
Cytosolic BNIP3 Dimer Interacts with Mitochondrial BAX Forming Heterodimers in the Mitochondrial Outer Membrane under Basal Conditions


 and
and 
Abstract
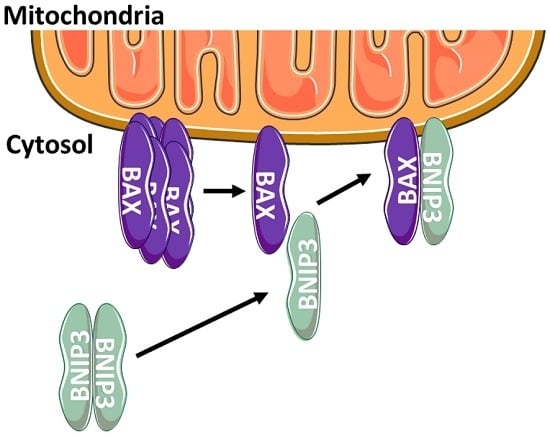
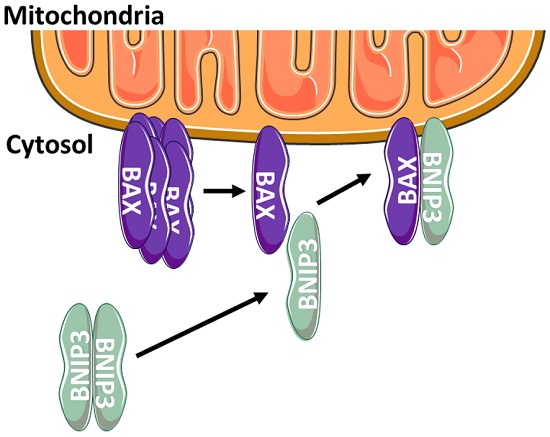
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
