
Genetics of Congenital Anomalies of the Kidney and Urinary Tract: The Current State of Play


Abstract
:1. Introduction
2. Pathogenesis
3. Research Strategies Overview
3.1. Candidate Gene Approach
3.2. Linkage Analysis
3.3. Genetic Isolates
3.4. Next-Generation Sequencing (NGS) (Whole Genome Sequencing, Whole Exome Sequencing and Targeted Resequencing)
3.5. Genome-Wide Association Studies
3.6. Structural Variants
4. Genetic Results
4.1. Syndromic Forms
4.2. Non-Syndromic Forms
4.3. Genomic Imbalance
5. Future Perspectives
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Vivante, A.; Kohl, S.; Hwang, D.; Dworschak, G.; Hildebrandt, F. Single-gene causes of congential anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr. Nephrol. 2014, 29, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Dressler, G. The cellular basis of kidney development. Annu. Rev. Cell Dev. Biol. 2006, 22, 509–529. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, I.; Kuwayama, F.; Pope, J.; Stephens, F.; Miyazaki, Y. Paradigm shift from classic anatomic theories to contemporary cell biological views of CAKUT. Kidney Int. 2002, 61, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Sanna-Cherchi, S.; Caridi, G.; Weng, P.; Scolari, F.; Perfumo, F.; Gharavi, A.; Ghiggeri, G. Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr. Nephrol. 2007, 22, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Birth Defects Monitoring Program (BDMP)/Commission on Professional and Hospital Activities (CPHA) surveillance data 1988–1991. Teratology 1993, 48, 658–675. [Google Scholar]
- Loane, M.; Dolk, H.; Kelly, A.; Teljeur, C.; Greenlees, R.; Densem, J. EUROCAT working group paper 4: EUROCAT statistical monitoring: Identification and investigation of ten year trends of congenital anomalies in Europe. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 31–43. [Google Scholar] [CrossRef] [PubMed]
- North American Pediatric Renal Transplant Cooperative Study (NAPRTCS). 2008 Annual Report. Available online: https://web.emmes.com/study/ped/annlrept/Annual%20Report%20-2008.pdf (accessed on 10 April 2017).
- Ardissino, G.; Daccò, V.; Testa, S.; Bonaudo, R.; Claris-Appiani, A.; Taioli, E.; Marra, G.; Edefonti, A.; Sereni, F. Epidemiology of chronic renal failure in children: Data from the ItalKid project. Pediatrics 2003, 111, 382–387. [Google Scholar] [CrossRef]
- Sanna-Cherchi, S.; Ravani, P.; Corbani, V.; Parodi, S.; Haupt, R.; Piaggio, G.; Innocenti, M.; Somenzi, D.; Trivelli, A.; Caridi, G.; et al. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int. 2009, 76, 528–533. [Google Scholar] [CrossRef] [PubMed]
- La Scola, C.; Ammenti, A.; Puccio, G.; Lega, M.; de Mutiis, C.; Guiducci, C.; de Petris, L.; Perretta, R.; Venturoli, V.; Vergine, G.; et al. Congenital solitary kidney in children: Size matters. J. Urol. 2016, 196, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Harambat, J.; van Stralen, K.; Kim, J.; Tizard, E. Epidemiology of chronic kidney disease in children. Pediatr. Nephrol. 2012, 27, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Pope, J.; Brock, J.; Adams, M.; Stephens, F.; Ichikawa, I. How they begin and how they end: Classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J. Am. Soc. Nephrol. 1999, 10, 2018–2028. [Google Scholar] [PubMed]
- Hiraoka, M.; Tsukahara, H.; Ohshima, Y.; Kasuga, K.; Ishihara, Y.; Mayumi, M. Renal aplasia is the predominant cause of congenital solitary kidneys. Kidney Int. 2002, 61, 1840–1844. [Google Scholar] [CrossRef] [PubMed]
- Woolf, A.; Price, K.; Scambler, P.; Winyard, P. Evolving concepts in human renal dysplasia. J. Am. Soc. Nephrol. 2004, 15, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Sanyanusin, P.; Schimmenti, L.; McNoe, L.; Ward, T.; Pierpont, M.; Sullivan, M.; Dobyns, W.; Eccles, M. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat. Genet. 1995, 9, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Njolstad, P.; Horikawa, Y.; Bostad, L.; Bell, G.; Sovik, O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1β. Hum. Mol. Genet. 