
CoQ10 Deficiency May Indicate Mitochondrial Dysfunction in Cr(VI) Toxicity

Abstract
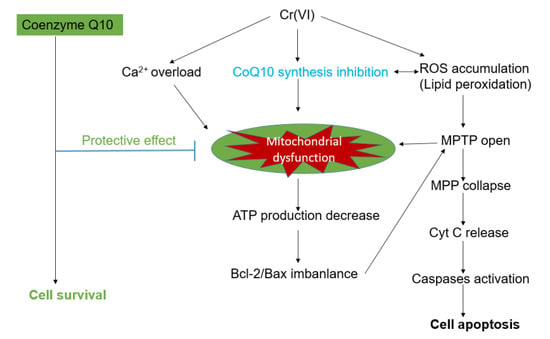
:
1. Introduction
2. Results
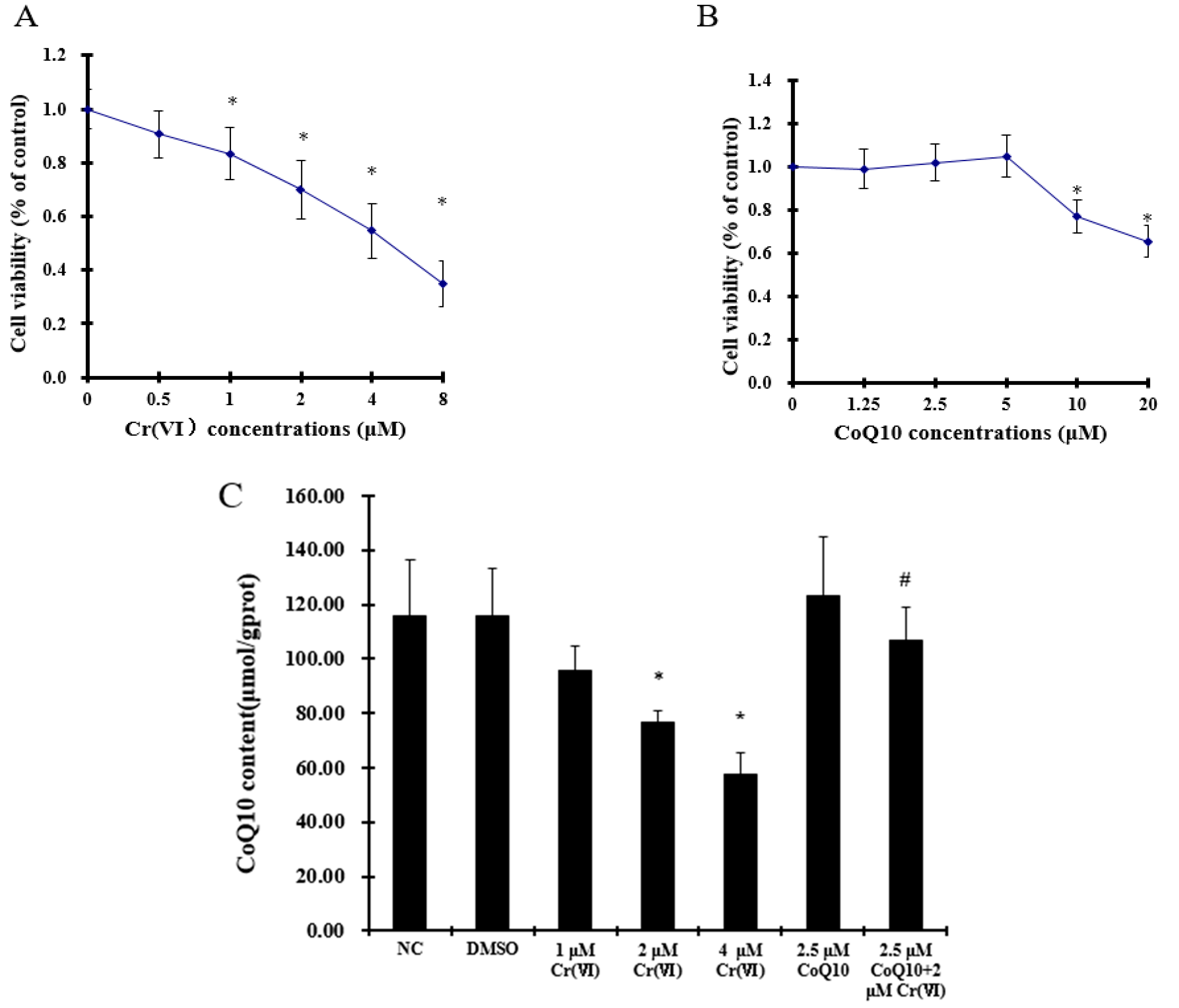
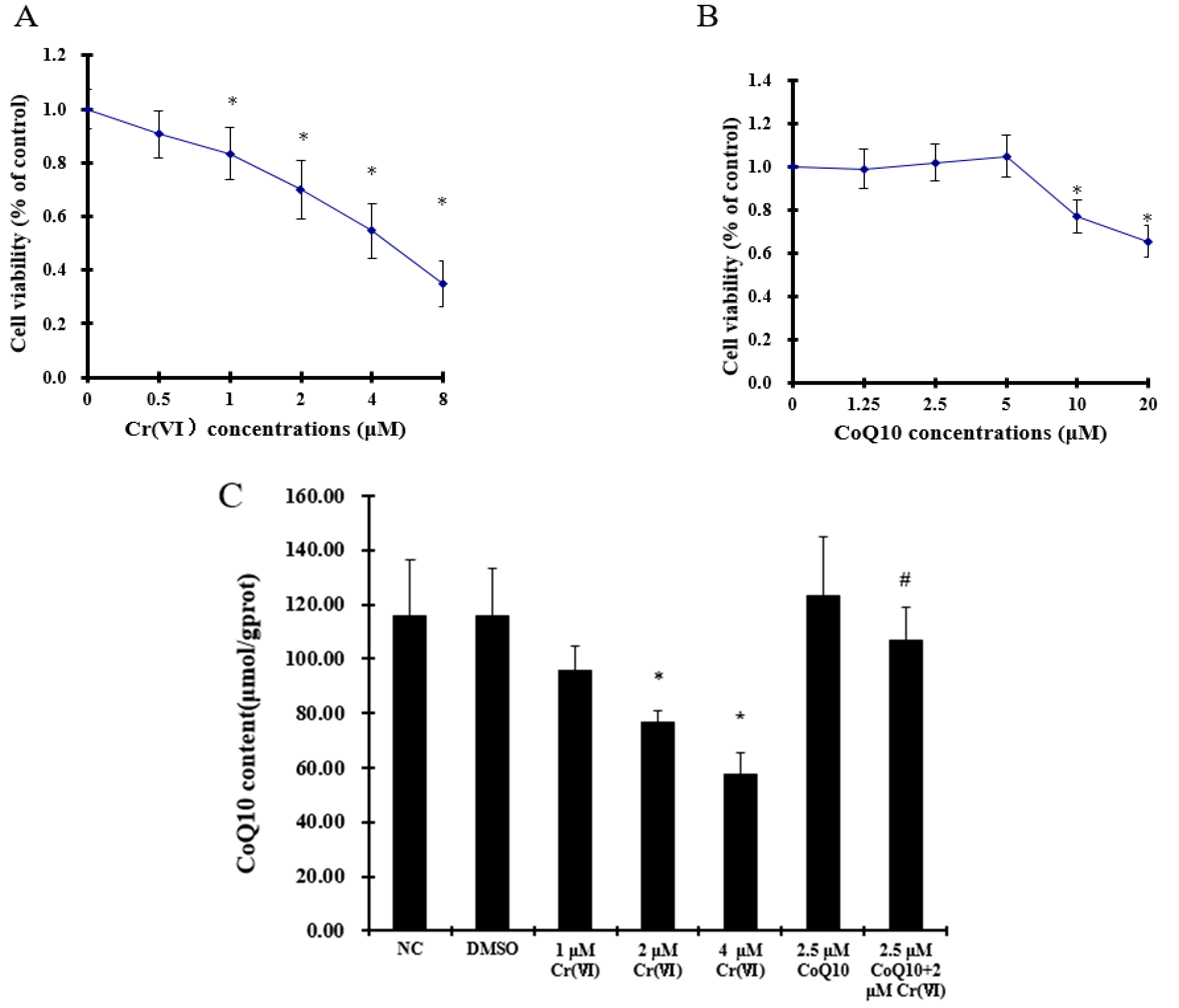
2.1. Effect of Coenzyme Q10 and Cr(VI) on L-02 Hepatocyte Viability
2.2. Cr(VI) Decreases CoQ10 Content in L-02 Hepatocytes
2.3. Effect of Cr(VI) on the Expression of Genes Involved in the CoQ10 Synthesis Pathway
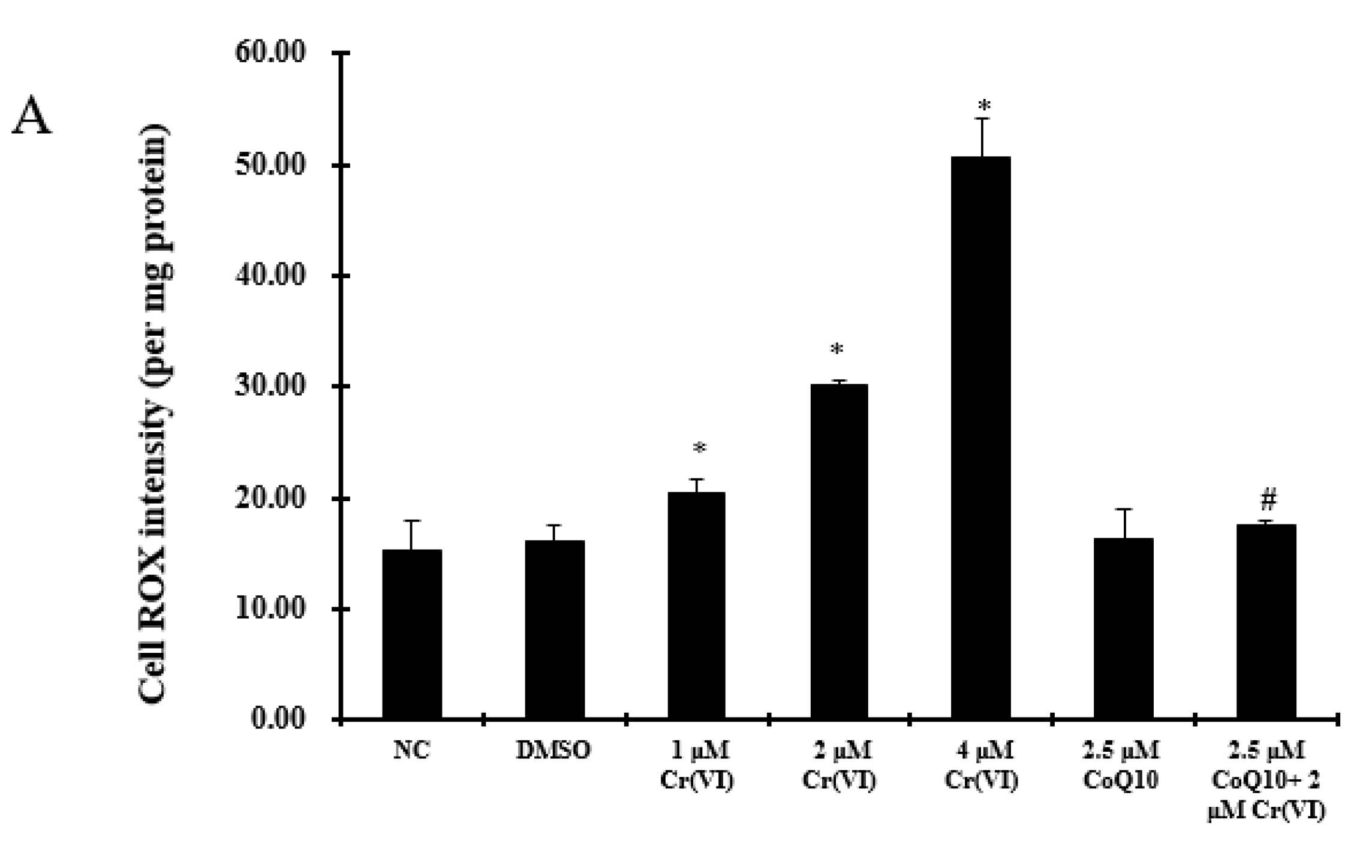
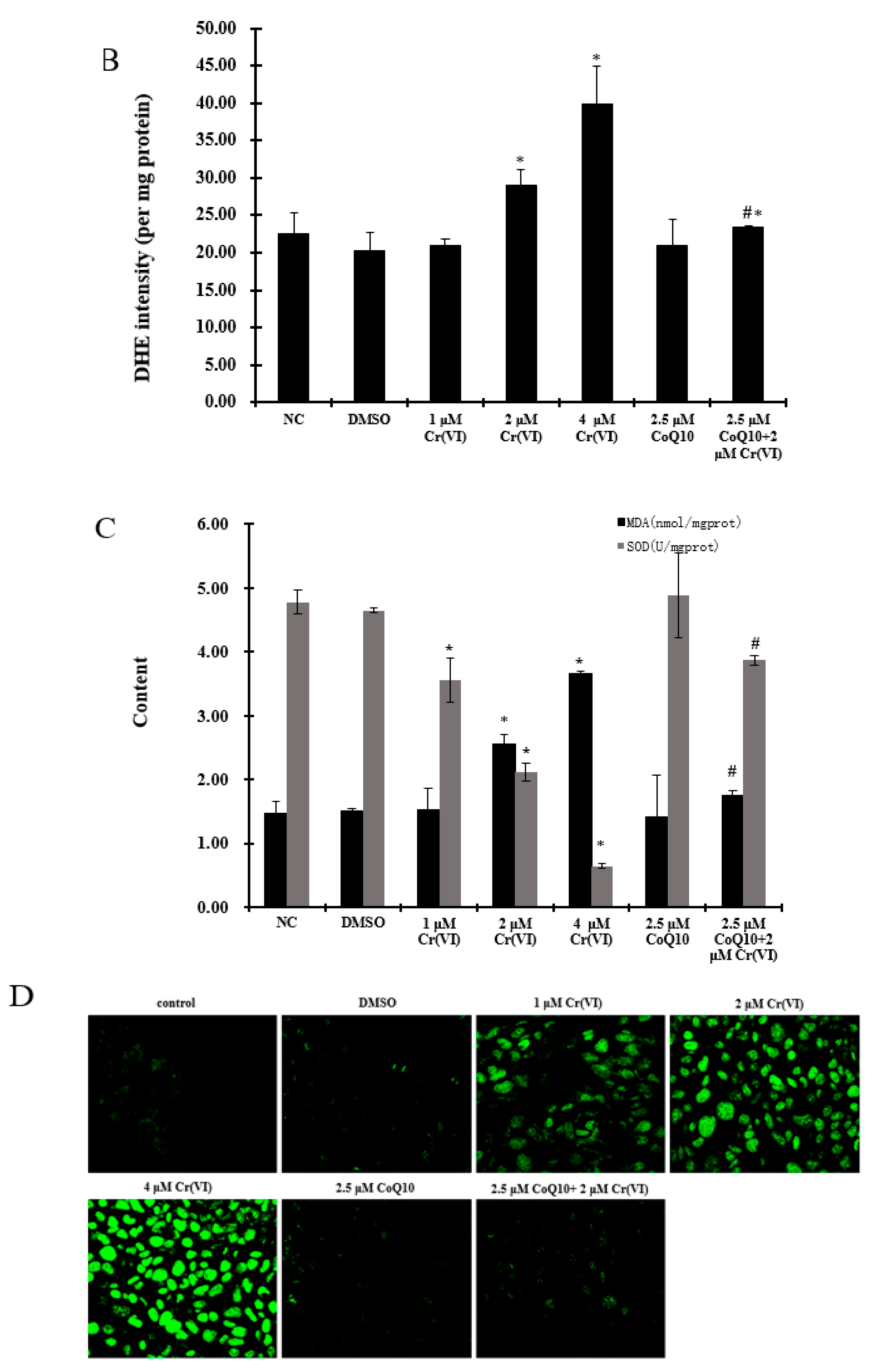
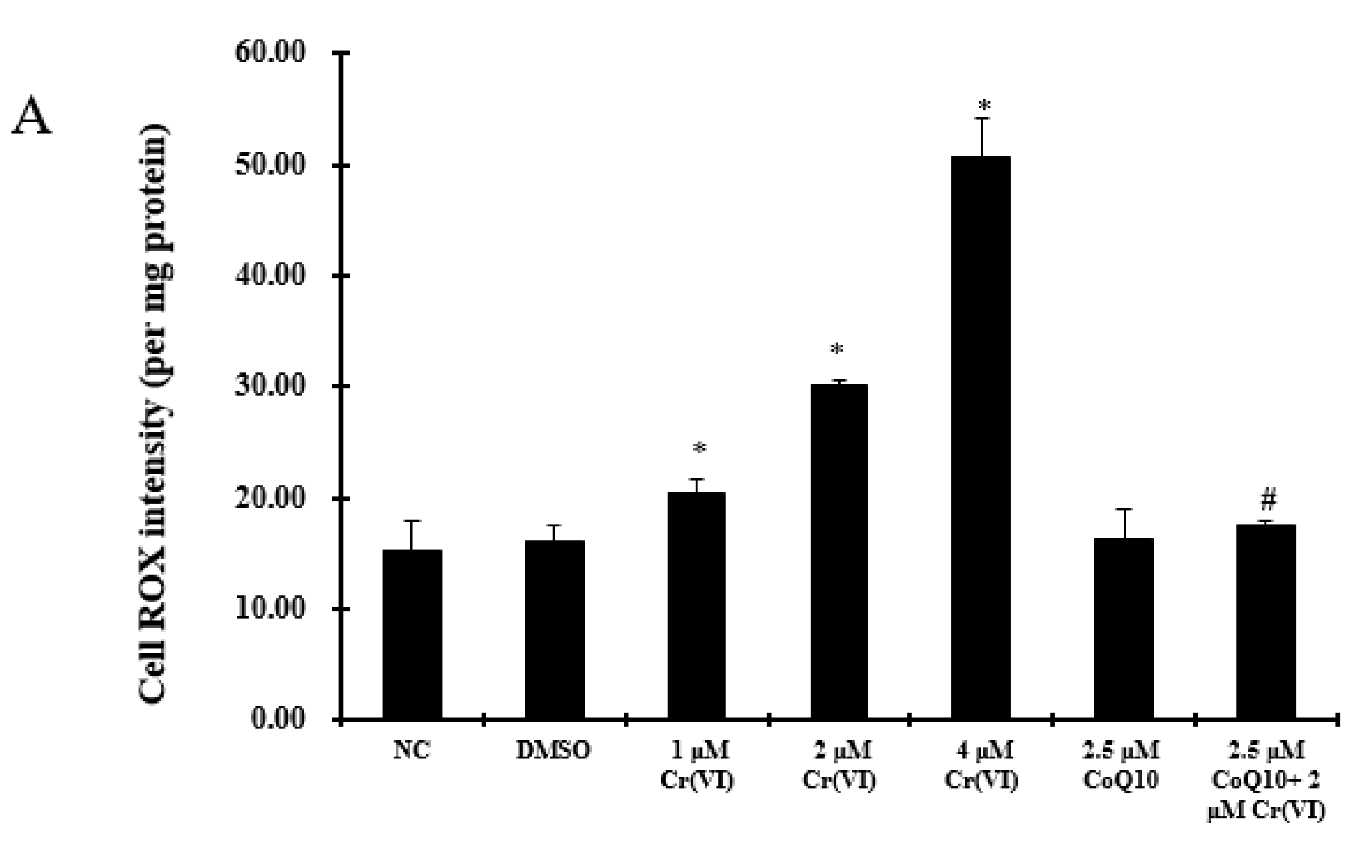
2.4. Oxidative Damage Induced by Cr(VI) Is Reduced by CoQ10
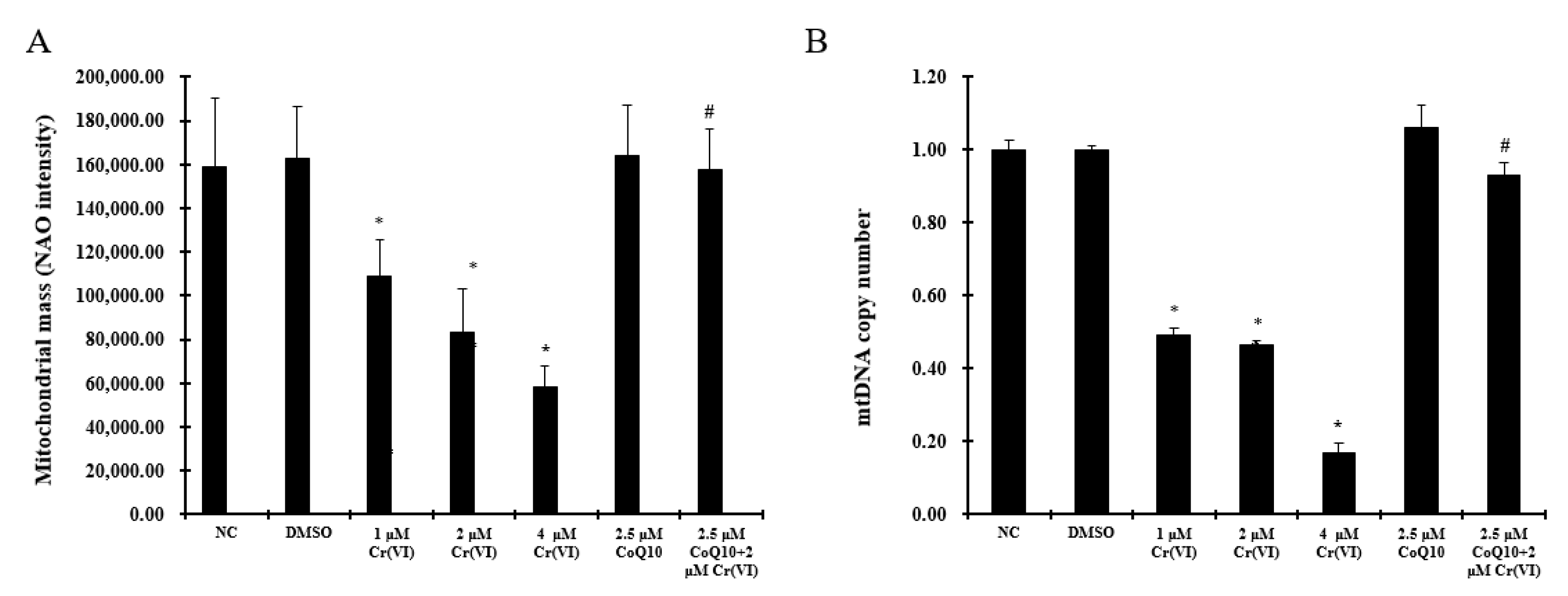
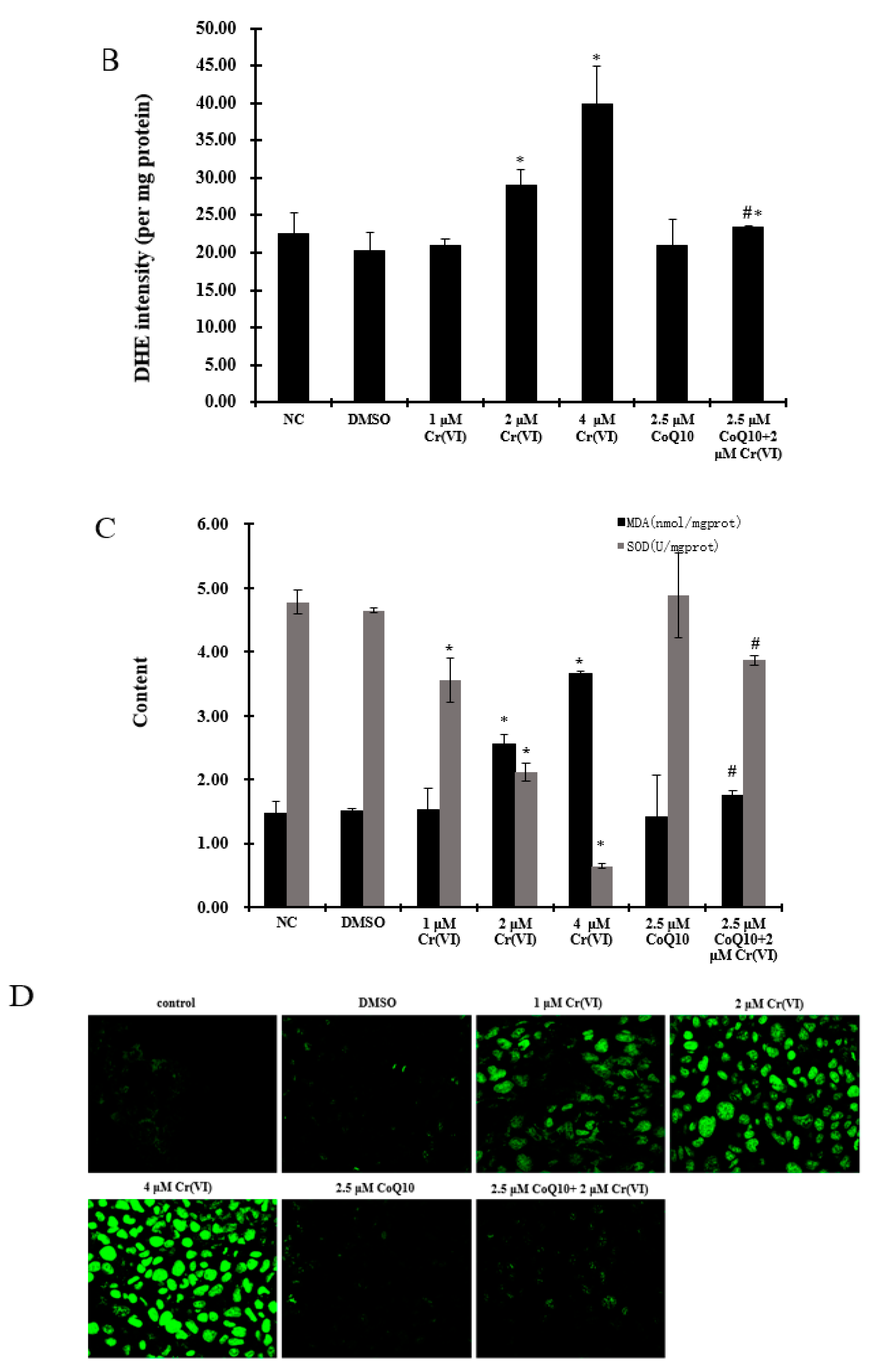
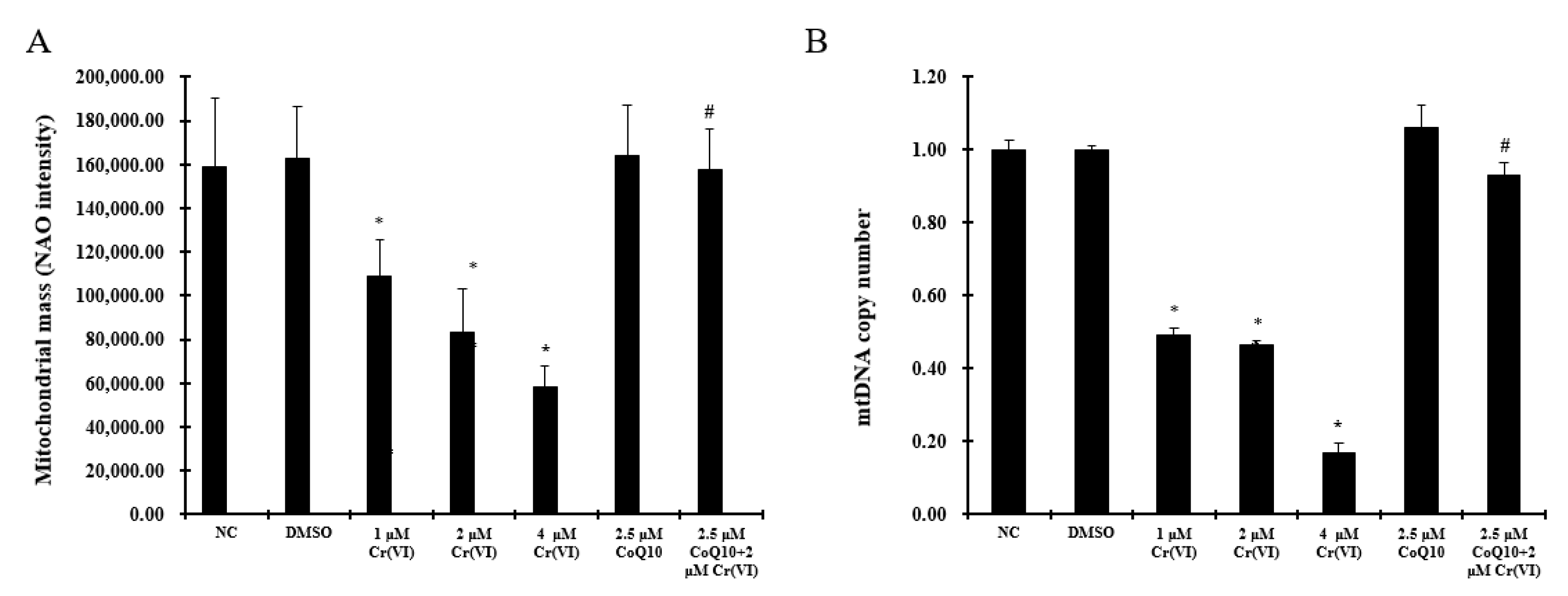
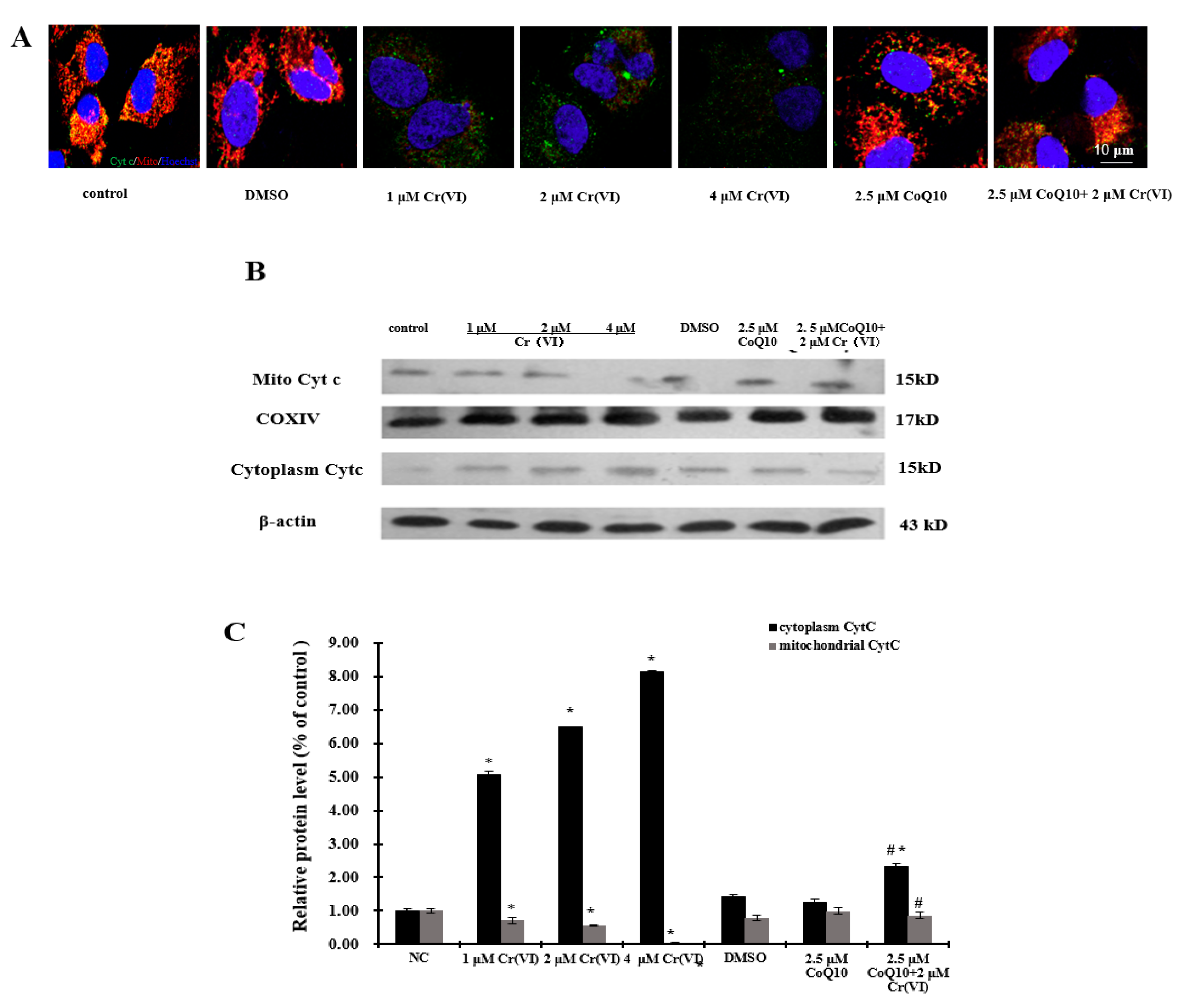
2.5. Induction of Mitochondrial Loss by Cr(VI) Can Be Counteracted by Supplementation with CoQ10
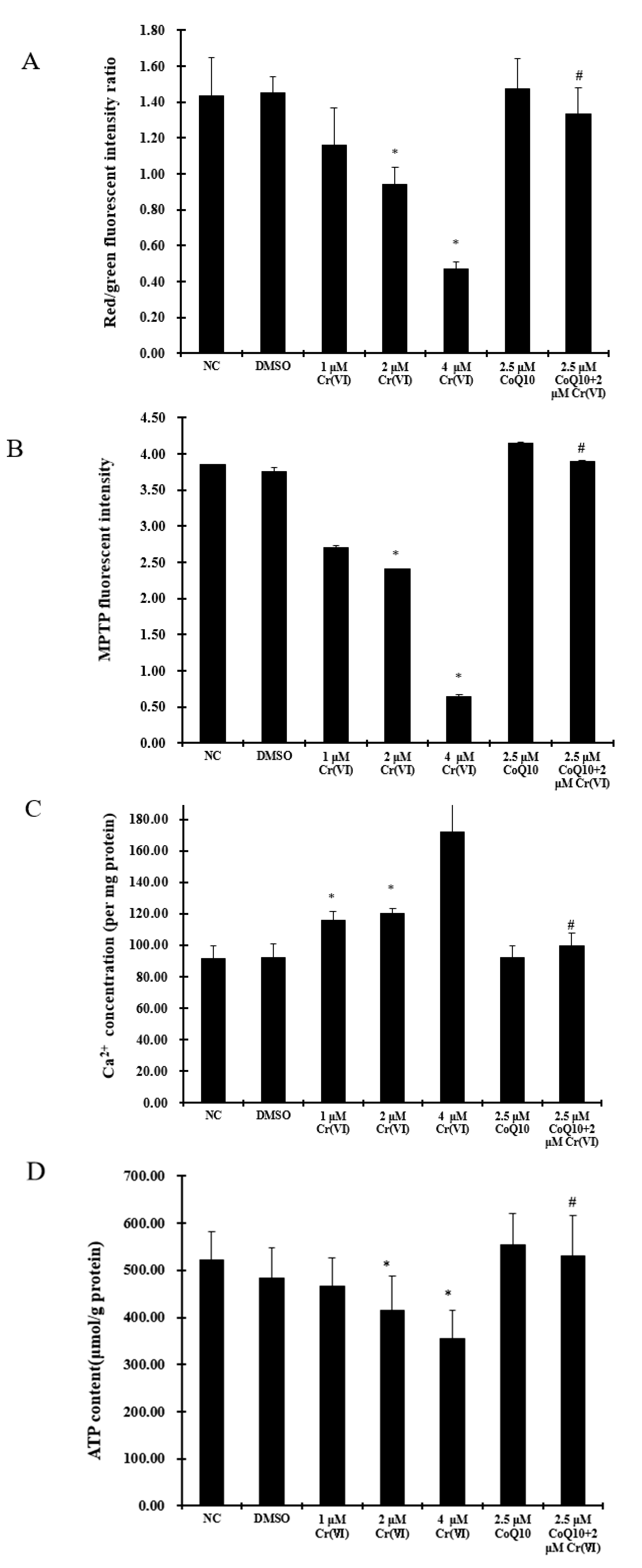
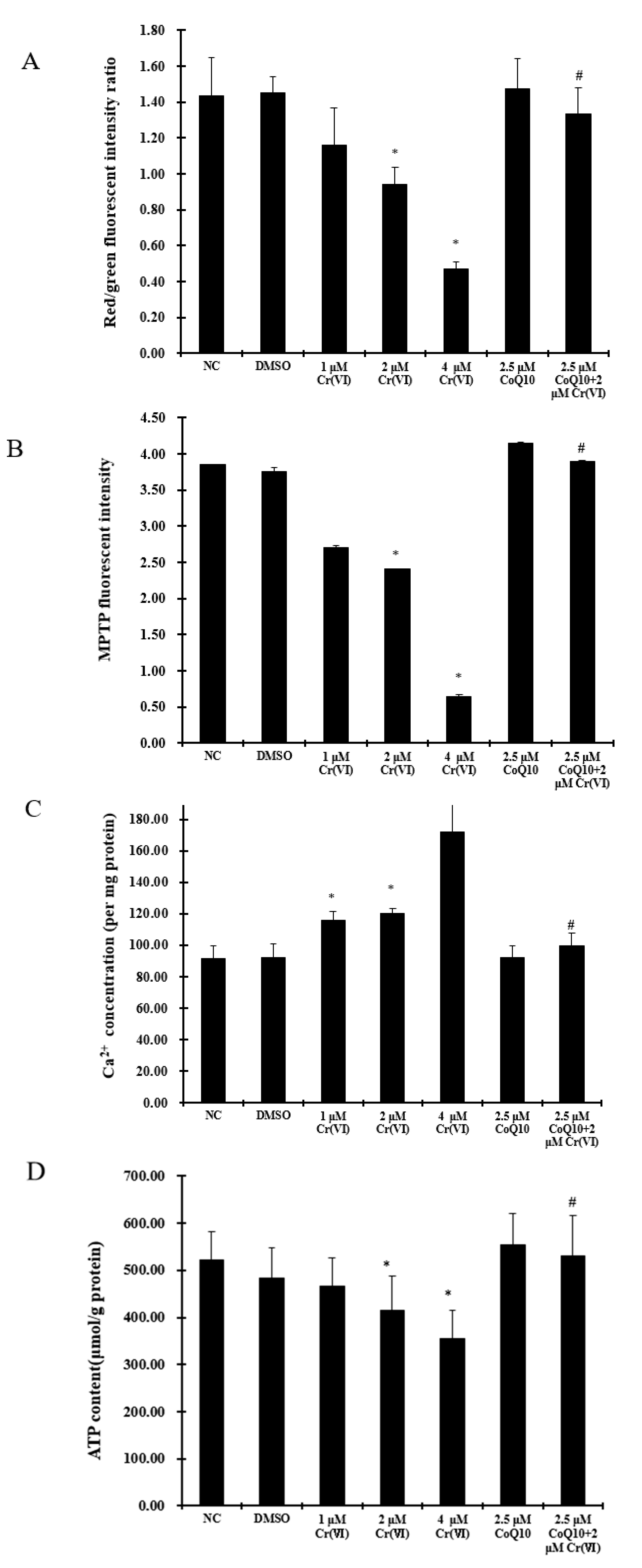
2.6. Cr(VI) Induces Mitochondrial Depolarization, MPTP Opening, Ca2+ Overload, and Decreased ATP Levels, and These Outcomes Are Attenuated by CoQ10
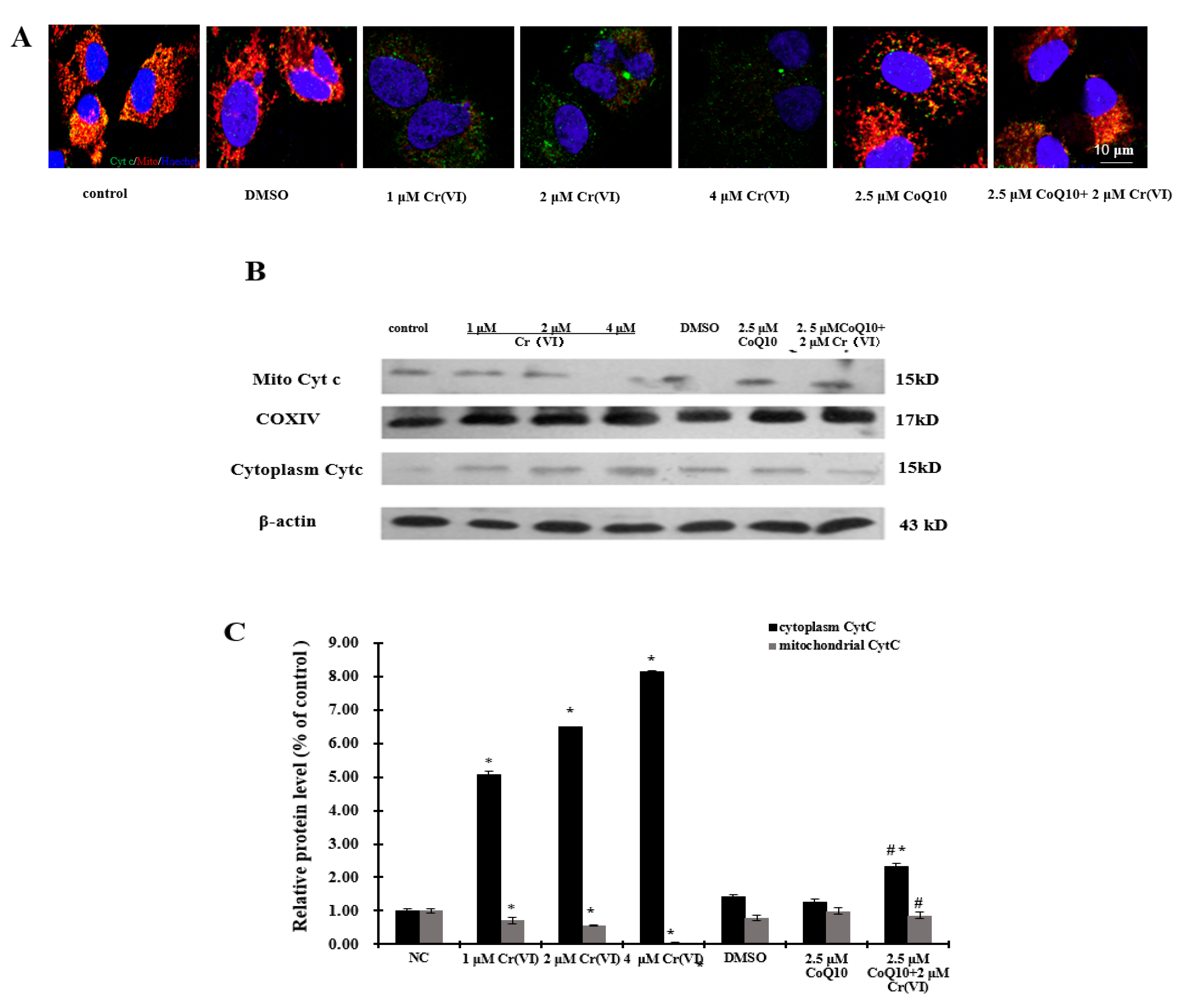
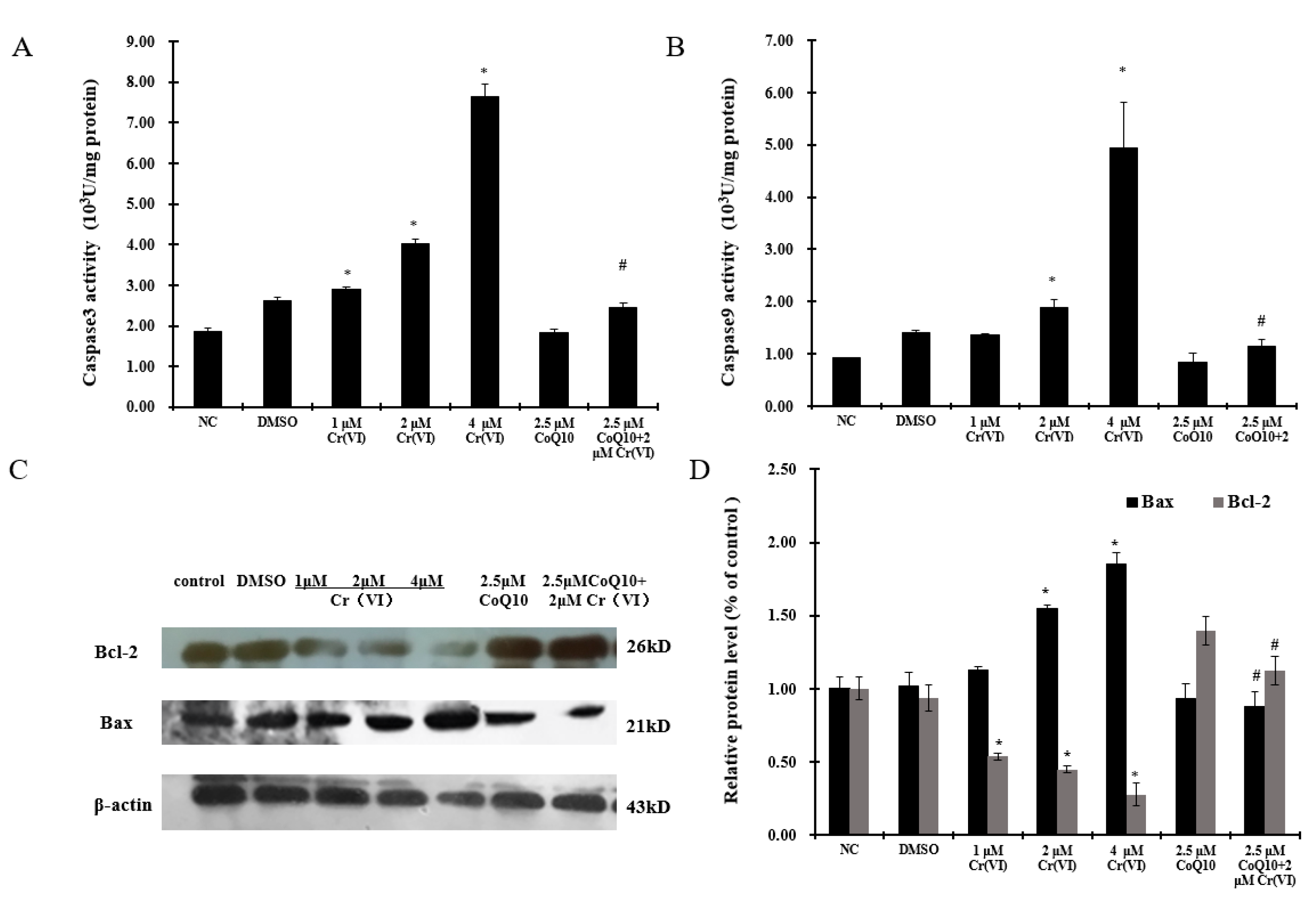
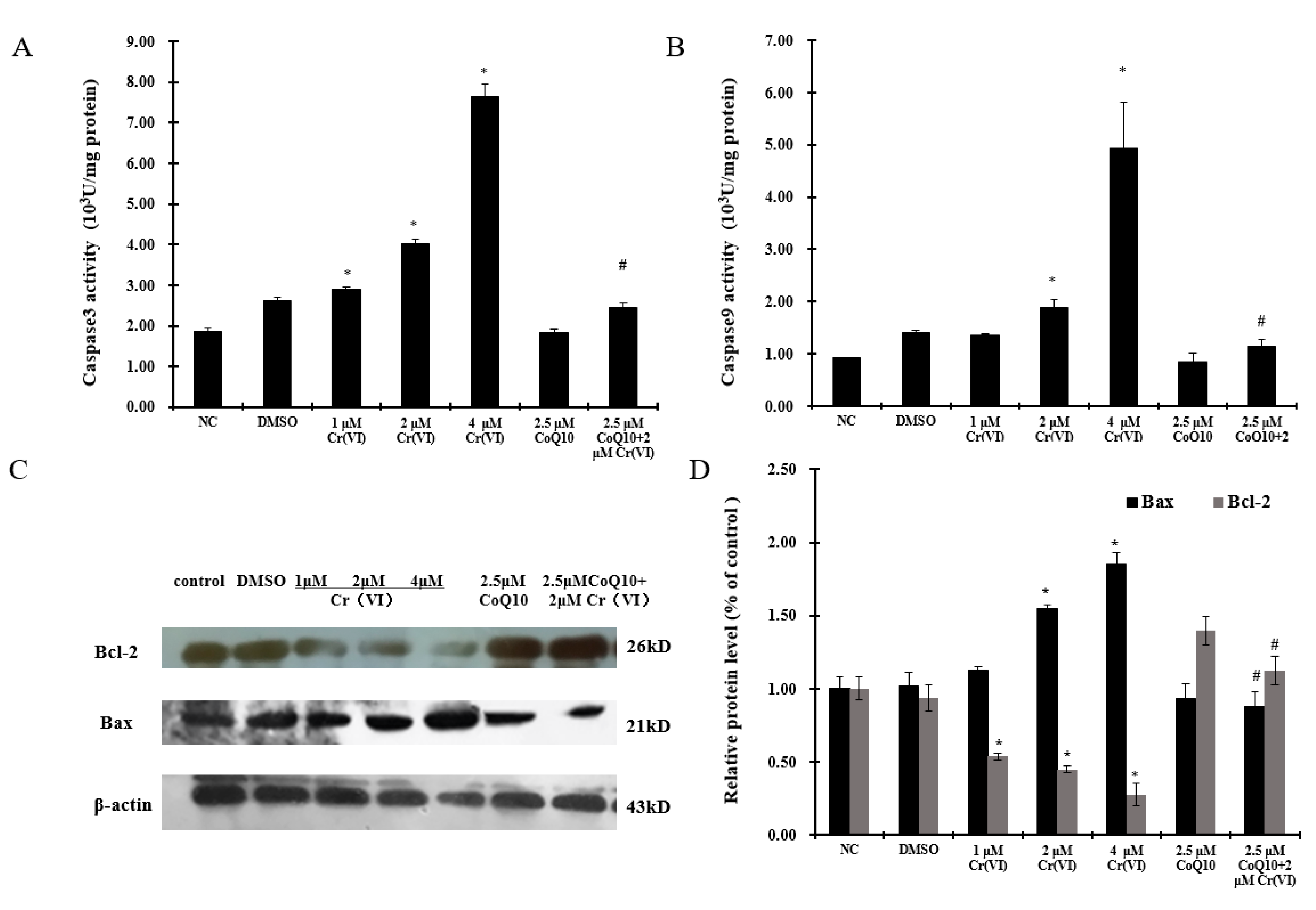
2.7. Cr(VI) Induces Cyt c Release, Caspase-3 and Caspase-9 Activation, and Unbalanced Bcl-2/Bax Expression in Response to Apoptotic Stimuli, and CoQ10 Counteracts These Outcomes
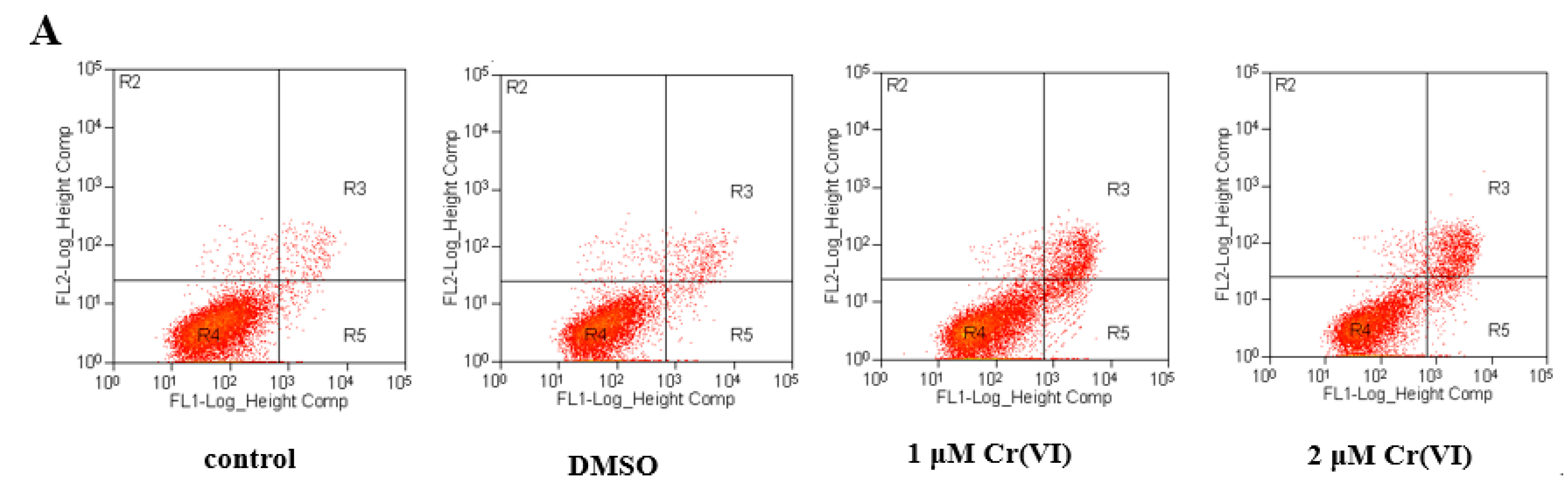
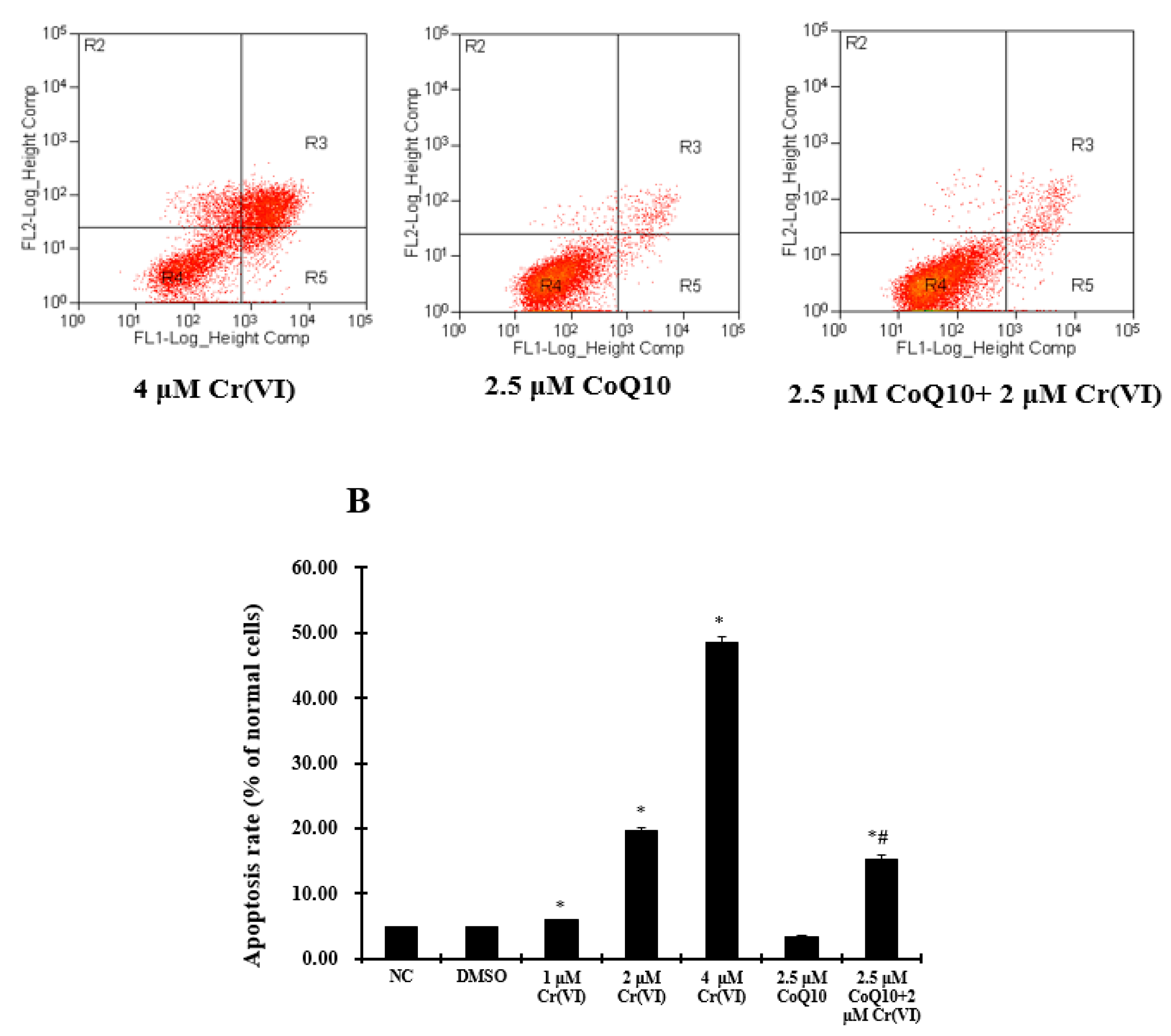
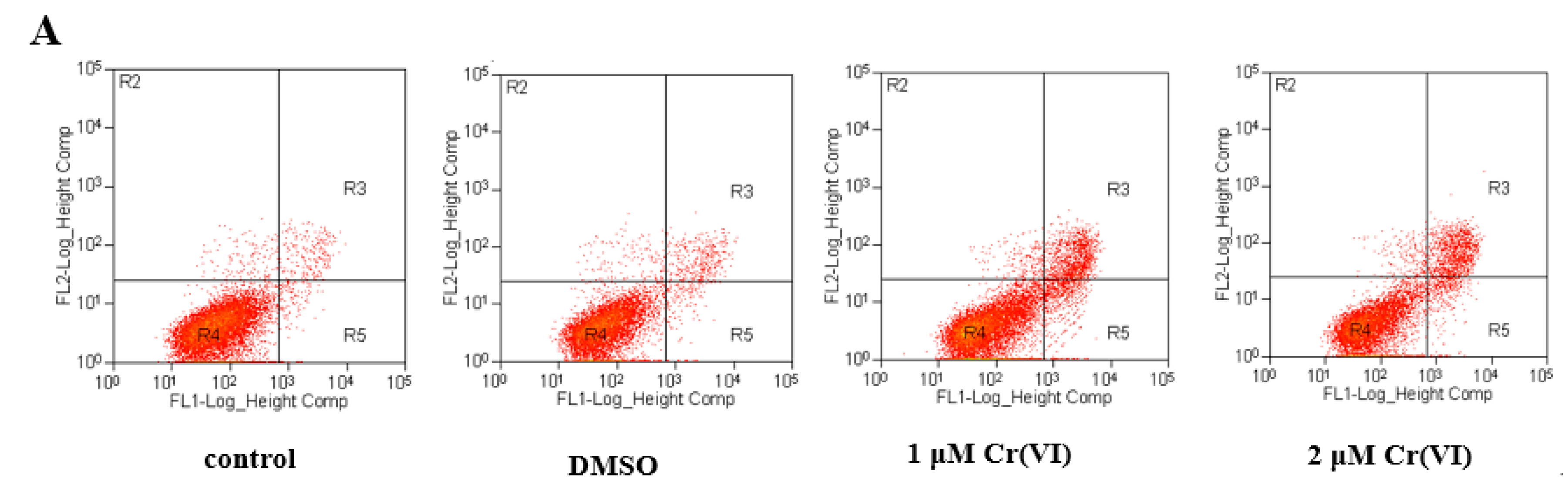
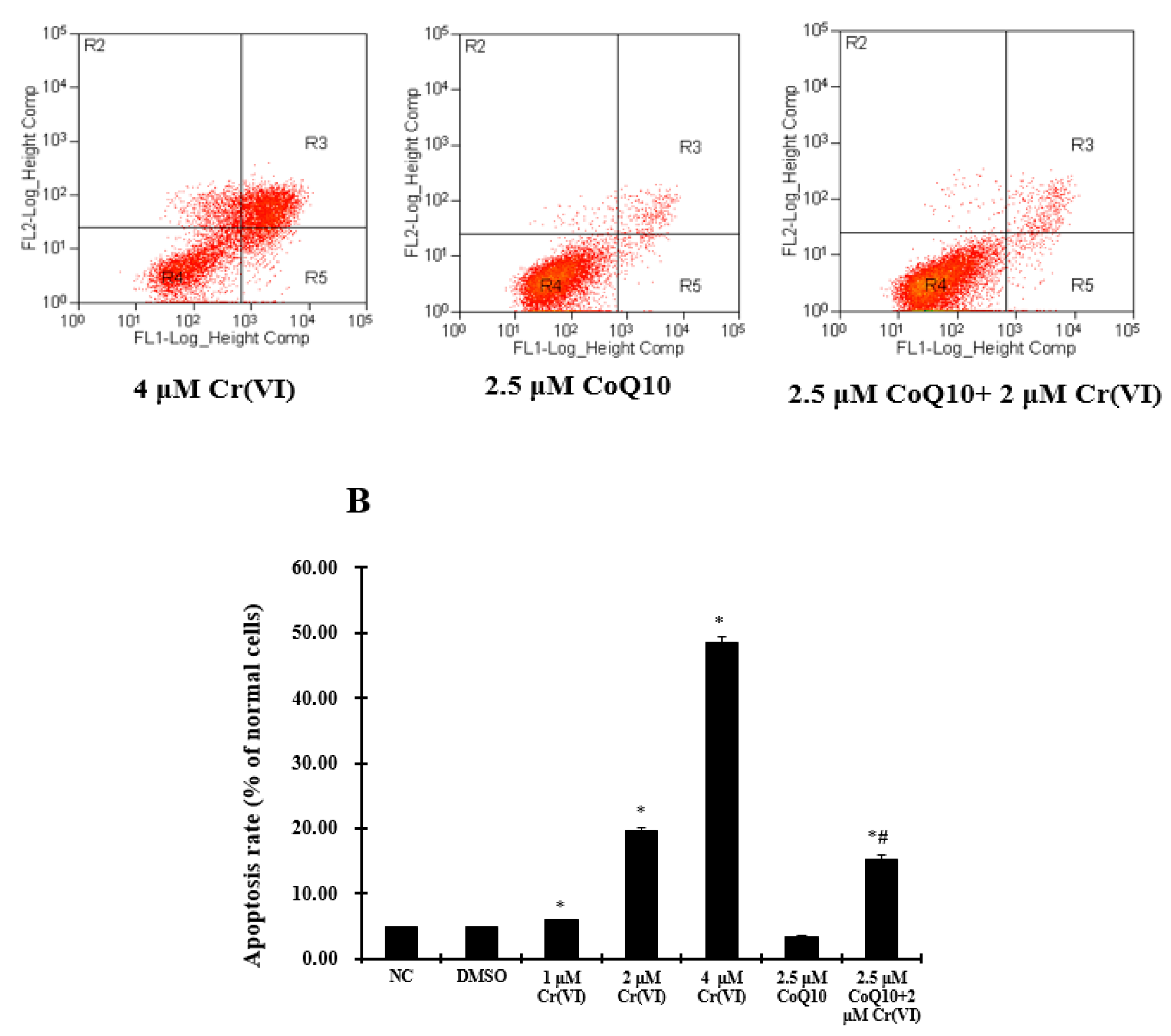
2.8. Cr(VI) Induces L-02 Hepatocyte Apoptosis in a Concentration-Dependent Manner, and CoQ10 Might Reduce the Rate of Apoptosis
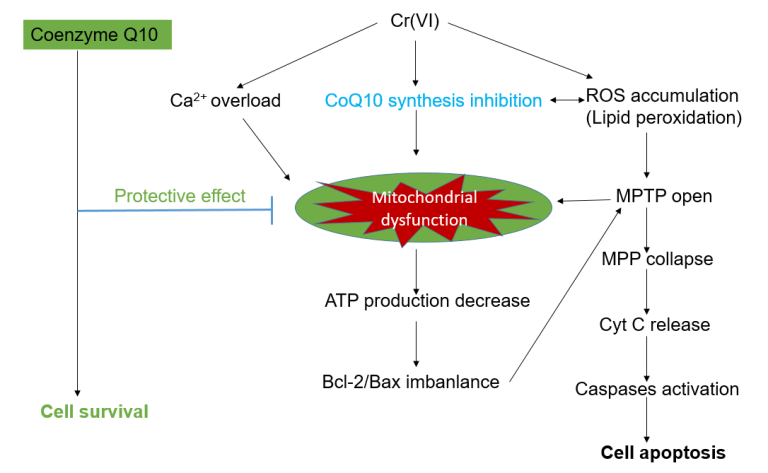
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Treatment of Cells with Cr(VI) and CoQ10
4.4. Cell Viability Assay
4.5. Preparation of Mitochondria
4.6. Extraction and Quantification of CoQ10
4.7. Gene Chip ANALYSIS
4.8. Real-Time PCR
4.9. Determination of Reactive Oxygen Species (ROS)
4.10. Measurement of Superoxide Anion (O2−)
4.11. Evaluation of Methane Dicarboxylic Aldehyde (MDA) and Superoxide Dismutase (SOD) Levels
4.12. Measurement of the Mitochondrial Mass
4.13. Measurement of the Mitochondrial Transmembrane Potential (MMP, Δψm) in Cells
4.14. Measurement of the MPTP Opening Degree
4.15. Measurement of Intracellular ATP Levels