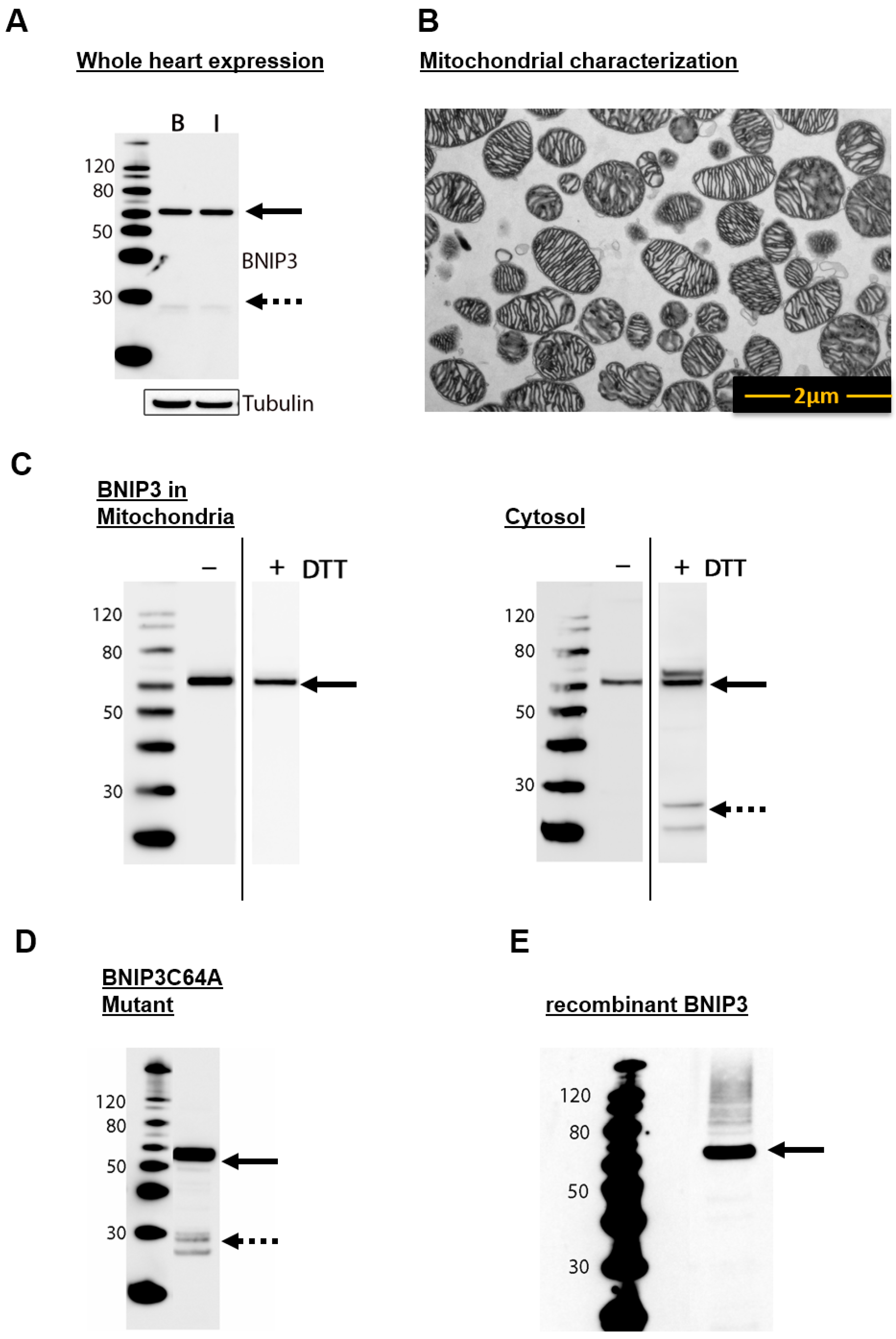
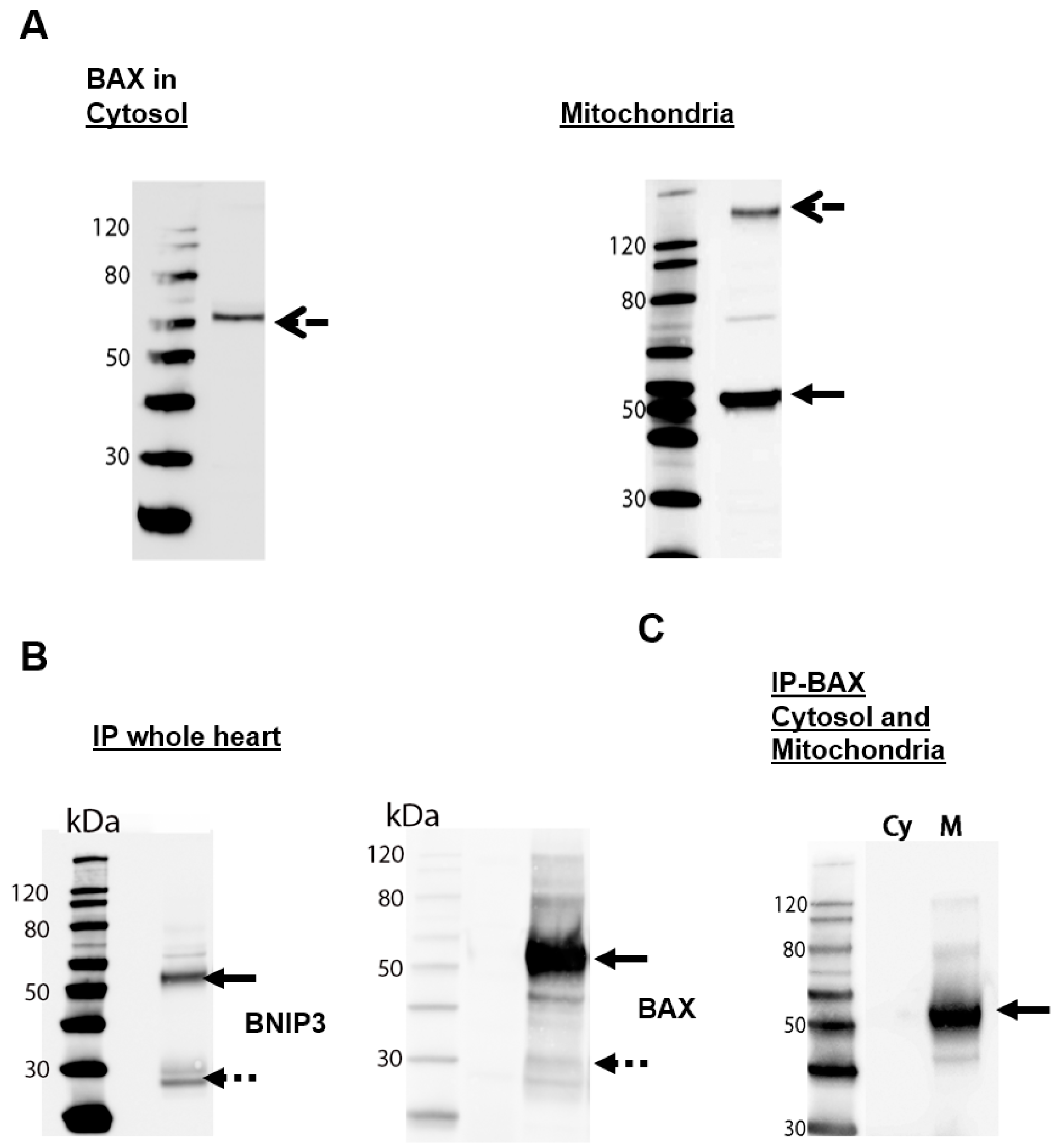
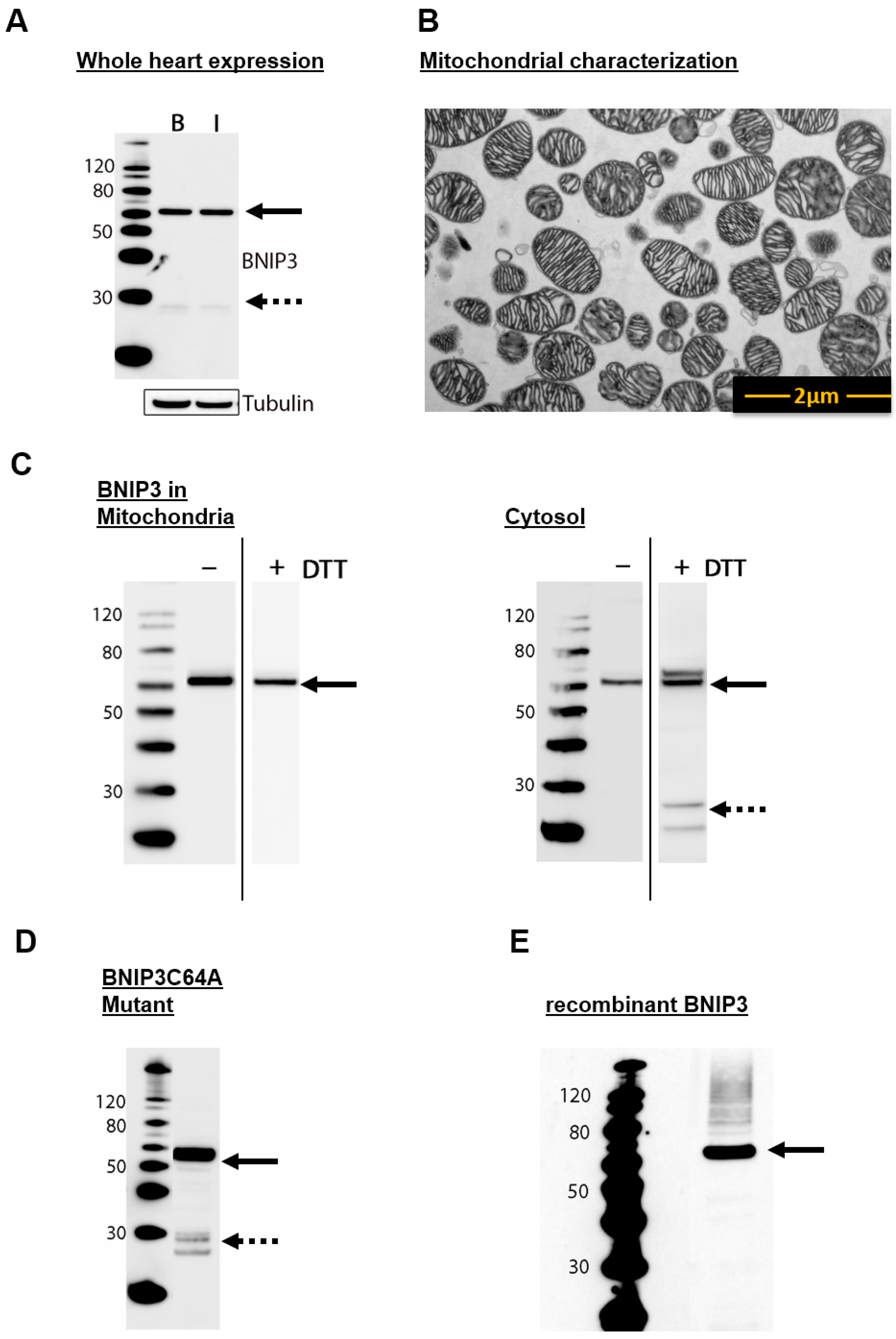