1999, 8, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Abdelhak, S.; Kalatzis, V.; Heilig, R.; Compain, S.; Samson, D.; Vincent, C.; Weil, D.; Cruaud, C.; Sahly, I.; Leibovici, M.; et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat. Genet. 1997, 15, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.; McCall, D.; Engelman, C.; Schrier, R. Congenital renal agenesis: Case-control analysis of birth characteristics. Am. J. Kidney Dis. 2002, 39, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.; Hernandez-Diaz, S.; Arbogast, P.; Dudley, J.; Dyer, S.; Gideon, P.; Hall, K.; Ray, W. Major congenital malformations after first-trimester exposure to ACE inhibitors. N. Engl. J. Med. 2006, 354, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Bulum, B.; Ozçakar, Z.; Ustüner, E.; Düşünceli, E.; Kavaz, A.; Duman, D.; Walz, K.; Fitoz, S.; Tekin, M.; Yalçınkaya, F. High frequency of kidney and urinary tract anomalies in asymptomatic first-degree relatives of patients with CAKUT. Pediatr. Nephrol. 2013, 28, 2143–2147. [Google Scholar] [CrossRef] [PubMed]
- Renkema, K.; Winyard, P.; Skovorodkin, I.; Levtchenko, E.; Hindryckx, A.; Jeanpierre, C.; Weber, S.; Salomon, R.; Antignac, C.; Vainio, S.; et al. Novel perspectives for investigating congenital anomalies of the kidney and urinary tract (CAKUT). Nephrol. Dial. Transplant. 2011, 26, 3843–3851. [Google Scholar] [CrossRef] [PubMed]
- Weber, S. Novel genetic aspects of congenital anomalies of kidney and urinary tract. Curr. Opin. Pediatr. 2012, 24, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Roodhooft, A.; Birnholz, J.; Holmes, L. Familial nature of congenital absence and severe dysgenesis of both kidneys. N. Engl. J. Med. 1984, 310, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- McPherson, E.; Carey, J.; Kramer, A.; Hall, J.; Pauli, R.; Schimke, R.; Tasin, M. Dominantly inherited renal adysplasia. Am. J. Med. Genet. 1987, 26, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.; Evans, K.; Pescia, G. A family study of renal agenesis. J. Med. Genet. 1979, 16, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Pasch, A.; Hoefele, J.; Grimminger, H.; Hacker, H.; Hildebrandt, F. Multiple urinary tract malformations with likely recessive inheritance in a large Somalian kindred. Nephrol. Dial. Transplant. 2004, 19, 3172–3175. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, B.; Milner, L.; Jequier, S.; Kaplan, P.; de Chadarevian, J. Autosomal dominant inheritance of small kidneys. Am. J. Med. Genet. 1989, 32, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Doray, B.; Gasser, B.; Reinartz, I.; Stoll, C. Hereditary renal adysplasia in a three generations family. Genet. Couns. 1999, 10, 251–257. [Google Scholar] [PubMed]
- Nicolaou, N.; Renkema, K.; Bongers, E.; Giles, R.; Knoers, N. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat. Rev. Nephrol. 2015, 11, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Schedl, A. Renal abnormalities and their developmental origin. Nat. Rev. Genet. 2007, 8, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Dressler, G. Advances in early kidney specification, development and patterning. Development 2009, 136, 3863–3874. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.; Rosenblum, N. Cell and molecular biology of kidney development. Semin. Nephrol. 2009, 29, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Faa, G.; Gerosa, C.; Fanni, D.; Monga, G.; Zaffanello, M.; van Eyken, P.; Fanos, V. Morphogenesis and molecular mechanisms involved in human kidney development. J. Cell Physiol. 2012, 227, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Vainio, S.; Lin, Y. Coordinating early kidney development: Lessons from gene targeting. Nat. Rev. Genet. 2002, 3, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Woolf, A. Embriology in Pediatric Nephrology, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Kohl, S.; Chen, J.; Vivante, A.; Hwang, D.; Shril, S.; Dworschak, G.; van der Ven, A.; Sanna-Cherchi, S.; Bauer, S.; Lee, R.; et al. Targeted sequencing of 96 renal developmental microRNAs in 1213 individuals from 980 families with congenital anomalies of the kidney and urinary tract. Nephrol. Dial. Transplant. 2016, 31, 1280–1283. [Google Scholar] [CrossRef] [PubMed]