4.16. Measurement of the Cellular Calcium Concentration (Ca2+)
4.17. Caspase Activity Assay
4.18. Western Blot Analysis
4.19. Immunofluorescence
4.20. FITC Annexin V/propidium Iodide (PI) Staining for Apoptotic Cells
4.21. Protein Assay
4.22. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Cr(VI) | hexavalent chromium |
| CoQ10 | coenzyme Q10 |
| ROS | reactive oxygen species |
| MMP | mitochondrial membrane potential |
| Cyt c | cytochrome c |
| MPTP | mitochondrial permeability transition pore |
| MDA | methane dicarboxylic aldehyde |
| SOD | superoxide dismutase |
| PDSS1 | prenyl(decaprenyl) diphosphate synthase, subunit 1 |
| PDSS2 | prenyl (decaprenyl) diphosphate synthase, subunit 2 |
| COQ2 | 4-hydroxybenzoate polyprenyltransferase |
| COQ3 | coenzyme Q3 methyltransferase |
| COQ4 | coenzyme Q4 |
| COQ5 | coenzyme Q5 methyltransferase |
| COQ6 | coenzyme Q6 monooxygenase |
| COQ7 | coenzyme Q7 homolog |
| ADCK3 | aarF domain-containing kinase 3 |
| ADCK4 | aarF domain-containing kinase 4 |
| COQ9 | coenzyme Q9 |
| COQ10A | coenzyme Q10A |
| COA10B | coenzyme Q10B |
| APTX | aprataxin |
| ETFDH | electron transfer flavoprotein dehydrogenase |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase |
| PMVK | phosphomevalonate kinase |
| MVD | mevalonate diphosphate decarboxylase |
| MVK | mevalonate kinase |
References
- Krumschnabel, G.; Nawaz, M. Acute toxicity of hexavalent chromium in isolated teleost hepatocytes. Aquat. Toxicol. 2004, 70, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.M.; Kirman, C.R.; Proctor, D.M.; Haws, L.C.; Suh, M.; Hays, S.M.; Hixon, J.G.; Harris, M.A. A chronic oral reference dose for hexavalent chromium-induced intestinal cancer. J. Appl. Toxicol. 2014, 34, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Stout, M.D.; Herbert, R.A.; Kissling, G.E.; Collins, B.J.; Travlos, G.S.; Witt, K.L.; Melnick, R.L.; Abdo, K.M.; Malarkey, D.E.; Hooth, M.J. Hexavalent chromium is carcinogenic to F344/N rats and B6C3F1 mice after chronic oral exposure. Environ. Health Perspect. 2009, 117, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Costa, M. Toxicity and carcinogenicity of Cr(VI) in animal models and humans. Crit. Rev. Toxicol. 1997, 27, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Felter, S.P.; Dourson, M.L. Hexavalent chromium-contaminated soils: Options for risk assessment and risk management. Regul. Toxicol. Pharmacol. 