
Sphingosine-1-Phosphate Metabolism and Its Role in the Development of Inflammatory Bowel Disease



Abstract
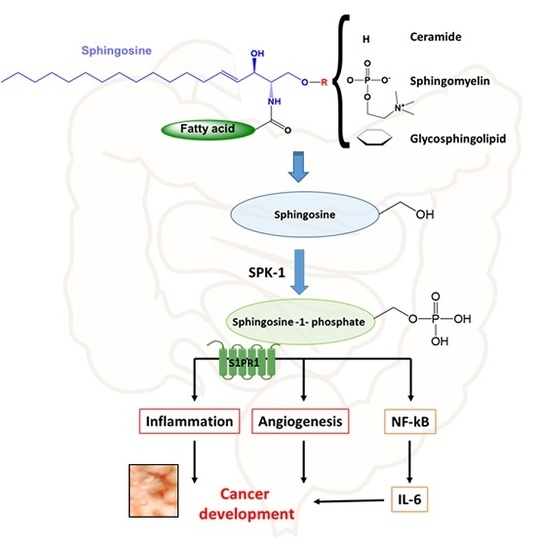
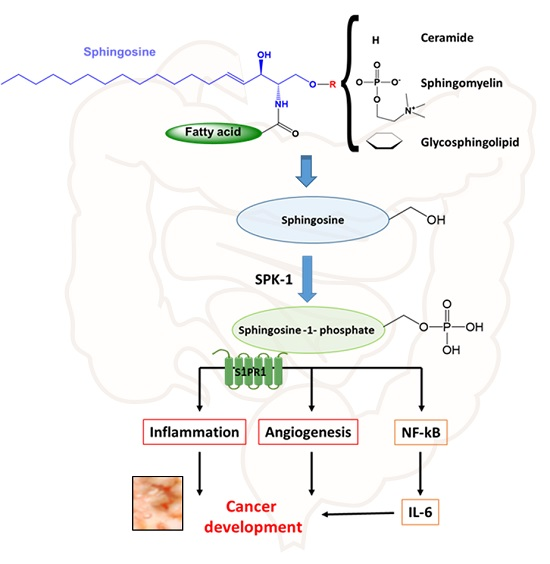{kind=link}
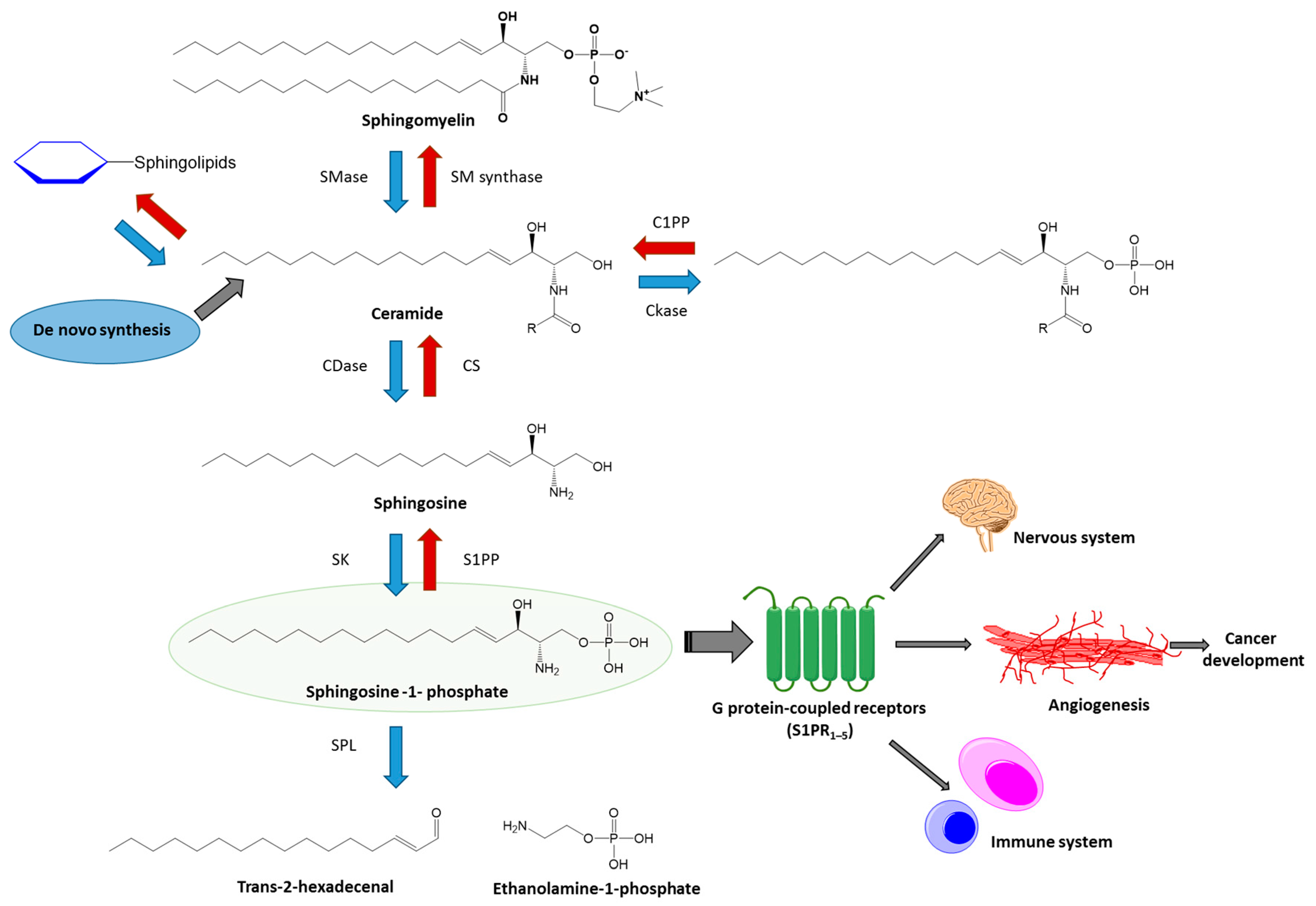{kind=link}
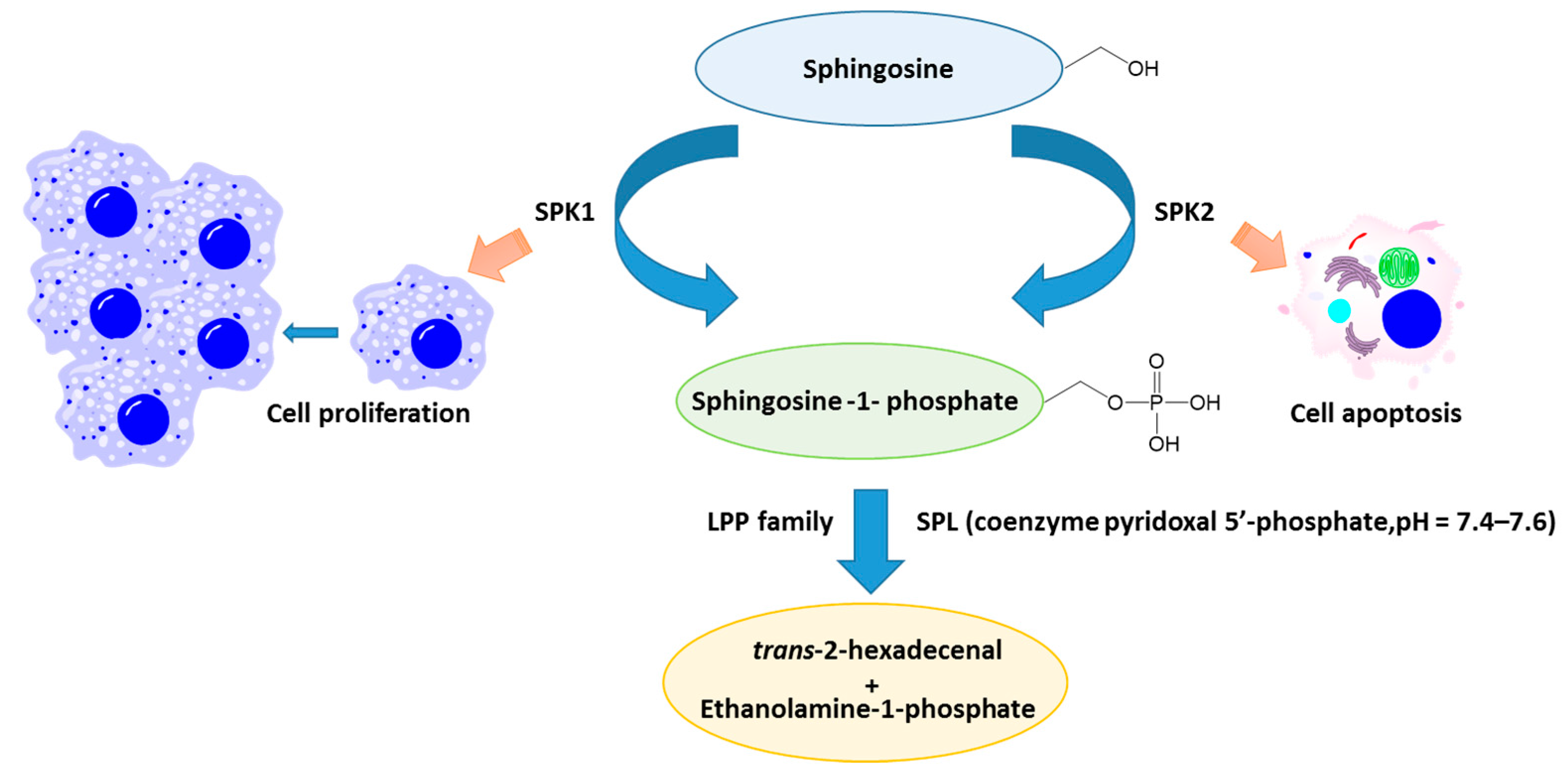{kind=link}
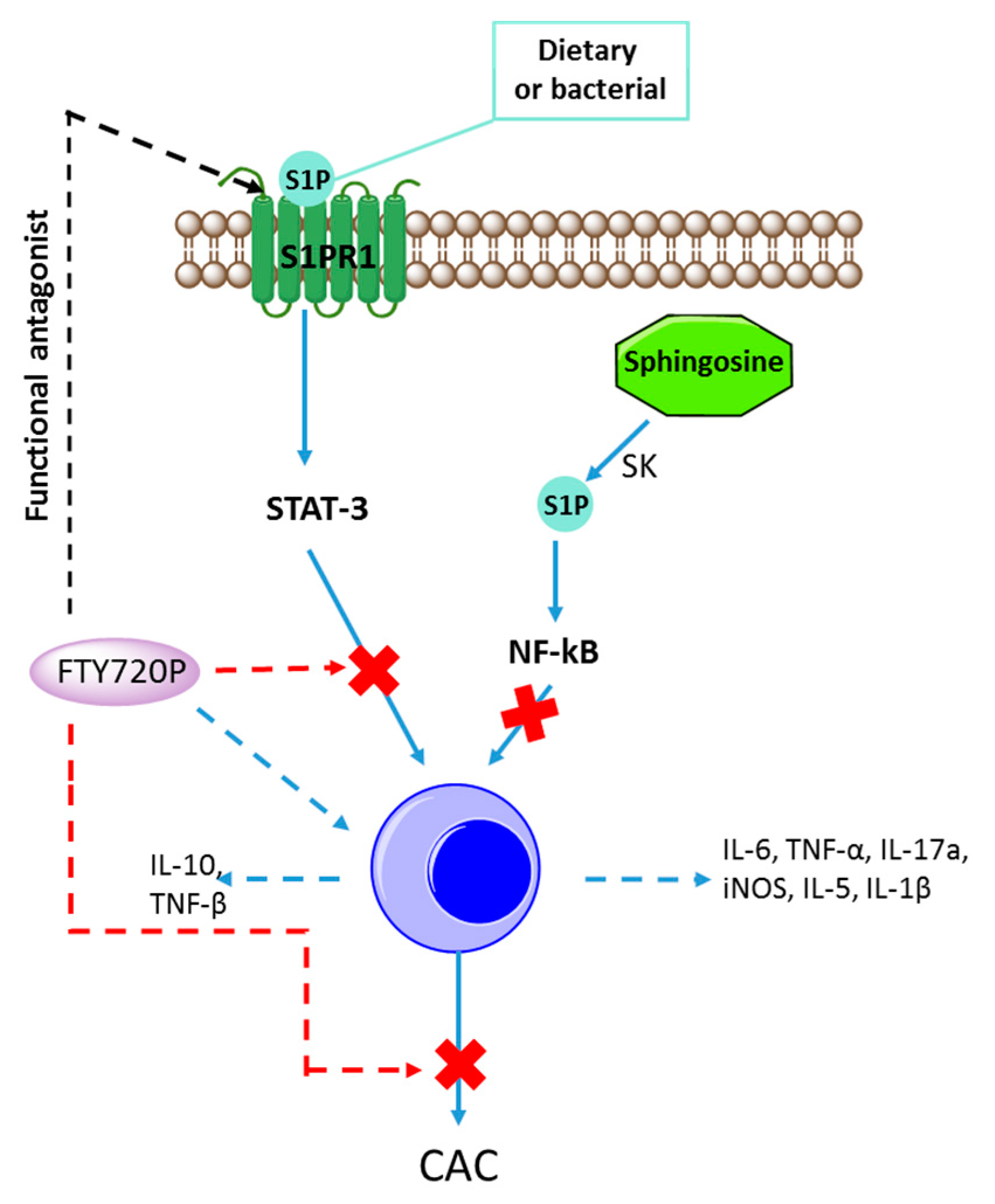{kind=link}
1. Introduction
2. Sphingolipids Metabolism
3. S1P
4. Autoimmune Disease and S1P
5. Inflammation in IBD and S1P
5.1. Role of SPK/S1P in Inflammation
5.2. In Vitro Experiments
5.3. In Vivo Experiments
6. Cancer Associated with IBD
7. Sphingolipids in Diet
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yamada, A.; Arakaki, R.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Role of regulatory T cell in the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 2195–2205. [Google Scholar] [PubMed]
- Degagne, E.; Saba, J.D. S1pping fire: Sphingosine-1-phosphate signaling as an emerging target in inflammatory bowel disease and colitis-associated cancer. Clin. Exp. Gastroenterol. 2014, 7, 205–214. [Google Scholar] [PubMed]
- Ilan, Y. Oral immune therapy: Targeting the systemic immune system via the gut immune system for the treatment of inflammatory bowel disease. Clin. Transl. Immunol. 2016, 5, e60. [Google Scholar] [CrossRef] [PubMed]
- Tai, E.K.; Wu, W.K.; Wong, H.P.; Lam, E.K.; Yu, L.; Cho, C.H. A new role for cathelicidin in ulcerative colitis in mice. Exp. Biol. Med. 2007, 232, 799–808. [Google Scholar]
- Bamias, G.; Clark, D.J.; Rivera-Nieves, J. Leukocyte traffic blockade as a therapeutic strategy in inflammatory bowel disease. Curr. Drug Targets 2013, 14, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.M.; Han, J.; Ginsberg, M.H. α4 integrins and the immune response. Immunol. Rev. 2002, 186, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Pribila, J.T.; Quale, A.C.; Mueller, K.L.; Shimizu, Y. Integrins and T cell-mediated immunity. Annu. Rev Immunol. 2004, 22, 157–180. [Google Scholar] [CrossRef] [PubMed]
- Divecha, N.; Irvine, R.F. Phospholipid signaling. Cell 1995, 80, 269–278. [Google Scholar] [CrossRef]
- Blumberg, R.S. Inflammation in the intestinal tract: Pathogenesis and treatment. Digit. Distrib. 2009, 27, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Medzhitov, R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 2011, 140, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Blumberg, R.S. Adaptive immunity in inflammatory bowel disease: State of the art. Curr. Opin. Gastroenterol. 