- Mackie, G.; Stephens, F. Duplex kidneys: A correlation of renal dysplasia with position of the ureteral orifice. J. Urol. 1975, 114, 274–280. [Google Scholar] [PubMed]
- Grieshammer, U.; le Ma, P.A.; Wang, F.; Tessier-Lavigne, M.; Martin, G. SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev. Cell 2004, 6, 709–717. [Google Scholar] [CrossRef]
- Yu, J.; McMahon, A.; Valerius, M. Recent genetic studies of mouse kidney development. Curr. Opin. Genet. Dev. 2004, 14, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Oshima, K.; Fogo, A.; Ichikawa, I. Evidence that bone morphogenetic protein 4 has multiple biological functions during kidney and urinary tract development. Kidney Int. 2003, 63, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Durbec, P.; Marcos-Gutierrez, C.; Kilkenny, C.; Grigoriou, M.; Wartiowaara, K.; Suvanto, P.; Smith, D.; Ponder, B.; Costantini, F.; Saarma, M.; et al. GDNF signalling through the RET receptor tyrosine kinase. Nature 1996, 381, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Gómez-Pardo, E.; Dressler, G.; Gruss, P. Pax-2 controls multiple steps of urogenital development. Development 1995, 121, 4057–4065. [Google Scholar] [PubMed]
- Schmidt-Ott, K.; Lan, D.; Hirsh, B.; Barasch, J. Dissecting stages of mesenchymal-to-epithelial conversion during kidney development. Nephron Physiol. 2006, 104, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Moriniere, V.; Knüppel, T.; Charbit, M.; Dusek, J.; Ghiggeri, G.; Jankauskiené, A.; Mir, S.; Montini, G.; Peco-Antic, A.; et al. Prevalence of mutations in renal developmental genes in children with renal hypodisplasia: Results of the ESCAPE Study. J. Am. Soc. Nephrol. 2006, 17, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Nikali, K.; Suomalainen, A.; Terwilliger, J.; Koskinen, T.; Weissenbach, J.; Peltonen, L. Random search for shared chromosomal regions in four affected individuals: The assignment of a new hereditary ataxia locus. Am. J. Hum. Genet. 1995, 56, 1088–1095. [Google Scholar] [PubMed]
- Houwen, R.; Baharloo, S.; Blankenship, K.; Raeymaekers, P.; Juyn, J.; Sandkuijl, L.; Freimer, N. Genome screening by searching for shared segments: Mapping a gene for benign recurrent intrahepatic cholestasis. Nat. Genet. 1994, 8, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Gianfrancesco, F.; Esposito, T.; Ombra, M.; Forabosco, P.; Maninchedda, G.; Fattorini, M.; Casula, S.; Vaccargiu, S.; Casu, G.; Cardia, F.; et al. Identification of a novel gene and a common variant associated with uric acid nephrolithiasis in a Sardinian genetic isolate. Am. J. Hum. Genet. 2003, 72, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Izzi, C.; Sanna-Cherchi, S.; Prati, E.; Belleri, R.; Remedio, A.; Tardanico, R.; Foramitti, M.; Guerini, S.; Viola, B.; Movilli, E.; et al. Familial aggregation of primary glomerulonephritis in an Italian population isolate: Valtrompia study. KIdney Int. 2006, 69, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Morinière, V.; Dahan, K.; Hilbert, P.; Lison, M.; Lebbah, S.; Topa, A.; Bole-Feysot, C.; Pruvost, S.; Nitschke, P.; Plaisier, E.; et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J. Am. Soc. Nephrol. 2014, 25, 2740–2751. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.; de Ligt, J.; Gilissen, C.; Janssen, I.; Steehouwer, M.; de Vries, P.; van Lier, B.; Arts, P.; Wieskamp, N.; del Rosario, M.; et al. A de novo paradigm for mental retardation. Nat. Genet. 2010, 42, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Girard, S.; Gauthier, J.; Noreau, A.; Xiong, L.; Zhou, S.; Jouan, L.; Dionne-Laporte, A.; Spiegelman, D.; Henrion, E.; Diallo, O.; et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 2011, 43, 860–863. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.; Girirajan, S.; Karakoc, E.; Mackenzie, A.; Ng, S.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef] [PubMed]
- The International HapMap Consortium. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar]