1997, 25, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Megharaj, M.; Avudainayagam, S.; Naidu, R. Toxicity of hexavalent chromium and its reduction by bacteria isolated from soil contaminated with tannery waste. Curr. Microbial. 2003, 47, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Nudler, S.I.; Quinteros, F.A.; Miler, E.A.; Cabilla, J.P.; Ronchetti, S.A.; Duvilanski, B.H. Chromium VI administration induces oxidative stress in hypothalamus and anterior pituitary gland from male rats. Toxicol. Lett. 2009, 185, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Michie, C.A.; Hayhurst, M.; Knobel, G.J.; Stokol, J.M.; Hensley, B. Poisoning with a traditional remedy containing potassium dichromate. Hum. Exp. Toxicol. 1991, 10, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.J.; Stout, M.D.; Levine, K.E.; Kissling, G.E.; Melnick, R.L.; Fennell, T.R.; Walden, R.; Abdo, K.; Pritchard, J.B.; Fernando, R.A.; et al. Exposure to hexavalent chromium resulted in significantly higher tissue chromium burden compared with trivalent chromium following similar oral doses to male F344/N rats and female B6C3F1 mice. Toxicol. Sci. 2010, 118, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Proctor, D.M.; Otani, J.M.; Finley, B.L.; Paustenbach, D.J.; Bland, J.A.; Speizer, N.; Sargent, E.V. Is hexavalent chromium carcinogenic via ingestion? A weight-of-evidence review. J. Toxicol. Environ. Health Part A 2002, 65, 701–746. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Feng, X.; Zeng, M.; Guan, L.; Hu, Q.; Zhong, C. Hexavalent chromium induces energy metabolism disturbance and p53-dependent cell cycle arrest via reactive oxygen species in L-02 hepatocytes. Mol. Cell. Biochem. 2012, 371, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Linos, A.; Petralias, A.; Christophi, C.A.; Christoforidou, E.; Kouroutou, P.; Stoltidis, M.; Veloudaki, A.; Tzala, E.; Makris, K.C.; Karagas, M.R. Oral ingestion of hexavalent chromium through drinking water and cancer mortality in an industrial area of Greece—An ecological study. Environ. Health 2011, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, J.J.; Sedman, R.M.; Reynolds, S.D.; Sherman, C.D.; Li, L.H.; Howd, R.A.; Sandy, M.S.; Zeise, L.; Alexeeff, G.V. Cancer mortality in a chinese population exposed to hexavalent chromium in drinking water. Epidemiology 2008, 19, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, H.; Xiang, X.H.; Liu, F.Y. Outline of occupational chromium poisoning in china. Bull. Environ. Contam. Toxicol. 