2.1. BNIP3 Forms Different Dimers in the Cytosol and in the MOM in Mouse Heart Cells
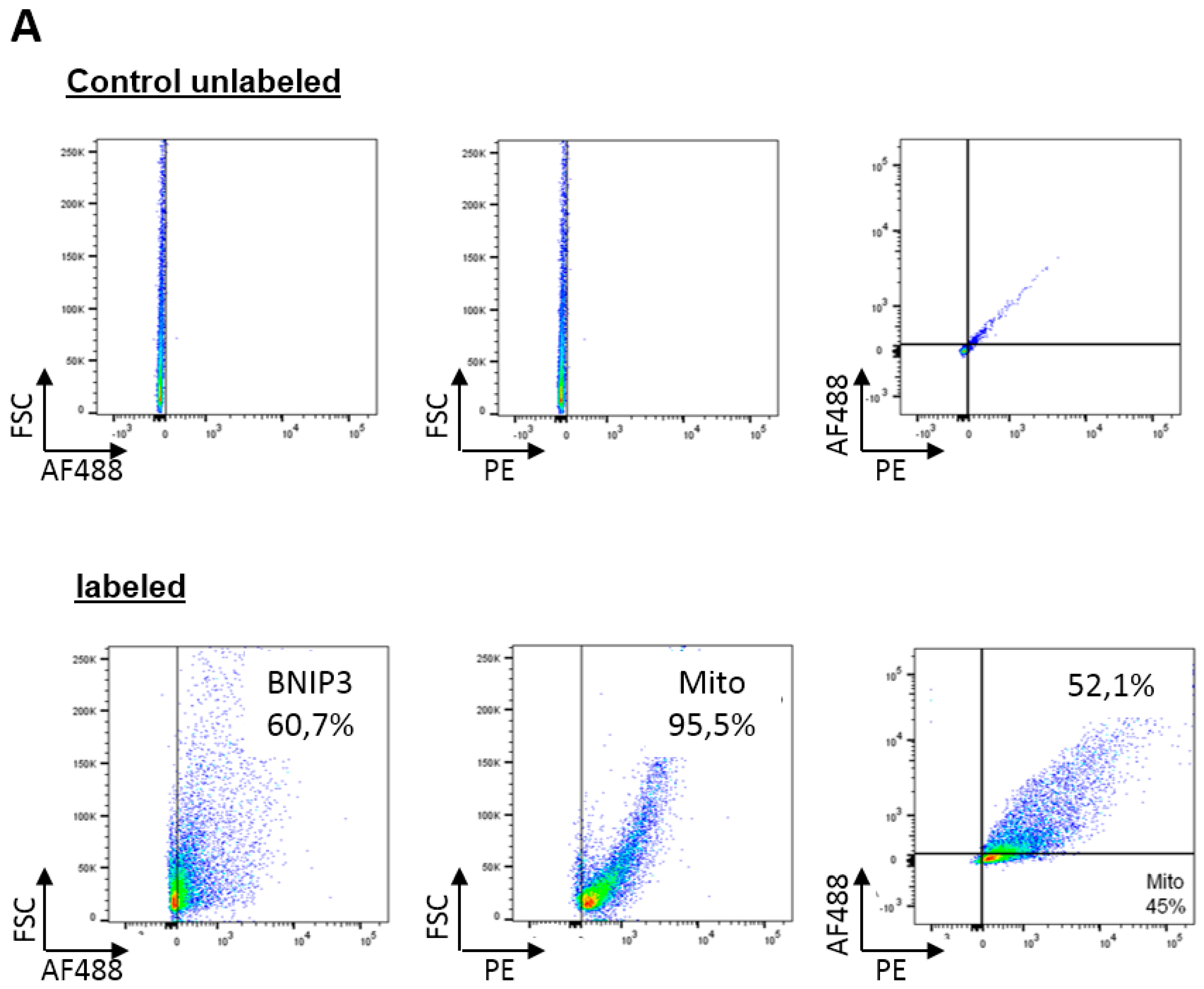