- Alkan, C.; Coe, B.; Eichler, E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Khaja, R.; Zhang, J.; MacDonald, J.; He, Y.; Joseph-George, A.; Wei, J.; Rafiq, M.; Qian, C.; Shago, M.; Pantano, L.; et al. Genome assembly comparison identifies structural variants in the human genome. Nat. Genet. 2006, 38, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Redon, R.; Ishikawa, S.; Fitch, K.; Feuk, L.; Perry, G.; Andrews, T.; Fiegler, H.; Shapero, M.; Carson, A.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Rosenfeld, J.; Coe, B.; Parikh, S.; Friedman, N.; Goldstein, A.; Filipink, R.; McConnell, J.; Angle, B.; Meschino, W.; et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012, 367, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.; Coe, B.; Girirajan, S.; Rosenfeld, J.; Vu, T.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; McClellan, J.; McCarthy, S.; Addington, A.; Pierce, S.; Cooper, G.; Nord, A.; Kusenda, M.; Malhotra, D.; Bhandari, A.; et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008, 320, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Rujescu, D.; Cichon, S.; Pietiläinen, O.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buizer-Voskamp, J.; et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008, 455, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Soemedi, R.; Wilson, I.; Bentham, J.; Darlay, R.; Töpf, A.; Zelenika, D.; Cosgrove, C.; Setchfield, K.; Thornborough, C.; Granados-Riveron, J.; et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 2012, 91, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Hitz, M.; Lemieux-Perreault, L.; Marshall, C.; Feroz-Zada, Y.; Davies, R.; Yang, S.; Lionel, A.; D’Amours, G.; Lemyre, E.; Cullum, R.; et al. Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet. 2012, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Berg, J.; Scaglia, F.; Belmont, J.; Bacino, C.; Sahoo, T.; Lalani, S.; Graham, B.; Lee, B.; Shinawi, M.; et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008, 40, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Osoegawa, K.; Vessere, G.; Utami, K.; Mansilla, M.; Johnson, M.; Riley, B.; L’Heureux, J.; Pfundt, R.; Staaf, J.; van der Vliet, W.; et al. Identification of novel candidate genes associated with cleft lip and palate using array comparative genomic hybridisation. J. Med. Genet. 2008, 45, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Greenway, S.; Pereira, A.; Lin, J.; DePalma, S.; Israel, S.; Mesquita, S.; Ergul, E.; Conta, J.; Korn, J.; McCarroll, S.; et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009, 41, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Golzio, C.; Katsanis, N. Genetic architecture of reciprocal CNVs. Curr. Opin. Genet. Dev. 2013, 23, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Uetani, N.; Bouchard, M. Plumbing in the embryo: Developmental defects of the urinary tracts. Clin. Genet. 2009, 75, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, A.; Vainio, S.; Kispert, A.; McMahon, J.; McMahon, A. Wnt11 and RET/GDNF pathways cooperate in regulating ureteric branching during metanephric kidney development. Development 2003, 130, 3175–3185. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F.; Shakya, R. GDNF/RET signaling and the development of the kidney. Bioessays 2006, 28, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Limwongse, C. Syndromes and malformations of the urinary tract. Pediatr. Nephrol. 2004, 121–156. [Google Scholar] [CrossRef]
- Turnpenny, P.; Ellard, S. Alagille syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2012, 20, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Ruf, R.; Xu, P.; Silvius, D.; Otto, E.; Beekmann, F.; Muerb, U.; Kumar, S.; Neuhaus, T.; Kemper, M.; Raymond, R.J.; et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc. Natl. Acad. Sci. USA 2004, 101, 8090–8095. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, B.; Cramer, C.; Silvius, D.; Zou, D.; Raymond, R.; Orten, D.; Kimberling, W.; Smith, R.; Weil, D.; Petit, C.; et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am. J. Hum. Genet. 2007, 80, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Kohlhase, J.; Wischermann, A.; Reichenbach, H.; Froster, U.; Engel, W. Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat. Genet. 1998, 18, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Bower, M.; Salomon, R.; Allanson, J.; Antignac, C.; Benedicenti, F.; Benetti, E.; Binenbaum, G.; Jensen, U.; Cochat, P.; DeCramer, S.; et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 2012, 33, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Thépot, D.; Babinet, C.; Yaniv, M.; Barra, J. Essential role for the homeoprotein vHNF1/HNF1β in visceral endoderm differentiation. Development 1999, 21, 4785–4794. [Google Scholar]