2008, 24, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Iskandar, H.N.; Ciorba, M.A. Biomarkers in inflammatory bowel disease: Current practices and recent advances. Transl. Res. 2012, 159, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.D. Physiological functions and clinical implications of sphingolipids in the gut. J. Dig. Dis. 2011, 12, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Nagahashi, M.; Terracina, K.P.; Takabe, K. Emerging Role of Sphingosine-1-phosphate in Inflammation, Cancer, and Lymphangiogenesis. Biomolecules 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Thudichum, J.L.W. A Treatise on the Chemical Constitution of the Brain: Based throughout upon Original Researches; Baillière, Tindall, and Cox: London, UK, 1884. [Google Scholar]
- Hannun, Y.A.; Linardic, C.M. Sphingolipid breakdown products: Anti-proliferative and tumor-suppressor lipids. Biochim. Biophys. Acta 1993, 1154, 223–236. [Google Scholar] [CrossRef]
- Sabbadini, R.A. Targeting sphingosine-1-phosphate for cancer therapy. Br. J. Cancer 2006, 95, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.; Wadham, C.; Gamble, J.; Xia, P. Sphingosine-1-phosphate receptor 1 transmits estrogens’ effects in endothelial cells. Steroids 2015, 104, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wada, R.; Yamashita, T.; Mi, Y.; Deng, C.X.; Hobson, J.P.; Rosenfeldt, H.M.; Nava, V.E.; Chae, S.S.; Lee, M.J.; et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Investig. 2000, 106, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Chen, Z.; Xu, Y.; Zhao, Y.; Zha, R.; Huang, S.; Liu, L.; Chen, T.; Li, J.; Tu, H.; He, X. Sphingosine kinase 1 promotes tumour cell migration and invasion via the S1P/EDG1 axis in hepatocellular carcinoma. Liver Int. 2012, 32, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.; Proia, R.L.; Olivera, A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat. Rev. Immunol. 2008, 8, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Burns, T.A.; Luberto, C. Sphingolipid metabolism and leukemia: A potential for novel therapeutic approaches. Anticancer Agents Med. Chem. 2011, 11, 863–881. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim. Biophys. Acta 2002, 1585, 193–201. [Google Scholar] [CrossRef]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Obeid, L.M. Ceramidases: Regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta 2008, 1781, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Takabe, K.; Kapitonov, D.; Milstien, S.; Spiegel, S. Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Luberto, C.; Argraves, K.M. Enzymes of sphingolipid metabolism: From modular to integrative signaling. Biochemistry 2001, 40, 4893–4903. [Google Scholar] [CrossRef] [PubMed]
