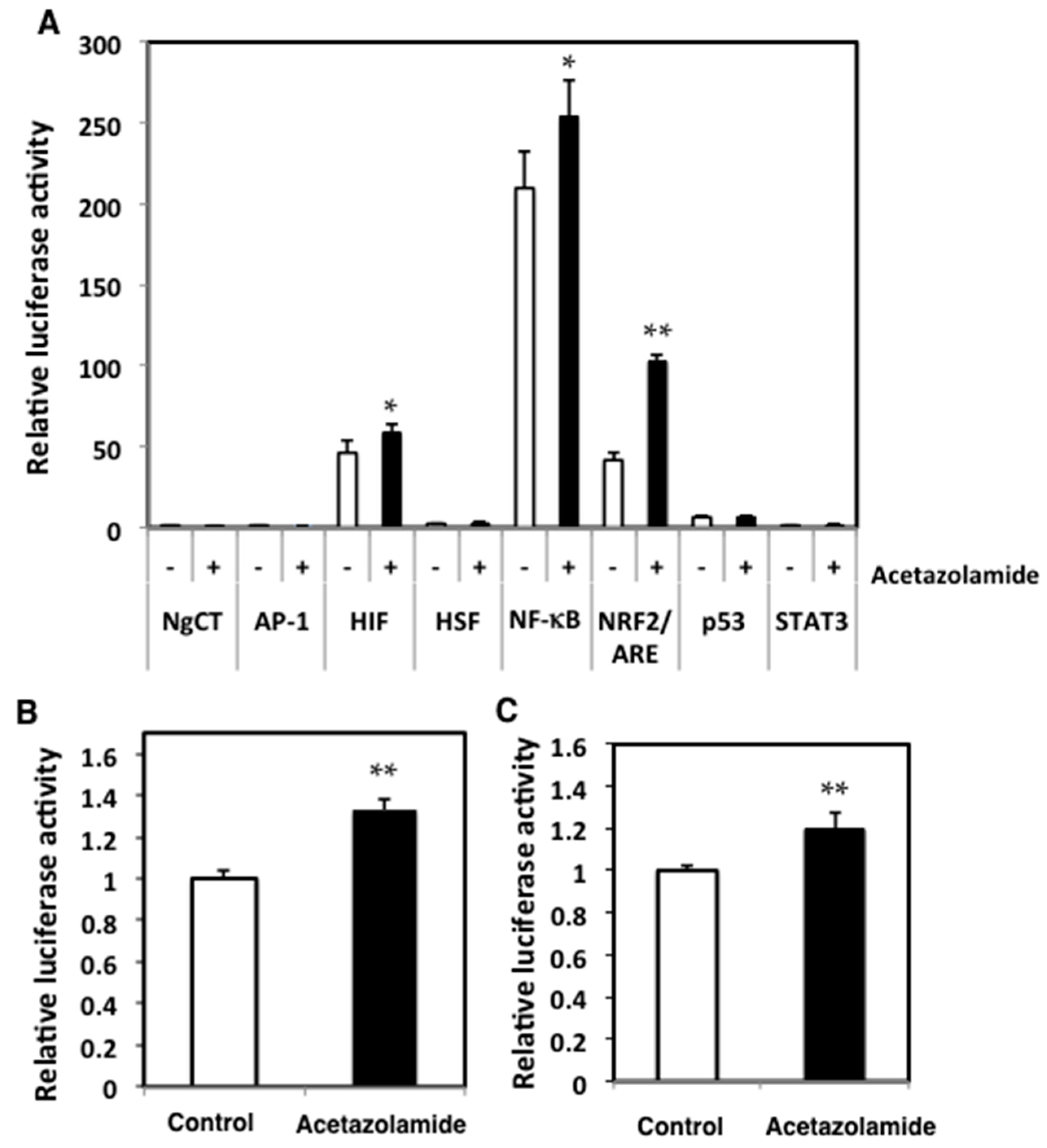
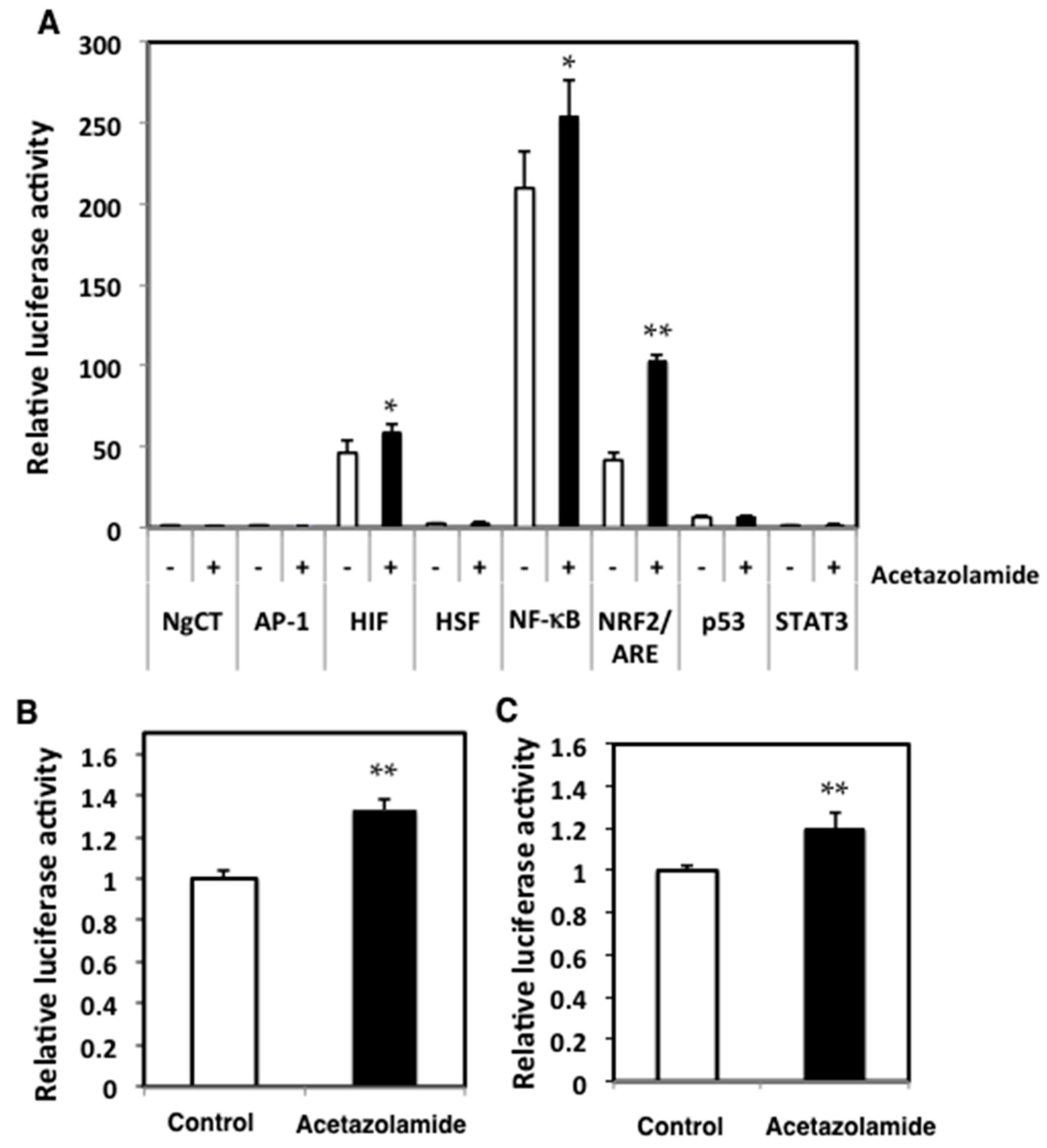
2.3. Effects of Acetazolamide on Seven Oxidative Stress-Related Transcriptional Activities in Human Colorectal Cancer Cells
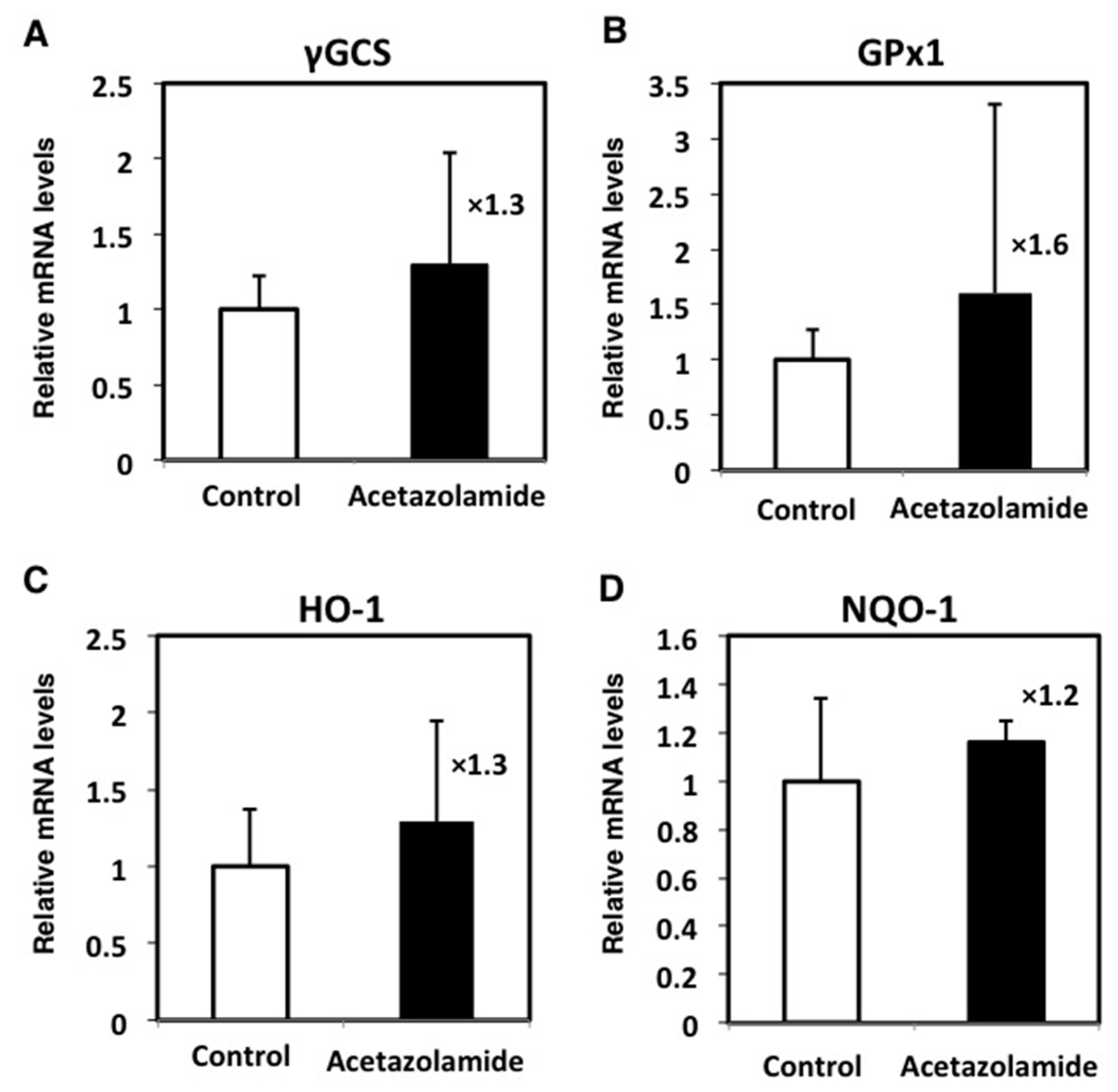
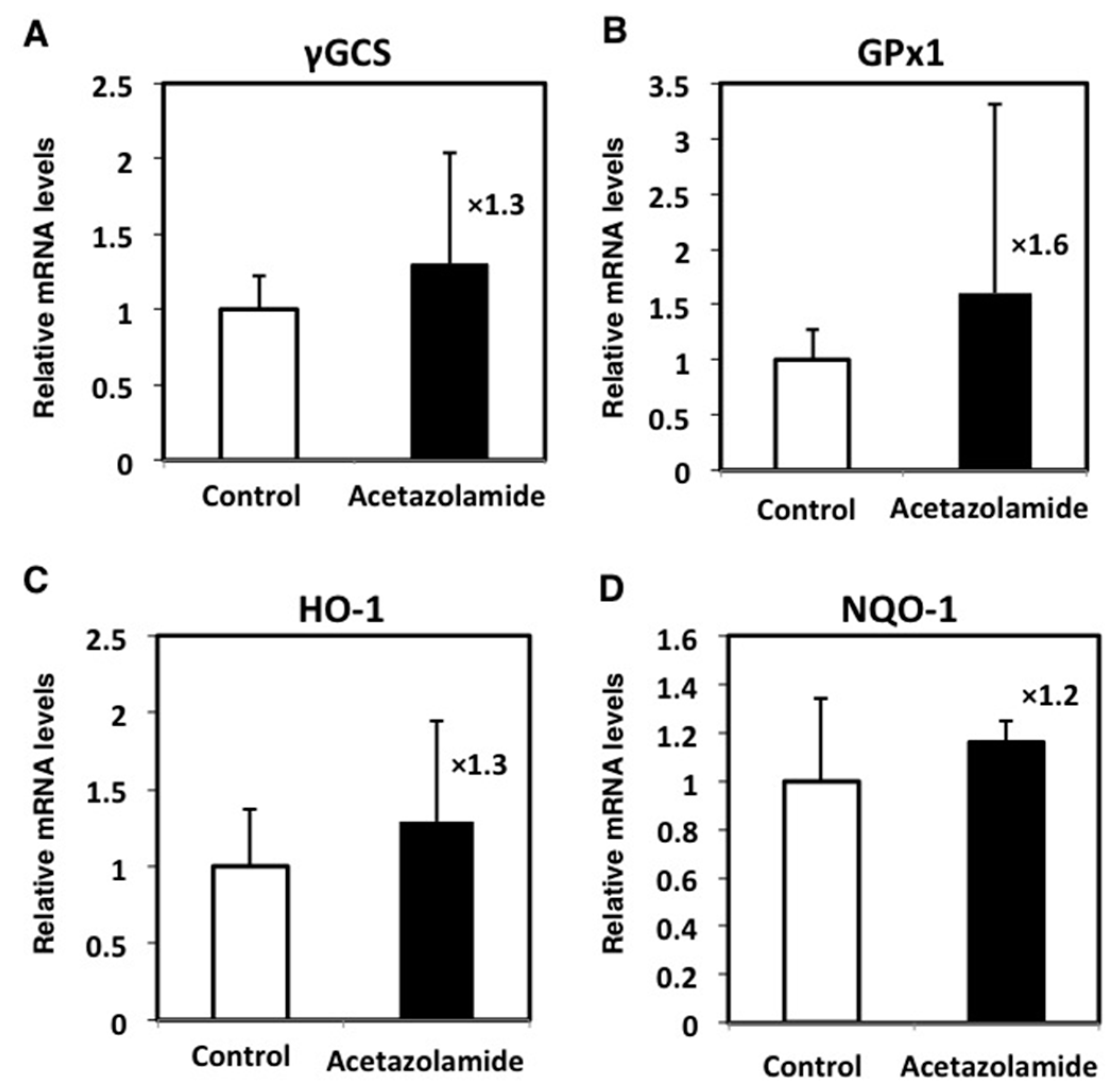
2.4. Effects of the Expression Levels of NRF2-Related Factor mRNA in the Intestinal Polyps
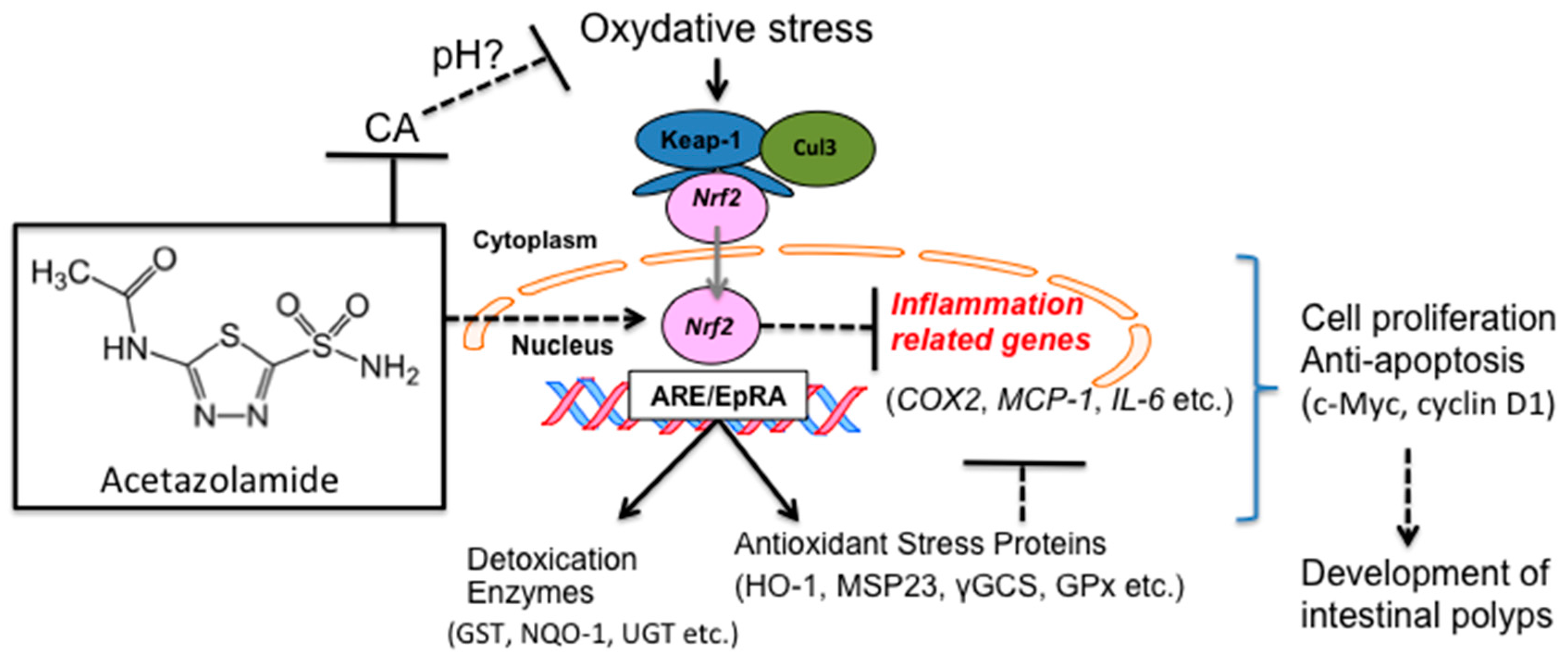
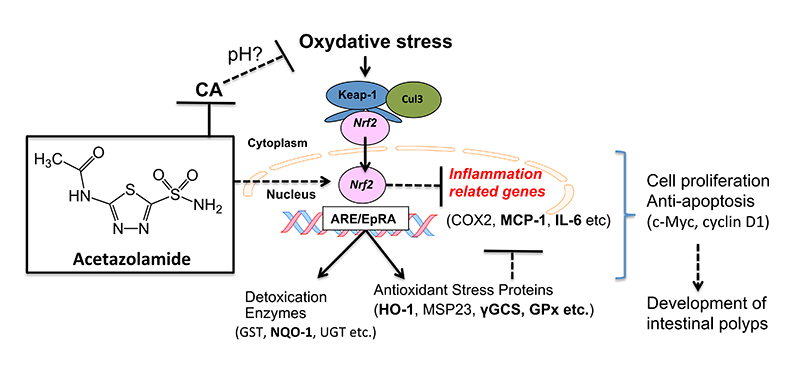
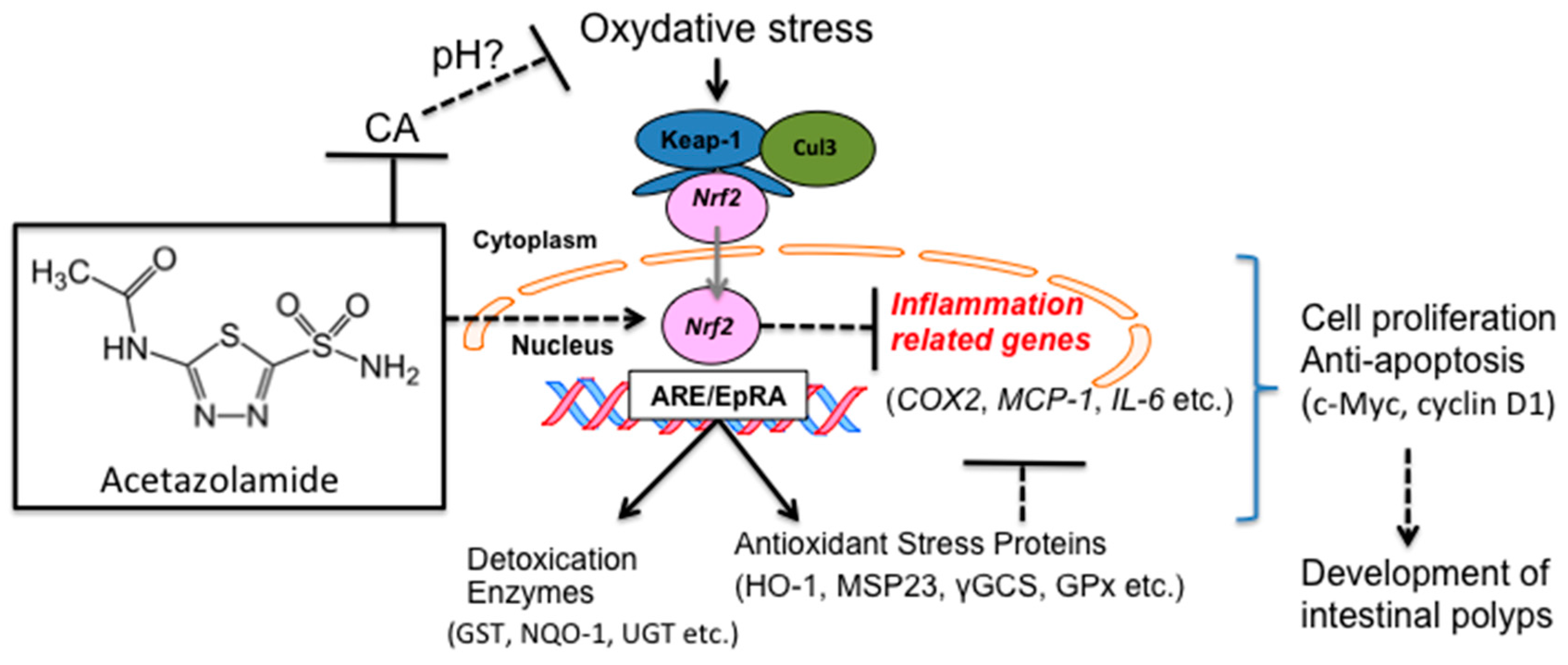
3. Discussion
4. Materials and Methods
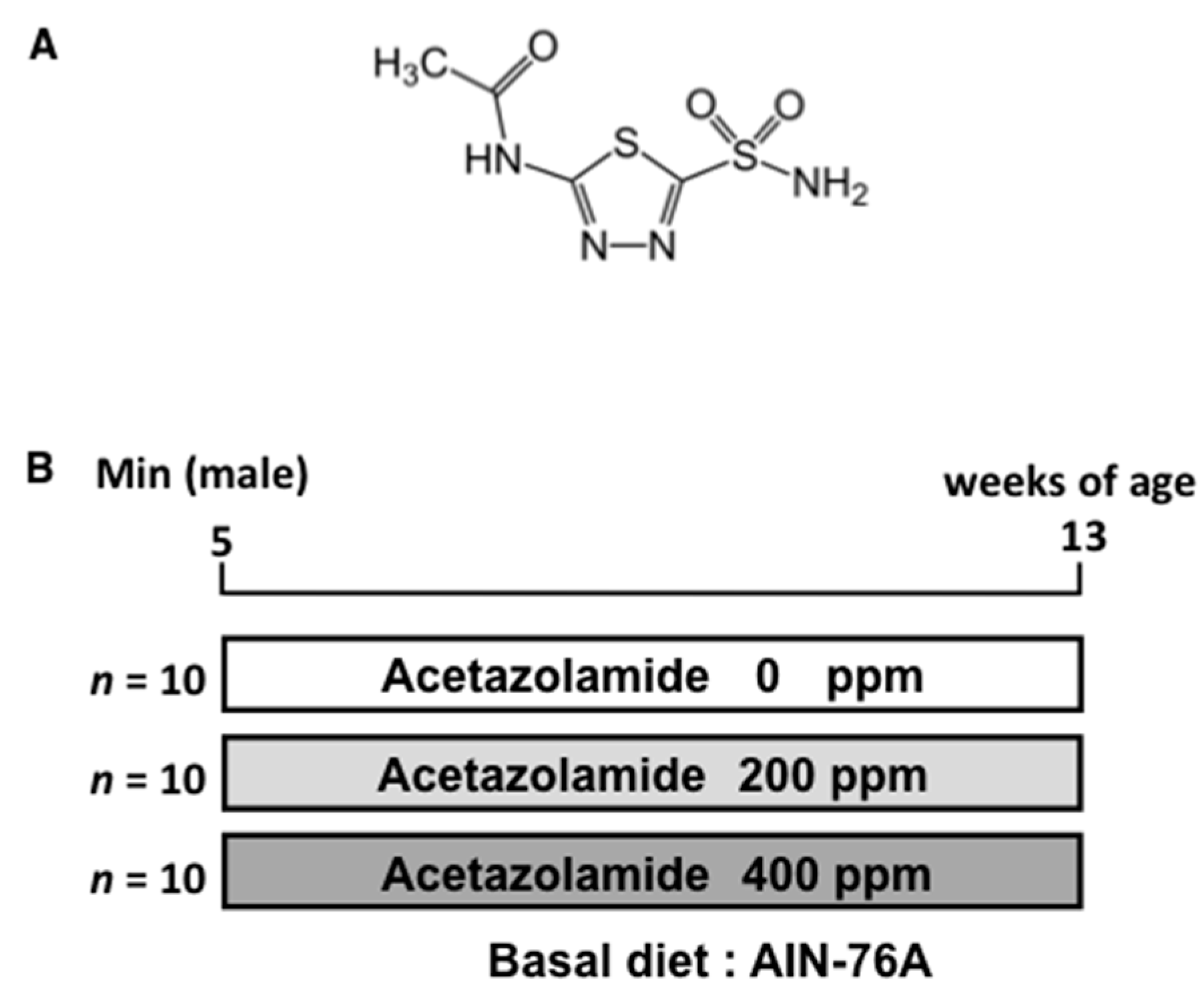
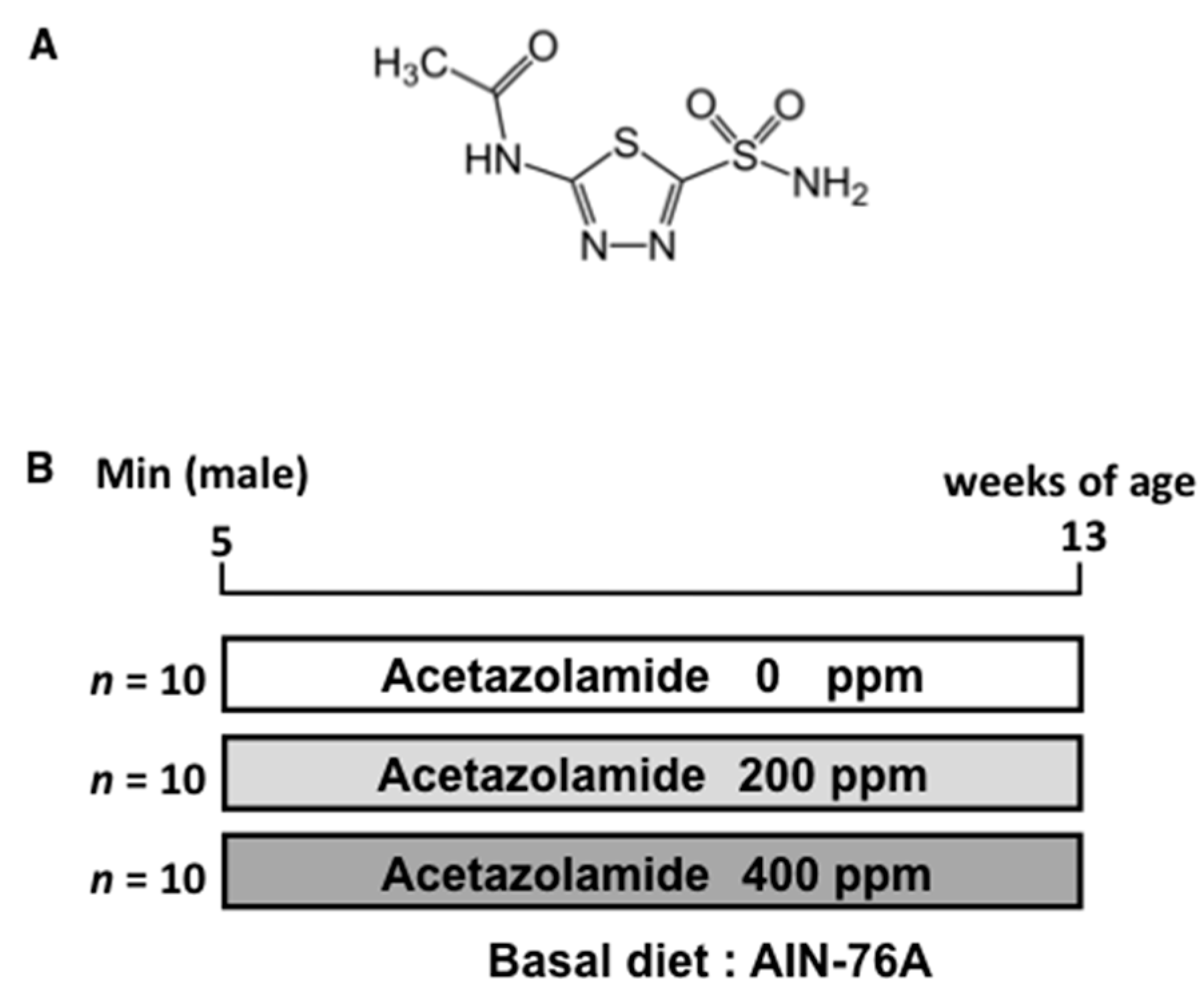
4.1. Chemicals
4.2. Cell Culture
4.3. Animals
4.4. Animal Experiment Protocols
4.5. Immunohistochemical Staining of Intestinal Polyps in Min Mice
4.6. Quantitative Real-Time Polymerase Chain Reaction (PCR) Analyses
4.7. Luciferase Assays for AP-1, HIF, HSF, NF-κB, NRF2, p53, and STAT3 Transcriptional Activity
4.8. Enzyme-Linked Immunosorbent Assay (ELISA) for Measuring Murine Serum IL-6 Levels in Mice