- Horikawa, Y.; Iwasaki, N.; Hara, M.; Furuta, H.; Hinokio, Y.; Cockburn, B.; Lindner, T.; Yamagata, K.; Ogata, M.; Tomonaga, O.; et al. Mutation in hepatocyte nuclear factor-1β gene (TCF2) associated with MODY. Nat. Genet. 1997, 17, 384–385. [Google Scholar] [CrossRef] [PubMed]
- Edghill, E.; Bingham, C.; Ellard, S.; Hattersley, A. Mutations in hepatocyte nuclear factor-1β and their related phenotypes. J. Med. Genet. 2006, 43, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Moreno-De-Luca, D.; Mulle, J.G.; Kaminsky, E.B.; Sanders, S.J.; Myers, S.M.; Adam, M.P.; Pakula, A.T.; Eisenhauer, N.J.; Uhas, K.; Weik, L. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am. J. Hum. Genet. 2010, 87, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Loirat, C.; Bellanné-Chantelot, C.; Husson, I.; Deschênes, G.; Guigonis, V.; Chabane, N. Autism in three patients with cystic or hyperechogenic kidneys and chromosome 17q12 deletion. Nephrol. Dial. Transplant. 2010, 25, 3430–3433. [Google Scholar] [CrossRef] [PubMed]
- Hiesberger, T.; Bai, Y.; Shao, X.; McNally, B.; Sinclair, A.; Tian, X.; Somlo, S.; Igarashi, P. Mutation of hepatocyte nuclear factor-1β inhibits PKHD1 gene expression and produces renal cysts in mice. J. Clin. Investig. 2004, 113, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Sanna-Cherchi, S.; Warady, B.; Furth, S.; Kaskel, F.; Gharavi, A. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr. Nephrol. 2011, 26, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Madariaga, L.; Morinière, V.; Jeanpierre, C.; Bouvier, R.; Loget, P.; Martinovic, J.; Dechelotte, P.; Leporrier, N.; Thauvin-Robinet, C.; Jensen, U.; et al. Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin. J. Am. Soc. Nephrol. 2013, 8, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Ulinski, T.; Lescure, S.; Beaufils, S.; Guigonis, V.; Decramer, S.; Morin, D.; Clauin, S.; Deschênes, G.; Bouissou, F.; Bensman, A.; et al. Renal phenotypes related to hepatocyte nuclear factor-1β (TCF2) mutations in a pediatric cohort. J. Am. Soc. Nephrol. 2006, 17, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Taylor, J.; Winyard, P.; Baker, K.; Sullivan-Brown, J.; Schild, R.; Knüppel, T.; Zurowska, A.; Caldas-Alfonso, A.; Litwin, M.; et al. SIX2 and BMP4 mutations associate with anomalous kidney development. J. Am. Soc. Nephrol. 2008, 19, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaeifar, M.; Schlingmann, K.; Litwin, M.; Emre, S.; Bakkaloglu, A.; Mehls, O.; Antignac, C.; Schaefer, F.; Weber, S.; ESCAPE Trial Group. Functional analysis of BMP4 mutations identified in pediatric CAKUT patients. Pediatr. Nephrol. 2009, 24, 2361–2368. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Carlomagno, F.; Romano, A.; Bottaro, D.; Dathan, N.; Grieco, M.; Fusco, A.; Vecchio, G.; Matoskova, B.; Kraus, M. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science 1995, 267, 381–383. [Google Scholar] [CrossRef] [PubMed]
- Romeo, G.; Ronchetto, P.; Luo, Y.; Barone, V.; Seri, M.; Ceccherini, I.; Pasini, B.; Bocciardi, R.; Lerone, M.; Kääriäinen, H. Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung’s disease. Nature 1994, 367, 377–378. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.; Safford, S.; Reeves, J.; Jackson, M.; Freemerman, A. Renal aplasia in humans is associated with RET mutations. Am. J. Hum. Genet. 2008, 82, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Jeanpierre, C.; Macé, G.; Parisot, M.; Morinière, V.; Pawtowsky, A.; Benabou, M.; Martinovic, J.; Amiel, J.; Attié-Bitach, T.; Delezoide, A.; et al. RET and GDNF mutations are rare in fetuses with renal agenesis or other severe kidney development defect. J. Med. Genet. 