- Fyrst, H.; Saba, J.D. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat. Chem. Biol. 2010, 6, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.; Wang, T.C. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell 2009, 15, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Lundgreen, A.; Kadlubar, S.A.; Bondurant, K.L.; Wolff, R.K. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol. Carcinog. 2013, 52, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Corvinus, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufmann, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Lai, R.; Chirieac, L.R.; Li, C.; Thomazy, V.A.; Grammatikakis, I.; Rassidakis, G.Z.; Zhang, W.; Fujio, Y.; Kunisada, K.; et al. Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: Inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am. J. Pathol. 2005, 167, 969–980. [Google Scholar] [CrossRef]
- Santandreu, F.M.; Valle, A.; Oliver, J.; Roca, P. Resveratrol potentiates the cytotoxic oxidative stress induced by chemotherapy in human colon cancer cells. Cell. Physiol. Biochem. 2011, 28, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jin, H.; Xu, R.; Mei, Q.; Fan, D. Triptolide downregulates Rac1 and the JAK/STAT3 pathway and inhibits colitis-related colon cancer progression. Exp. Mol. Med. 2009, 41, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; de Haar, C.; Chen, M.; Deuring, J.; Gerrits, M.M.; Smits, R.; Xia, B.; Kuipers, E.J.; van der Woude, C.J. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut 2010, 59, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Oskouian, B.; Saba, J. Sphingosine-1-phosphate metabolism and intestinal tumorigenesis: Lipid signaling strikes again. Cell Cycle 2007, 6, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Furuya, H.; Shimizu, Y.; Kawamori, T. Sphingolipids in cancer. Cancer Metastasis Rev. 2011, 30, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Naser, S.A.; Arce, M.; Khaja, A.; Fernandez, M.; Naser, N.; Elwasila, S.; Thanigachalam, S. Role of ATG16L, NOD2 and IL23R in Crohn’s disease pathogenesis. World J. Gastroenterol. 2012, 18, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Kohama, T.; Olivera, A.; Edsall, L.; Nagiec, M.M.; Dickson, R.; Spiegel, S. Molecular cloning and functional characterization of murine sphingosine kinase. J. Biol. Chem. 1998, 273, 23722–23728. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sugiura, M.; Nava, V.E.; Edsall, L.C.; Kono, K.; Poulton, S.; Milstien, S.; Kohama, T.; Spiegel, S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 2000, 275, 19513–19520. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Hartung, H.P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Toman, R.E.; Goparaju, S.K.; Maceyka, M.; Nava, V.E.; Sankala, H.; Payne, S.G.; Bektas, M.; Ishii, I.; Chun, J.; et al. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J. Biol. Chem. 2003, 278, 40330–40336. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Maceyka, M.; Hait, N.C.; Paugh, S.W.; Sankala, H.; Milstien, S.; Spiegel, S. Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 2005, 579, 5313–5317. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, A.; Saba, J.D. Truth and consequences of sphingosine-1-phosphate lyase. Adv. Biol. Regul. 2012, 52, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Murata, N.; Sato, K.; Kon, J.; Tomura, H.; Okajima, F. Quantitative measurement of sphingosine 1-phosphate by radioreceptor-binding assay. Anal. Biochem. 2000, 282, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Al Fadel, F.; Fayyaz, S.; Japtok, L.; Kleuser, B. Involvement of Sphingosine 1-Phosphate in Palmitate-Induced Non-Alcoholic Fatty Liver Disease. Cell. Physiol. Biochem. 2016, 40, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.D.; Nara, F.; Bielawska, A.; Garrett, S.; Hannun, Y.A. The BST1 gene of Saccharomyces cerevisiae is the sphingosine-1-phosphate lyase. J. Biol. Chem. 1997, 272, 26087–26090. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Schwab, S.R.; Pereira, J.P.; Matloubian, M.; Xu, Y.; Huang, Y.; Cyster, J.G. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005, 309, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Donoviel, M.S.; Read, R.; Hansen, G.M.; Hazlewood, J.; Anderson, S.J.; Sun, W.; Swaffield, J.; Oravecz, T. Incomplete inhibition of sphingosine 1-phosphate lyase modulates immune system function yet prevents early lethality and non-lymphoid lesions. PLoS ONE 2009, 4, e4112. [Google Scholar] [CrossRef] [PubMed]
- Breart, B.; Ramos-Perez, W.D.; Mendoza, A.; Salous, A.K.; Gobert, M.; Huang, Y.; Adams, R.H.; Lafaille, J.J.; Escalante-Alcalde, D.; Morris, A.J.; et al. Lipid phosphate phosphatase 3 enables efficient thymic egress. J. Exp. Med. 2011, 208, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Chun, J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann. Neurol. 2011, 69, 759–777. [Google Scholar] [CrossRef] [PubMed]