4.9. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AP-1 | Activator protein-1 |
| ARE | Antioxidant response element |
| Bcl-2 | B-cell lymphoma 2 |
| CA | Carbonic anhydrase |
| CRC | Colorectal cancer |
| FBS | Fetal bovine serum |
| γGCS | γ-Glutamylcysteine synthetase |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GPx1 | Glutathione peroxidase 1 |
| HIF | Hypoxia inducible factor |
| HNPCC | Hereditary nonpolyposis colorectal cancer |
| HO-1 | Heme oxygenase-1 |
| HSF | Histamine sensitizing factor |
| IL-6 | Interleukine-6 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| NF-κB | Nuclear factor-κB |
| NQO-1 | NAD(P)H:quinone oxidoreductase-1 |
| NRF2 | NF-E2-related factor 2 |
| Pai-1 | Plasminogen activator inhibitor-1 |
| PCNA | Proliferating cell nuclear antigen |
| PCR | Polymerase chain reaction |
| ssDNA | Single-stranded DNA |
| STAT3 | Signal transducer and activator of transcription 3 |
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Suh, N. Chemoprevention: An essential approach to controlling cancer. Nat. Rev. Cancer 2002, 2, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases—An overview. Curr. Pharm. Des. 2008, 14, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Jarvela, S.; Parkkila, S.; Bragge, H.; Kahkonen, M.; Parkkila, A.K.; Soini, Y.; Pastorekova, S.; Pastorek, J.; Haapasalo, H. Carbonic anhydrase IX in oligodendroglial brain tumors. BMC Cancer 2008, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Ilc, K.; Laferriere, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouyssegur, J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Niemela, A.M.; Hynninen, P.; Mecklin, J.P.; Kuopio, T.; Kokko, A.; Aaltonen, L.; Parkkila, A.K.; Pastorekova, S.; Pastorek, J.; Waheed, A.; et al. Carbonic anhydrase IX is highly expressed in hereditary nonpolyposis colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Cianchi, F.; Vinci, M.C.; Supuran, C.T.; Peruzzi, B.; de Giuli, P.; Fasolis, G.; Perigli, G.; Pastorekova, S.; Papucci, L.; Pini, A.; et al. Selective