2011, 48, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Prato, A.P.; Musso, M.; Ceccherini, I.; Mattioli, G.; Giunta, C.; Ghiggeri, G.; Jasonni, V. Hirschsprung disease and congenital anomalies of the kidney and urinary tract (CAKUT): A novel syndromic association. Medicine 2009, 88, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sanna-Cherchi, S.; Sampogna, R.; Papeta, N.; Burgess, K.; Nees, S.; Perry, B.; Choi, M.; Bodria, M.; Liu, Y.; Weng, P.; et al. Mutations in DSTYK and dominant urinary tract malformations. N. Engl. J. Med. 2013, 369, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Ma, W.; O’Brien, L.; Chung, E.; Guo, J.; Cheng, J.; Valerius, M.; McMahon, J.; Wong, W.; McMahon, A. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev. Cell 2012, 23, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Vivante, A.; Mark-Danieli, M.; Davidovits, M.; Harari-Steinberg, O.; Omer, D.; Gnatek, Y.; Cleper, R.; Landau, D.; Kovalski, Y.; Weissman, I.; et al. Renal hypodysplasia associates with a WNT4 variant that causes aberrant canonical WNT signaling. J. Am. Soc. Nephrol. 2013, 24, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Gribouval, O.; Gonzales, M.; Neuhaus, T.; Aziza, J.; Bieth, E.; Laurent, N.; Bouton, J.; Feuillet, F.; Makni, S.; Amar, H.B.; et al. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive tubular dysgenesis. Nat. Genet. 2005, 37, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Yerkes, E.; Hohenfellner, K.; Miyazaki, Y.; Ma, J.; Hunley, T.; Yoshida, H.; Ichiki, T.; Threadgill, D.; Phillips, J.; et al. Role of the angiotensin type 2 receptor gene in congenital anomalies of the kidney and urinary tract, CAKUT, of mice and men. Mol. Cell 1999, 3, 1–10. [Google Scholar] [CrossRef]
- Okubo, S.; Niimura, F.; Matsusaka, T.; Fogo, A.; Hogan, B.; Ichikawa, I. Angiotensinogen gene null-mutant mice lack homeostatic regulation of glomerular filtration and tubular reabsorption. Kidney Int. 1998, 53, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Tamm, I.; Horsfall, F.J. Characterization and separation of an inhibitor of viral hemagglutination present in urine. Proc. Soc. Exp. Biol. Med. 1959, 74, 106–108. [Google Scholar] [CrossRef]
- Hart, T.; Gorry, M.; Hart, P.; Woodard, A.; Shihabi, Z.; Sandhu, J.; Shirts, B.; Xu, L.; Zhu, H.; Barmada, M.; et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J. Med. Genet. 2002, 39, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Zlabinger, G.; Säemann, M. The multiple functions of Tamm-Horsfall protein in human health and disease: A mystery clears up. Wien. Klin. Wochenschr. 2005, 117, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Hoskins, B.; Beck, B.; Hoppe, B.; Tasic, V.; Otto, E.; Hildebrandt, F. Mutation analysis of the Uromodulin gene in 96 individuals with urinary tract anomalies (CAKUT). Pediatr. Nephrol. 2009, 24, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Dworschak, G.; Kohl, S.; Saisawat, P.; Vivante, A.; Hilger, A.; Reutter, H.; Soliman, N.; Bogdanovic, R.; Kehinde, E.; et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 2014, 85, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Heidet, L.; Decramer, S.; Pawtowski, A.; Morinière, V.; Bandin, F.; Knebelmann, B.; Lebre, A.; Faguer, S.; Guigonis, V.; Antignac, C.; et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin. J. Am. Soc. Nephrol. 2010, 5, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, N.; Pulit, S.; Nijman, I.; Monroe, G.; Feitz, W.; Schreuder, M.; van Eerde, A.; de Jong, T.; Giltay, J.; van der Zwaag, B.; et al. Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney Int. 2016, 89, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Saisawat, P.; Tasic, V.; Vega-Warner, V.; Kehinde, E.; Günther, B.; Airik, R.; Innis, J.; Hoskins, B.; Hoefele, J.; Otto, E.; et al. Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney Int. 2012, 81, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, R.; Ramos, E.; Hoffman, M.; VanWinkle, J.; Martin, D.; Davis, T.; Hoshi, M.; Hmiel, S.; Beck, A.; Hruska, K.; et al. Traditional and targeted exome sequencing reveals common, rare and novel functional deleterious variants in RET-signaling complex in a cohort of living US patients with urinary tract malformations. Hum. Genet. 2012, 131, 1725–1738. [Google Scholar] [CrossRef] [PubMed]