- Kulakowska, A.; Zendzian-Piotrowska, M.; Baranowski, M.; Kononczuk, T.; Drozdowski, W.; Gorski, J.; Bucki, R. Intrathecal increase of sphingosine 1-phosphate at early stage multiple sclerosis. Neurosci. Lett. 2010, 477, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Groves, A.; Kihara, Y.; Chun, J. Fingolimod: Direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. Neurol. Sci. 2013, 328, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Bucki, R.; Kulakowska, A.; Byfield, F.J.; Zendzian-Piotrowska, M.; Baranowski, M.; Marzec, M.; Winer, J.P.; Ciccarelli, N.J.; Gorski, J.; Drozdowski, W.; et al. Plasma gelsolin modulates cellular response to sphingosine 1-phosphate. Am. J. Physiol. Cell Physiol. 2010, 299, C1516–C1523. [Google Scholar] [CrossRef] [PubMed]
- Kulakowska, A.; Drozdowski, W.; Sadzynski, A.; Bucki, R.; Janmey, P.A. Gelsolin concentration in cerebrospinal fluid from patients with multiple sclerosis and other neurological disorders. Eur. J. Neurol. 2008, 15, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Gamble, J.R.; Wang, L.; Pitson, S.M.; Moretti, P.A.; Wattenberg, B.W.; D’Andrea, R.J.; Vadas, M.A. An oncogenic role of sphingosine kinase. Curr. Biol. 2000, 10, 1527–1530. [Google Scholar] [CrossRef]
- Xia, P.; Wang, L.; Moretti, P.A.; Albanese, N.; Chai, F.; Pitson, S.M.; D’Andrea, R.J.; Gamble, J.R.; Vadas, M.A. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J. Biol. Chem. 2002, 277, 7996–8003. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Wu, L.; Acharya, S.; Arac, A.; Blaho, V.A.; Huang, Y.; Moon, B.S.; Axtell, R.C.; Ho, P.P.; Steinberg, G.K.; et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat. Immunol. 2013, 14, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Okamoto, Y.; Inoki, I.; Yoshioka, K.; Du, W.; Qi, X.; Takuwa, N.; Gonda, K.; Yamamoto, Y.; Ohkawa, R.; et al. Sphingosine-1-phosphate receptor-2 deficiency leads to inhibition of macrophage proinflammatory activities and atherosclerosis in apoE-deficient mice. J. Clin. Investig. 2010, 120, 3979–3995. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Sato, K.; Kuwabara, A.; Tomura, H.; Ishiwara, M.; Kobayashi, I.; Ui, M.; Okajima, F. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J. Biol. Chem. 2001, 276, 31780–31785. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Matsuyuki, H.; Maeda, Y.; Sugahara, K. Role of sphingosine 1-phosphate receptor type 1 in lymphocyte egress from secondary lymphoid tissues and thymus. Cell. Mol. Immunol. 2006, 3, 11–19. [Google Scholar] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Festen, E.A.; Szperl, A.M.; Weersma, R.K.; Wijmenga, C.; Wapenaar, M.C. Inflammatory bowel disease and celiac disease: Overlaps in the pathology and genetics, and their potential drug targets. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Erdman, S.E.; Poutahidis, T. Roles for inflammation and regulatory T cells in colon cancer. Toxicol. Pathol. 2010, 38, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Maeda, S.; Nakagawa, H.; Hikiba, Y.; Shibata, W.; Sakamoto, K.; Yanai, A.; Hirata, Y.; Ogura, K.; Muto, S.; et al. Effectiveness of IkappaB kinase inhibitors in murine colitis-associated tumorigenesis. J. Gastroenterol. 2009, 44, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed]