inhibition of carbonic anhydrase IX decreases cell proliferation and induces ceramide-mediated apoptosis in human cancer cells. J. Pharmacol. Exp. Ther. 2010, 334, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Akocak, S.; Alam, M.R.; Shabana, A.M.; Sanku, R.K.; Vullo, D.; Thompson, H.; Swenson, E.R.; Supuran, C.T.; Ilies, M.A. PEGylated bis-sulfonamide carbonic anhydrase inhibitors can efficiently control the growth of several carbonic anhydrase IX-expressing carcinomas. J. Med. Chem. 2016, 59, 5077–5088. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ma, B.; Li, T.; Yu, H.M.; Li, X.J. Acetazolamide suppresses tumor metastasis and related protein expression in mice bearing Lewis lung carcinoma. Acta Pharmacol. Sin. 2002, 23, 745–751. [Google Scholar] [PubMed]
- Moser, A.R.; Pitot, H.C.; Dove, W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990, 247, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A. Signal transduction of inflammatory cytokines and tumor development. Cancer Sci. 2006, 97, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Teraoka, N.; Mutoh, M.; Takasu, S.; Ueno, T.; Yamamoto, M.; Sugimura, T.; Wakabayashi, K. Inhibition of intestinal polyp formation by pitavastatin, a HMG-CoA reductase inhibitor. Cancer Prev. Res. 2011, 4, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Onuma, W.; Tomono, S.; Miyamoto, S.; Fujii, G.; Hamoya, T.; Fujimoto, K.; Miyoshi, N.; Fukai, F.; Wakabayashi, K.; Mutoh, M. Irsogladine maleate, a gastric mucosal protectant, suppresses intestinal polyp development in Apc-mutant mice. Oncotarget 2016, 7, 8640–8652. [Google Scholar] [PubMed]
- Hamoya, T.; Miyamoto, S.; Tomono, S.; Fujii, G.; Nakanishi, R.; Komiya, M.; Tamura, S.; Toshima, J.; Wakabayashi, K.; Mutoh, M. Chemopreventive effects of a low-side-effect antibiotic drug, erythromycin, on mouse intestinal tumors. JCBN 2017, in press. [Google Scholar] [CrossRef]