2013, 90, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.M.; Antholine, W.E.; Myers, C.R. The intracellular redox stress caused by hexavalent chromium is selective for proteins that have key roles in cell survival and thiol redox control. Toxicology 2011, 281, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Fang, K.M.; Yang, C.S.; Tzeng, S.F. Reactive oxygen species-induced cell death of rat primary astrocytes through mitochondria-mediated mechanism. J. Cell. Biochem. 2009, 107, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Banu, S.K.; Stanley, J.A.; Lee, J.; Stephen, S.D.; Arosh, J.A.; Hoyer, P.B.; Burghardt, R.C. Hexavalent chromium-induced apoptosis of granulosa cells involves selective sub-cellular translocation of Bcl-2 members, ERK1/2 and p53. Toxicol. Appl. Pharmacol. 2011, 251, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Marouani, N.; Tebourbi, O.; Mokni, M.; Yacoubi, M.T.; Sakly, M.; Benkhalifa, M.; Rhouma, K.B. Hexavalent chromium-induced apoptosis in rat uterus: Involvement of oxidative stress. Arch. Environ. Occup. Health 2015, 70, 189–195. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.; Somayajulu, M.; Sikorska, M.; Borowy-Borowski, H.; Pandey, S. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water-soluble coenzyme Q10. Toxicol. Appl. Pharmacol. 2004, 201, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme Q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [CrossRef] [PubMed]
- Doimo, M.; Desbats, M.A.; Cerqua, C.; Cassina, M.; Trevisson, E.; Salviati, L. Genetics of coenzyme Q10 deficiency. Mol. Syndromol. 2014, 5, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Kawamukai, M. Biosynthesis of coenzyme Q in eukaryotes. Biosci. Biotechnol. Biochem. 2015, 80, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Lopez, L.C.; Naini, A.; DiMauro, S.; Hirano, M. Human COQ10 deficiencies. BioFactors 2008, 32, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Ben-Meir, A.; Burstein, E.; Borrego-Alvarez, A.; Chong, J.; Wong, E.; Yavorska, T.; Naranian, T.; Chi, M.; Wang, Y.; Bentov, Y.; et al. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell 2015, 14, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Lopez, L.C.; Gilkerson, R.W.; Dorado, B.; Coku, J.; Naini, A.B.; Lagier-Tourenne, C.; Schuelke, M.; Salviati, L.; Carrozzo, R.; et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of COQ10 deficiency. FASEB J. 2010, 24, 3733–3743. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Emmanuele, V.; Hirano, M. Clinical presentations of coenzyme Q10 deficiency syndrome. Mol. Syndromol. 2014, 5, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Potgieter, M.; Pretorius, E.; Pepper, M.S. Primary and secondary coenzyme Q10 deficiency: The role of therapeutic supplementation. Nutr. Rev. 2013, 71, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Kooncumchoo, P.; Sharma, S.; Porter, J.; Govitrapong, P.; Ebadi, M. Coenzyme Q10 provides neuroprotection in iron-induced apoptosis in dopaminergic neurons. J. Mol. Neurosci. 2006, 28, 125–141. [Google Scholar] [CrossRef]
- Abdallah, G.M.; El-Sayed el, S.M.; Abo-Salem, O.M. Effect of lead toxicity on coenzyme Q levels in rat tissues. Food Chem. Toxicol. 2010, 48, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.H.; Hans, V.H.; Palmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.J.; Fonseca, L.V.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochim. Biophys. Acta. 2016, 1857, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Liu, C.C.; Yen, H.C. Detection of suppressed maturation of the human COQ5 protein in the mitochondria following mitochondrial uncoupling by an antibody recognizing both precursor and mature forms of COQ5. Mitochondrion 2013, 13, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.C.; Liu, Y.C.; Kan, C.C.; Wei, H.J.; Lee, S.H.; Wei, Y.H.; Feng, Y.H.; Chen, C.W.; Huang, C.C. Disruption of the human COQ5-containing protein complex is associated with diminished coenzyme Q10 levels under two different conditions of mitochondrial energy deficiency. Biochim. Biophys. Acta 2016, 1860, 1864–1876. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, B.; Jia, Z.C. Preparation and characterization of human ADCK3, a putative atypical kinase. Protein Expr. Purif. 2015, 108, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, J.; Zhang, Y.; Fang, Y.; Wang, F.; Wang, J.; Zheng, X.; Yang, J. A systematic study on drug-response associated genes using baseline gene expressions of the cancer cell line encyclopedia. Sci. Rep. 2016, 6, 22811. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Li, Y.; Luo, L.; Xie, Y.; Zeng, M.; Wang, A.; Chen, H.; Zhong, C. Role of mitochondrial electron transport chain dysfunction in Cr(VI)-induced cytotoxicity in L-02 hepatocytes. Cell. Physiol. Biochem. 