- McGovern, D.P.; Gardet, A.; Torkvist, L.; Goyette, P.; Essers, J.; Taylor, K.D.; Neale, B.M.; Ong, R.T.; Lagace, C.; Li, C.; et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010, 42, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Skieceviciene, J.; Kiudelis, G.; Ellinghaus, E.; Balschun, T.; Jonaitis, L.V.; Zvirbliene, A.; Denapiene, G.; Leja, M.; Pranculiene, G.; Kalibatas, V.; et al. Replication study of ulcerative colitis risk loci in a Lithuanian-Latvian case-control sample. Inflamm. Bowel Dis. 2013, 19, 2349–2355. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.H.; Braegger, C.P.; Walker-Smith, J.A.; MacDonald, T.T. Location of tumour necrosis factor alpha by immunohistochemistry in chronic inflammatory bowel disease. Gut 1993, 34, 1705–1709. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.D.; Ayass, M.; Chensue, S. Tumor necrosis factor and IL-1 β expression in pediatric patients with inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 1993, 16, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Billich, A.; Bornancin, F.; Mechtcheriakova, D.; Natt, F.; Huesken, D.; Baumruker, T. Basal and induced sphingosine kinase 1 activity in A549 carcinoma cells: Function in cell survival and IL-1β and TNF-α induced production of inflammatory mediators. Cell Signal. 2005, 17, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Bielawski, J.; Porcelli, A.M.; Reames, D.L.; Johnson, K.R.; Morrow, J.; Chalfant, C.E.; Obeid, L.M.; Hannun, Y.A. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-α. FASEB J. 2003, 17, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Zhi, L.; Leung, B.P.; Melendez, A.J. Sphingosine kinase 1 regulates pro-inflammatory responses triggered by TNFα in primary human monocytes. J. Cell. Physiol. 2006, 208, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Snider, A.J.; Kawamori, T.; Bradshaw, S.G.; Orr, K.A.; Gilkeson, G.S.; Hannun, Y.A.; Obeid, L.M. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 2009, 23, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Feagan, B.G.; Wolf, D.C.; D’Haens, G.; Vermeire, S.; Hanauer, S.B.; Ghosh, S.; Smith, H.; Cravets, M.; Frohna, P.A.; et al. Ozanimod Induction and Maintenance Treatment for Ulcerative Colitis. N. Engl. J. Med. 2016, 374, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-mediated STAT3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Keku, T.O.; Martin, C.; Galanko, J.; Woosley, J.T.; Schroeder, J.C.; Satia, J.A.; Halabi, S.; Sandler, R.S. Circulating levels of inflammatory cytokines and risk of colorectal adenomas. Cancer Res. 2008, 68, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Gamble, J.R.; Rye, K.A.; Wang, L.; Hii, C.S.; Cockerill, P.; Khew-Goodall, Y.; Bert, A.G.; Barter, P.J.; Vadas, M.A. Tumor necrosis factor-α induces adhesion molecule expression through the sphingosine kinase pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 14196–14201. [Google Scholar] [CrossRef] [PubMed]
- Pitson, S.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Xia, P.; Vadas, M.A.; Wattenberg, B.W. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003, 22, 5491–5500. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Kawamori, T.; Osta, W.; Johnson, K.R.; Pettus, B.J.; Bielawski, J.; Tanaka, T.; Wargovich, M.J.; Reddy, B.S.; Hannun, Y.A.; Obeid, L.M.; et al. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J. 2006, 20, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deng, J.; Wang, L.; Lee, H.; Armstrong, B.; Scuto, A.; Kowolik, C.; Weiss, L.M.; Forman, S.; Yu, H. S1PR1 is an effective target to block STAT3 signaling in activated B cell-like diffuse large B-cell lymphoma. Blood 2012, 120, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-κB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef] [PubMed]
- Kulakowska, A.; Byfield, F.J.; Zendzian-Piotrowska, M.; Zajkowska, J.M.; Drozdowski, W.; Mroczko, B.; Janmey, P.A.; Bucki, R. Increased levels of sphingosine-1-phosphate in cerebrospinal fluid of patients diagnosed with tick-borne encephalitis. J. Neuroinflamm. 2014, 11, 193. [Google Scholar] [CrossRef] [PubMed]
- Vesper, H.; Schmelz, E.M.; Nikolova-Karakashian, M.N.; Dillehay, D.L.; Lynch, D.V.; Merrill, A.H., Jr. Sphingolipids in food and the emerging importance of sphingolipids to nutrition. J. Nutr. 1999, 129, 1239–1250. [Google Scholar] [PubMed]
- Thaker, A.I.; Shaker, A.; Rao, M.S.; Ciorba, M.A. Modeling colitis-associated cancer with azoxymethane (AOM) and dextran sulfate sodium (DSS). J. Vis. Exp. 2012, 11, 4100. [Google Scholar] [CrossRef] [PubMed]
- Santiago, C.; Pagan, B.; Isidro, A.A.; Appleyard, C.B. Prolonged chronic inflammation progresses to dysplasia in a novel rat model of colitis-associated colon cancer. Cancer Res. 2007, 67, 10766–10773. [Google Scholar] [CrossRef] [PubMed]