- Yosypiv, I. Congenital anomalies of the kidney and urinary tract: A genetic disorder? Int. J. Nephrol. 2012, 2012, 909083. [Google Scholar] [CrossRef] [PubMed]
- Chen, F. Genetic and developmental basis for urinary tract obstruction. Pediatr. Nephrol. 2009, 24, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Barak, H.; Huh, S.; Chen, S.; Jeanpierre, C.; Martinovic, J.; Parisot, M.; Bole-Feysot, C.; Nitschké, P.; Salomon, R.; Antignac, C.; et al. FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev. Cell 2012, 22, 1191–1207. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, Q.; Xu, J.; Ding, K.; Wang, Y.; Wang, W.; Li, S.; Shen, Y. Mutation screening of BMP4, BMP7, HOXA4 and HOXB6 genes in Chinese patients with hypospadias. Eur. J. Hum. Genet. 2007, 15, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Groenen, P.M.; Vanderlinden, G.; Devriendt, K.; Fryns, J.P.; van de Ven, W.J. Rearrangement of the human CDC5L gene by a t(6;19)(p21;q13. 1) in a patient with multicystic renal dysplasia. Genomics 1998, 49, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Brockschmidt, A.; Chung, B.; Weber, S.; Fischer, D.; Kolatsi-Joannou, M.; Christ, L.; Heimbach, A.; Shtiza, D.; Klaus, G.; Simonetti, G.; et al. CHD1L: A new candidate gene for congenital anomalies of the kidneys and urinary tract (CAKUT). Nephrol. Dial. Transplant. 2012, 27, 2355–2364. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; van Eerde, A.; Fan, X.; Quintero-Rivera, F.; Kulkarni, S.; Ferguson, H.; Kim, H.; Fan, Y.; Xi, Q.; Li, Q.; et al. Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am. J. Hum. Genet. 2007, 80, 616–632. [Google Scholar] [CrossRef] [PubMed]
- Ruf, R.; Berkman, J.; Wolf, M.; Nurnberg, P.; Gattas, M.; Ruf, E.; Hyland, V.; Kromberg, J.; Glass, I.; Macmillan, J.; et al. A gene locus for branchio-otic syndrome maps to chromosome 14q21.3-q24.3. J. Med. Genet. 2003, 40, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Gimelli, S.; Caridi, G.; Beri, S.; McCracken, K.; Bocciardi, R.; Zordan, P.; Dagnino, M.; Fiorio, P.; Murer, L.; Benetti, E.; et al. Mutations in SOX17 are associated with congenital anomalies of the kidney and the urinary tract. Hum. Mutat. 2010, 31, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.; Bitner-Glindzicz, M.; Malcolm, S.; Hu, C.; Allison, J.; Winyard, P.; Gullett, A.; Thomas, D.; Belk, R.; Feather, S.; et al. De novo Uroplakin IIIa heterozygous mutations cause human renal adysplasia leading to severe kidney failure. J. Am. Soc. Nephrol. 2005, 16, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Groenen, P.; Nesbit, M.; Schuffenhauer, S.; Lichtner, P.; Vanderlinden, G.; Harding, B.; Beetz, R.; Bilous, R.; Holdaway, I.; et al. GATA3 haplo-insufficiency causes human HDR syndrome. Nature 2000, 406, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Gbadegesin, R.; Brophy, P.; Adeyemo, A.; Hall, G.; Gupta, I.; Hains, D.; Bartkowiak, B.; Rabinovich, C.; Chandrasekharappa, S.; Homstad, A.; et al. TNXB mutations can cause vesicoureteral reflux. J. Am. Soc. Nephrol. 2013, 24, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Hwang, D.; Dworschak, G.; Hilger, A.; Saisawat, P.; Vivante, A.; Stajic, N.; Bogdanovic, R.; Reutter, H.; Kehinde, E.; et al. Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. J. Am. Soc. Nephrol. 2014, 25, 1917–1922. [Google Scholar] [CrossRef] [PubMed]
- Saisawat, P.; Kohl, S.; Hilger, A.; Hwang, D.; Gee, H.Y.; Dworschak, G.; Tasic, V.; Pennimpede, T.; Natarajan, S.; Sperry, E.; et al. Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int. 2014, 85, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Landwehr, C.; Renkert, M.; Hoischen, A.; Wühl, E.; Denecke, J.; Radlwimmer, B.; Haffner, D.; Schaefer, F.; Weber, R. Mapping candidate regions and genes for congenital anomalies of the kidneys and urinary tract (CAKUT) by array-based comparative genomic hybridization. Nephrol. Dial. Transplant. 2011, 26, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Sanna-Cherchi, S.; Kiryluk, K.; Burgess, K.; Bodria, M.; Sampson, M.; Hadley, D.; Nees, S.; Verbitsky, M.; Perry, B.; Sterken, R.; et al. Copy-number disorders are a common cause of congenital kidney malformations. Am. J. Hum. Genet. 