2014, 33, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Montero, R.; Pineda, M.; Aracil, A.; Vilaseca, M.A.; Briones, P.; Sanchez-Alcazar, J.A.; Navas, P.; Artuch, R. Clinical, biochemical and molecular aspects of cerebellar ataxia and coenzyme Q10 deficiency. Cerebellum 2007, 6, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Martin, J.M.; Salviati, L.; Trevisson, E.; Montini, G.; DiMauro, S.; Quinzii, C.; Hirano, M.; Rodriguez-Hernandez, A.; Cordero, M.D.; Sanchez-Alcazar, J.A.; et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum. Mol. Genet. 2007, 16, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J.J.; Kagan, V.; Ackrell, B.A.; Serbinova, E.; Packer, L. Succinate-ubiquinone reductase linked recycling of α-tocopherol in reconstituted systems and mitochondria: Requirement for reduced ubiquinone. Arch. Biochem. Biophys. 1992, 292, 47–53. [Google Scholar] [CrossRef]
- Duberley, K.E.; Heales, S.J.R.; Abramov, A.Y.; Chalasani, A.; Land, J.M.; Rahman, S.; Hargreaves, I.P. Effect of coenzyme Q10 supplementation on mitochondrial electron transport chain activity and mitochondrial oxidative stress in coenzyme Q10 deficient human neuronal cells. Int. J. Biochem. Cell Biol. 2014, 50, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Maraver, J.; Cordero, M.D.; Oropesa-Avila, M.; Fernandez Vega, A.; de la Mata, M.; Delgado Pavon, A.; de Miguel, M.; Perez Calero, C.; Villanueva Paz, M.; Cotan, D.; et al. Coenzyme Q10 therapy. Mol. Syndromol. 2014, 5, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sevillano, M.A.; Garcia-Barrera, T.; Navarro, F.; Gomez-Ariza, J.L. Absolute quantification of superoxide dismutase in cytosol and mitochondria of mice hepatic cells exposed to mercury by a novel metallomic approach. Anal. Chim. Acta 2014, 842, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.L.; Wang, Z.H.; Li, G.R.; Liu, L.H. Protective effects of akebia saponin D against rotenone-induced hepatic mitochondria dysfunction. J. Pharmacol. Sci. 2014, 126, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Forster, M.J. Coenzyme Q, oxidative stress and aging. Mitochondrion 2007, 7, S103–S111. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Domijan, A.M.; Walker, M.C.; Abramov, A.Y. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 2014, 5, e1442. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Schweizer, M.; Cossarizza, A.; Franceschi, C. Control of apoptosis by the cellular ATP level. FEBS Lett. 1996, 378, 107–110. [Google Scholar] [CrossRef]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; di Lisa, F. The mitochondrial permeability transition, release of cytochrome C and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, T.; Wang, T.; Song, J.; Zhou, Z. Effects of apoptosis-related proteins caspase-3, Bax and Bcl-2 on cerebral ischemia rats. Biomed. Rep. 2013, 1, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Eghbal, M.A.; Abdoli, N.; Azarmi, Y. Efficiency of hepatocyte pretreatment with coenzyme Q10 against statin toxicity. Arh. Hig. Rada Toksikol. 2014, 65, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Liou, S.W.; Chen, C.C.; Chen, W.C.; Hu, F.R.; Wang, I.J.; Lin, S.J. Coenzyme Q10 reduces ethanol-induced apoptosis in corneal fibroblasts. PLoS ONE 2011, 6, e19111. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Li, H.B.; Gu, J.D.; Li, X.Y. Kinetics of the reduction of chromium VI by vitamin C. Environ. Toxicol. Chem. 2005, 24, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta Biomembr. 2004, 1660, 171–199. [Google Scholar] [CrossRef]
- Fontaine, E.; Bernardi, P. Progress on the mitochondrial permeability transition pore: Regulation by complex I and ubiquinone analogs. J. Bioenerg. Biomembr. 1999, 31, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Y.; Cai, N.; Ye, L.H.; Zhang, X.D. Transformation of human liver L-02 cells mediated by stable HBX transfection. Acta Pharmacol. Sin. 2009, 30, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.B.; Shi, Y.H.; Zhou, J.; Qiu, S.J.; Xu, Y.; Dai, Z.; Shi, G.M.; Wang, X.Y.; Ke, A.W.; Wu, B.; et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Turkowicz, M.J.; Karpinska, J. Analytical problems with the determination of coenzyme Q10 in biological samples. BioFactors 2013, 39, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Lass, A.; Sohal, R.S. Electron transport-linked ubiquinone-dependent recycling of α-tocopherol inhibits autooxidation of mitochondrial membranes. Arch. Biochem. Biophys. 1998, 352, 229–236. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Description | log2(Ratio) | p-Values |
|---|---|---|---|
| PDSS1 | prenyl (decaprenyl) diphosphate synthase, subunit 1 | 0.079217 | 0.728622 |
| PDSS2 | prenyl (decaprenyl) diphosphate synthase, subunit 2 | −0.917213 | 2.86 × 10−5 |