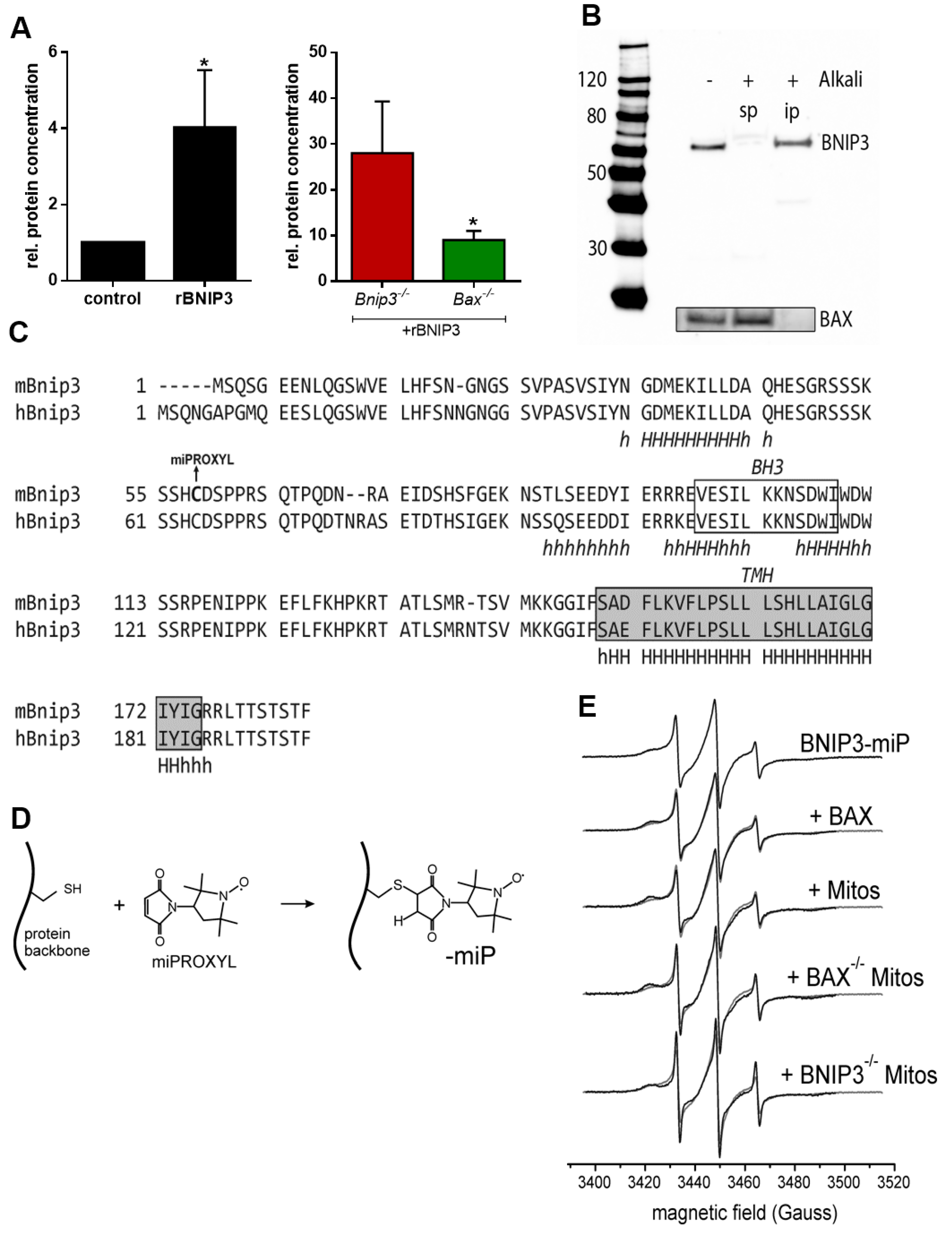
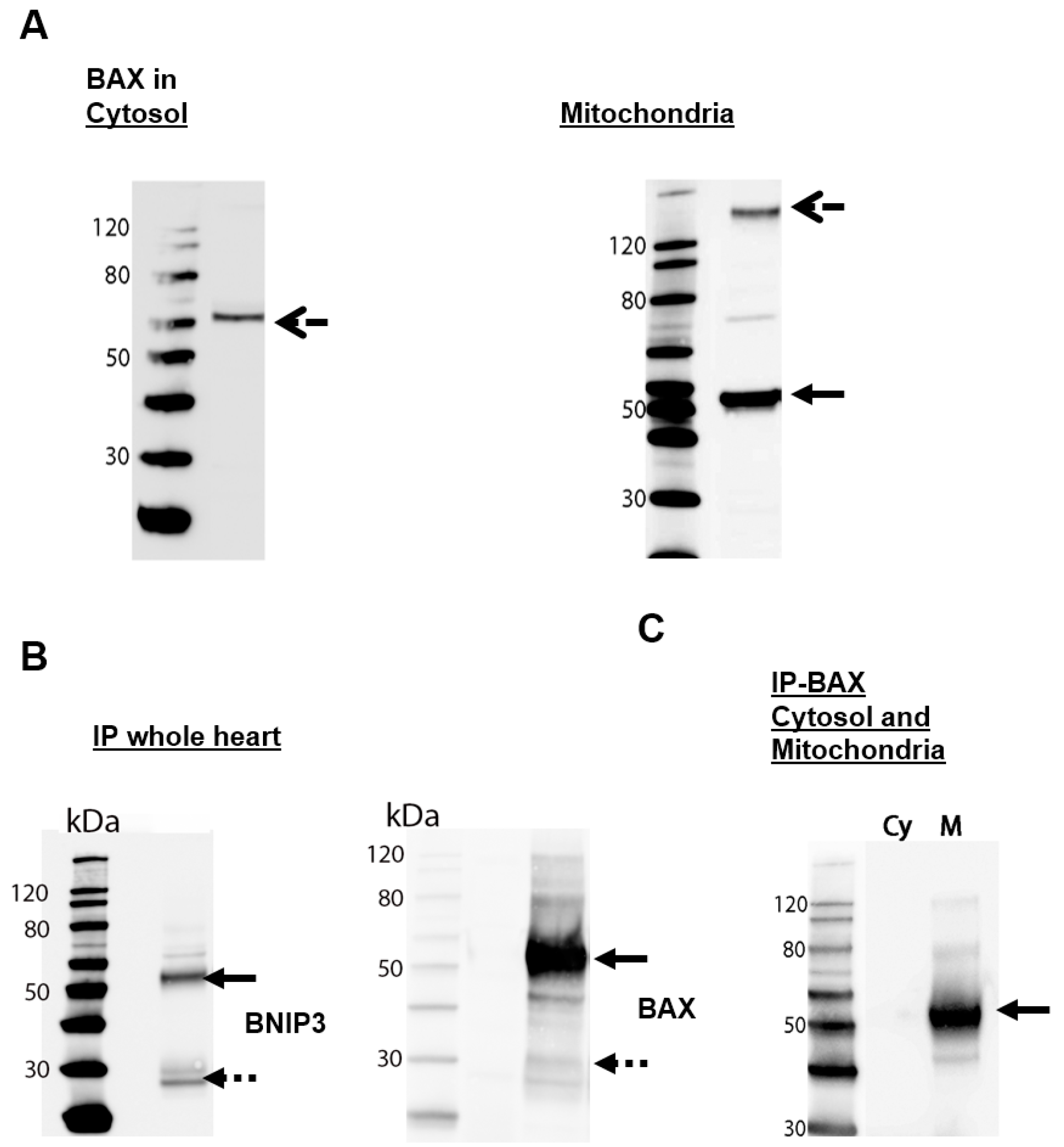
2.2. BNIP3 Interacts with Mitochondrial