- Symolon, H.; Schmelz, E.M.; Dillehay, D.L.; Merrill, A.H., Jr. Dietary soy sphingolipids suppress tumorigenesis and gene expression in 1,2-dimethylhydrazine-treated CF1 mice and ApcMin/+ mice. J. Nutr. 2004, 134, 1157–1161. [Google Scholar] [PubMed]
- Hamiza, O.O.; Rehman, M.U.; Tahir, M.; Khan, R.; Khan, A.Q.; Lateef, A.; Ali, F.; Sultana, S. Amelioration of 1,2 Dimethylhydrazine (DMH) induced colon oxidative stress, inflammation and tumor promotion response by tannic acid in Wistar rats. Asian Pac. J. Cancer Prev. 2012, 13, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, J.C.; Zhou, H.; Brayfield, B.P.; Hontecillas, R.; Bassaganya-Riera, J.; Schmelz, E.M. Suppression of intestinal inflammation and inflammation-driven colon cancer in mice by dietary sphingomyelin: Importance of peroxisome proliferator-activated receptor gamma expression. J. Nutr. Biochem. 2011, 22, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Fischbeck, A.; Leucht, K.; Frey-Wagner, I.; Bentz, S.; Pesch, T.; Kellermeier, S.; Krebs, M.; Fried, M.; Rogler, G.; Hausmann, M.; et al. Sphingomyelin induces cathepsin D-mediated apoptosis in intestinal epithelial cells and increases inflammation in DSS colitis. Gut 2011, 60, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Saba, J.D. Sphingosine-1-phosphate in inflammatory bowel disease and colitis-associated colon cancer: The fat’s in the fire. Transl. Cancer Res. 2015, 4, 469–483. [Google Scholar] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wollny, T.; Wątek, M.; Durnaś, B.; Niemirowicz, K.; Piktel, E.; Żendzian-Piotrowska, M.; Góźdź, S.; Bucki, R. Sphingosine-1-Phosphate Metabolism and Its Role in the Development of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2017, 18, 741. https://doi.org/10.3390/ijms18040741
Wollny T, Wątek M, Durnaś B, Niemirowicz K, Piktel E, Żendzian-Piotrowska M, Góźdź S, Bucki R. Sphingosine-1-Phosphate Metabolism and Its Role in the Development of Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2017; 18(4):741. https://doi.org/10.3390/ijms18040741
Chicago/Turabian StyleWollny, Tomasz, Marzena Wątek, Bonita Durnaś, Katarzyna Niemirowicz, Ewelina Piktel, Małgorzata Żendzian-Piotrowska, Stanisław Góźdź, and Robert Bucki. 2017. "Sphingosine-1-Phosphate Metabolism and Its Role in the Development of Inflammatory Bowel Disease" International Journal of Molecular Sciences 18, no. 4: 741. https://doi.org/10.3390/ijms18040741
APA StyleWollny, T., Wątek, M., Durnaś, B., Niemirowicz, K., Piktel, E., Żendzian-Piotrowska, M., Góźdź, S., & Bucki, R. (2017). Sphingosine-1-Phosphate Metabolism and Its Role in the Development of Inflammatory Bowel Disease. International Journal of Molecular Sciences, 18(4), 741. https://doi.org/10.3390/ijms18040741