2012, 91, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Caruana, G.; Wong, M.; Walker, A.; Heloury, Y.; Webb, N.; Johnstone, L.; James, P.; Burgess, T.; Bertram, J. Copy-number variation associated with congenital anomalies of the kidney and urinary tract. Pediatr. Nephrol. 2015, 30, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Materna-Kiryluk, A.; Kiryluk, K.; Burgess, K.; Bieleninik, A.; Sanna-Cherchi, S.; Gharavi, A.; Latos-Bielenska, A. The emerging role of genomics in the diagnosis and workup of congenital urinary tract defects: A novel deletion syndrome on chromosome 3q13.31-22.1. Pediatr. Nephrol. 2014, 29, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.; Clauin, S.; Sharp, A.; Moller, R.; Ullmann, R.; Kapur, R.; Pinkel, D.; Cooper, G.; Ventura, M.; Ropers, H.; et al. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am. J. Hum. Genet. 2007, 81, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Burtey, S. 22q11.2 microdeletion syndrome is a common cause of renal tract malformations. Nat. Clin. Pract. Nephrol. 2008, 4. [Google Scholar] [CrossRef] [PubMed]
- Shprintzen, R. Velocardiofacial syndrome and DiGeorge sequence. J. Med. Genet. 1994, 31, 423–424. [Google Scholar] [CrossRef] [PubMed]
- Westland, R.; Verbitsky, M.; Vukojevic, K.; Perry, B.; Fasel, D.; Zwijnenburg, P.; Bökenkamp, A.; Gille, J.; Saraga-Babic, M.; Ghiggeri, G.; et al. Copy number variation analysis identifies novel CAKUT candidate genes in children with a solitary functioning kidney. Kidney Int. 2015, 88, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, I.; Zivkovic, M.; Kostic, M.; Krstic, Z.; Djuric, T.; Kolic, I.; Alavantic, D.; Stankovic, A. Transcriptome-wide based identification of miRs in congenital anomalies of the kidney and urinary tract (CAKUT) in children: The significant upregulation of tissue miR-144 expression. J. Transl. Med. 2016, 3, 193. [Google Scholar] [CrossRef] [PubMed]
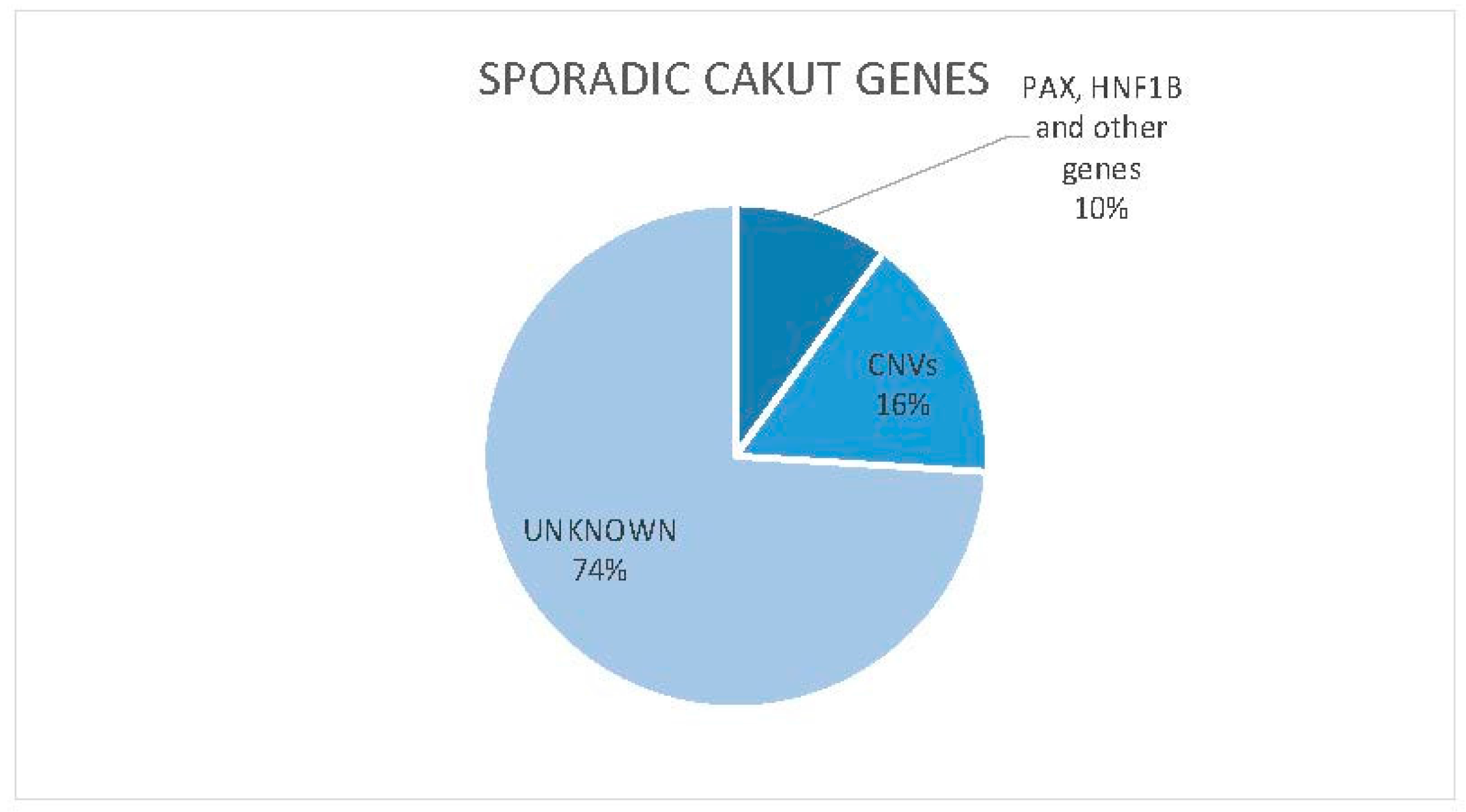

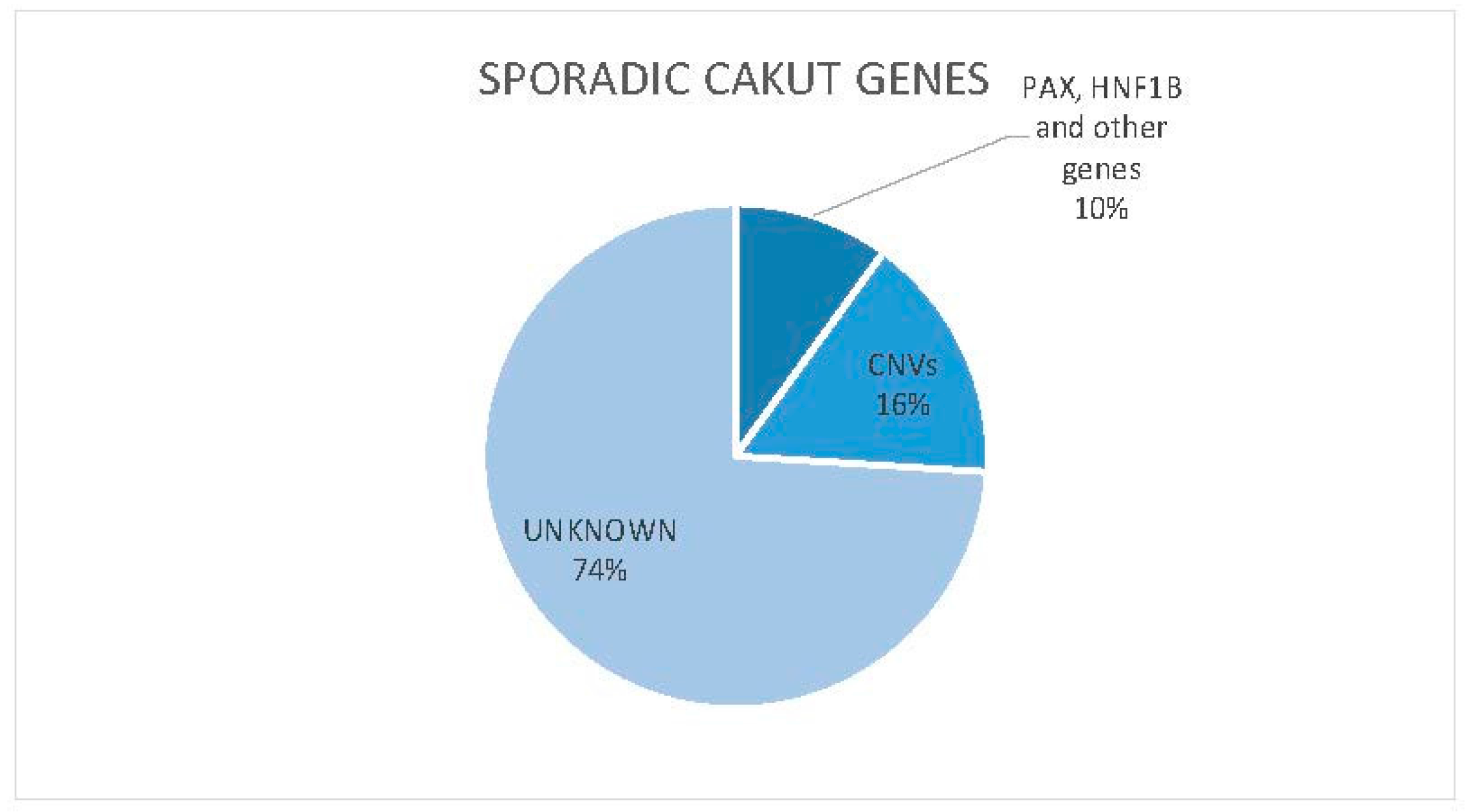{kind=link}
| Gene | Disease |
|---|---|
| ACE | Renal tubular dysgenesis |
| AGT | Renal tubular dysgenesis |
| AGTR1 | Renal tubular dysgenesis |
| BMP4 | CAKUT |
| DSTYK | CAKUT |
| EYA1 | Branchio-Oto-Renal Syndrome and renal hypoplasia |
| FRAS1 | Fraser Syndrome |
| FREM1 | Bifid nose, renal agenesis, anorectal malformations |
| FREM1 | Fraser Syndrome |
| GRIP1 | Fraser Syndrome |
| HNF1β | Multicystic dysplastic kidney, renal hypoplasia, renal cysts and diabetes Syndrome |
| NOTCH2 | Alagille syndrome, renal anomalies |
| PAX2 | Renal coloboma Syndrome and CAKUT |
| REN | Renal tubular dysgenesis |
| RET | Renal agenesis and Hirschsprung disease |
| SALL1 | Townes–Brocks Syndrome |
| SIX1 | Branchio-Oto-Renal Syndrome |
| SIX2 | Renal hypodysplasia |
| SIX5 | Branchio-Oto-Renal Syndrome |
| SOX17 | CAKUT |
| UPK3A | Renal dysplasia |
| WNT4 | Mullerian aplasia and hyperandrogenism |
| UMOD | Familial juvenile hyperuricemic nephropathy (FJHN), glomerulocystic kidney disease (GCKD), Autosomal dominant medullary cystic kidney disease 2 (MCKD2) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capone, V.P.; Morello, W.; Taroni, F.; Montini, G. Genetics of Congenital Anomalies of the Kidney and Urinary Tract: The Current State of Play. Int. J. Mol. Sci. 2017, 18, 796. https://doi.org/10.3390/ijms18040796
Capone VP, Morello W, Taroni F, Montini G. Genetics of Congenital Anomalies of the Kidney and Urinary Tract: The Current State of Play. International Journal of Molecular Sciences. 2017; 18(4):796. https://doi.org/10.3390/ijms18040796
Chicago/Turabian StyleCapone, Valentina P., William Morello, Francesca Taroni, and Giovanni Montini. 2017. "Genetics of Congenital Anomalies of the Kidney and Urinary Tract: The Current State of Play" International Journal of Molecular Sciences 18, no. 4: 796. https://doi.org/10.3390/ijms18040796
APA StyleCapone, V. P., Morello, W., Taroni, F., & Montini, G. (2017). Genetics of Congenital Anomalies of the Kidney and Urinary Tract: The Current State of Play. International Journal of Molecular Sciences, 18(4), 796. https://doi.org/10.3390/ijms18040796