| COQ2 | coenzyme Q2 homolog, prenyltransferase | −0.136864 | 0.434046 |
| COQ3 | coenzyme Q3 homolog, methyltransferase | −0.443767 | 0.002707 |
| COQ4 | coenzyme Q4 homolog | −0.646236 | 0.004524 |
| COQ5 | coenzyme Q5 homolog, methyltransferase | −0.986167 | 4.4 × 10−8 |
| COQ6 | coenzyme Q6 homolog, monooxygenase | −0.185303 | 0.267227 |
| COQ7 | coenzyme Q7 homolog, ubiquinone | −0.308896 | 0.011926 |
| COQ7 | coenzyme Q7 homolog, ubiquinone | −0.528129 | 0.115161 |
| ADCK3 | aarF domain containing kinase 3 | 0.970573 | 6.12 × 10−9 |
| ADCK4 | aarF domain containing kinase 4 | NA | NA |
| COQ9 | coenzyme Q9 homolog | −0.936477 | 3.1 × 10−18 |
| COQ10A | coenzyme Q10 homolog A | 0.239029 | 0.045092 |
| COQ10B | coenzyme Q10 homolog B | 0.59875 | 0.000958 |
| APTX | aprataxin | −0.648958 | 0.003698 |
| ETFDH | electron-transferring-flavoprotein dehydrogenase | −1.412843 | 4.43 × 10−7 |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase | −0.837738 | 0.008798 |
| PMVK | phosphomevalonate kinase | −0.146285 | 0.455768 |
| MVD | mevalonate (diphospho) decarboxylase | 0.842443 | 0.01009 |
| MVK | mevalonate kinase | 0.612375 | 0.000892 |
| Target | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|
| mtDNA | CAAACCTACGCCAAAATCCA | GAAATGAATGAGCCTACAGA |
| GAPDH | TGACAACAGCCTCAAGAT | GAGTCCTTCCACGATACC |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, X.; Yi, X.; Da Silveira e Sá, R.D.C.; Zhang, Y.; Liu, K.; Xiao, F.; Zhong, C. CoQ10 Deficiency May Indicate Mitochondrial Dysfunction in Cr(VI) Toxicity. Int. J. Mol. Sci. 2017, 18, 816. https://doi.org/10.3390/ijms18040816
Zhong X, Yi X, Da Silveira e Sá RDC, Zhang Y, Liu K, Xiao F, Zhong C. CoQ10 Deficiency May Indicate Mitochondrial Dysfunction in Cr(VI) Toxicity. International Journal of Molecular Sciences. 2017; 18(4):816. https://doi.org/10.3390/ijms18040816
Chicago/Turabian StyleZhong, Xiali, Xing Yi, Rita De Cássia Da Silveira e Sá, Yujing Zhang, Kaihua Liu, Fang Xiao, and Caigao Zhong. 2017. "CoQ10 Deficiency May Indicate Mitochondrial Dysfunction in Cr(VI) Toxicity" International Journal of Molecular Sciences 18, no. 4: 816. https://doi.org/10.3390/ijms18040816
APA StyleZhong, X., Yi, X., Da Silveira e Sá, R. D. C., Zhang, Y., Liu, K., Xiao, F., & Zhong, C. (2017). CoQ10 Deficiency May Indicate Mitochondrial Dysfunction in Cr(VI) Toxicity. International Journal of Molecular Sciences, 18(4), 816. https://doi.org/10.3390/ijms18040816