- Kwak, M.K.; Wakabayashi, N.; Kensler, T.W. Chemoprevention through the Keap1-Nrf2 signaling pathway by phase 2 enzyme inducers. Mutat. Res. 2004, 555, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Khor, T.O.; Huang, M.T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.N. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef] [PubMed]
- Aleksunes, L.M.; Manautou, J.E. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol. Pathol. 2007, 35, 459–473. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose (ppm) | Number of Mice | Number of Polyps/Mouse | ||||
|---|---|---|---|---|---|---|
| Small Intestine | Colon | Total | ||||
| Proximal | Middle | Distal | ||||
| 0 | 10 | 4.5 ± 1.5 | 17.0 ± 9.7 | 26.8 ± 9.8 | 0.5 ± 1.0 | 48.8 ± 18.8 |
| 200 | 10 | 3.9 ± 1.8 | 9.2 ± 3.2 * | 21.8 ± 6.3 | 0.3 ± 0.7 | 35.2 ± 9.2 |
| 400 | 10 | 2.7 ± 1.2 * | 8.0 ± 2.3 * | 15.4 ± 6.2 ** | 0.1 ± 0.3 | 26.2 ± 7.8 ** |
| Gene | Sequence | |
|---|---|---|
| c-Myc | Forward | GCT CGC CCA AAT CCT GTA CCC T |
| Reverse | TCT CCA CAG ACA CCA CAT CAA TTT C | |
| Cyclin D1 | Forward | TGA CTG CCG AGA AGT TGT GC |
| Reverse | CTC ATC CGC CTC TGG CAT T | |
| Bcl-2 | Forward | ACT TCG CAG AGA TGT CCA GTC A |
| Reverse | TGG CAA AGC GTC CCC TC | |
| IL-6 | Forward | TGT TCT CTG GGA AAT TCG TGG A |
| Reverse | AAG TGC ATC ATC GTT GTT CAT ACA | |
| MCP-1 | Forward | CAG CCA GAT GCA GTT AAC GC |
| Reverse | GCC TAC TCA TTG GGA TCA TCT TG | |
| Pai-1 | Forward | ACG TTG TGG AAC TGC CCT AC |
| Reverse | GCC AGG GTT GCA CTA AAC AT | |
| γGCS | Forward | CTA CCA CGC AGT CAA GGA CC |
| Reverse | CCT CCA TTC AGT AAC AAC TGG | |
| GPx1 | Forward | AAT GTC GCG TCT CTC TGA GG |
| Reverse | TCC GAA CTG ATT GCA CGG G | |
| HO-1 | Forward | GAT AGA GCG CAA CAA GCA GAA |
| Reverse | CAG TGA GGC CCA TAC CAG AAG | |
| NQO-1 | Forward | AGG ATG GGA GGT ACT CGA ATC |
| Reverse | TGC TAG AGA TGA CTC GGA AGG |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noma, N.; Fujii, G.; Miyamoto, S.; Komiya, M.; Nakanishi, R.; Shimura, M.; Tanuma, S.-i.; Mutoh, M. Impact of Acetazolamide, a Carbonic Anhydrase Inhibitor, on the Development of Intestinal Polyps in Min Mice. Int. J. Mol. Sci. 2017, 18, 851. https://doi.org/10.3390/ijms18040851
Noma N, Fujii G, Miyamoto S, Komiya M, Nakanishi R, Shimura M, Tanuma S-i, Mutoh M. Impact of Acetazolamide, a Carbonic Anhydrase Inhibitor, on the Development of Intestinal Polyps in Min Mice. International Journal of Molecular Sciences. 2017; 18(4):851. https://doi.org/10.3390/ijms18040851
Chicago/Turabian StyleNoma, Nobuharu, Gen Fujii, Shingo Miyamoto, Masami Komiya, Ruri Nakanishi, Misato Shimura, Sei-ichi Tanuma, and Michihiro Mutoh. 2017. "Impact of Acetazolamide, a Carbonic Anhydrase Inhibitor, on the Development of Intestinal Polyps in Min Mice" International Journal of Molecular Sciences 18, no. 4: 851. https://doi.org/10.3390/ijms18040851
APA StyleNoma, N., Fujii, G., Miyamoto, S., Komiya, M., Nakanishi, R., Shimura, M., Tanuma, S.-i., & Mutoh, M. (2017). Impact of Acetazolamide, a Carbonic Anhydrase Inhibitor, on the Development of Intestinal Polyps in Min Mice. International Journal of Molecular Sciences, 18(4), 851. https://doi.org/10.3390/ijms18040851