BAX in the MOM In Vivo under Basal Conditions
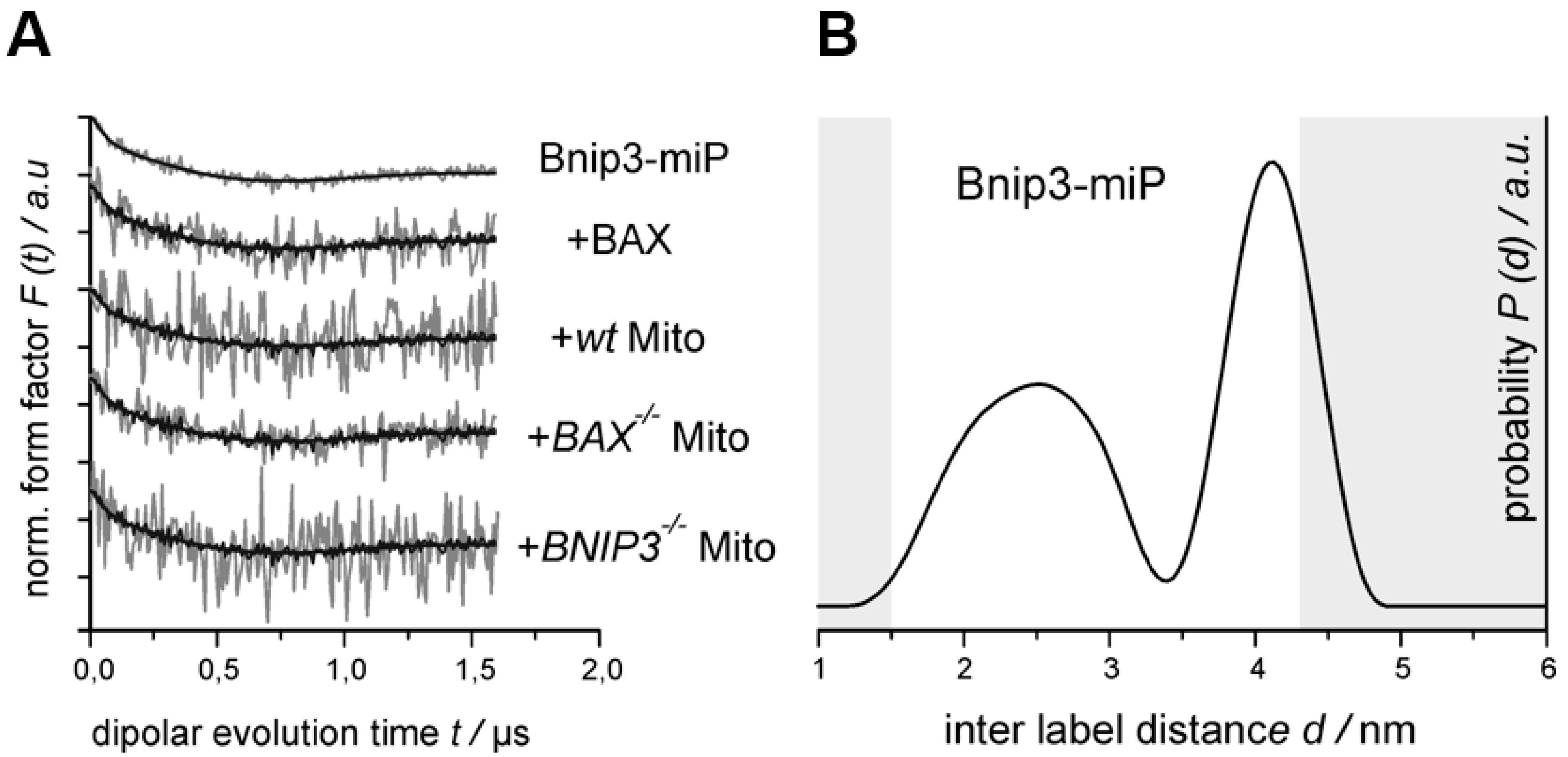
2.3. BNIP3 Homodimers Dissociate upon Interaction with BAX and Mitochondria
3. Materials and Methods
3.1. Animals
3.2. Induction of In Vivo Ischemia
3.3. Recombinant BNIP3 and BAX
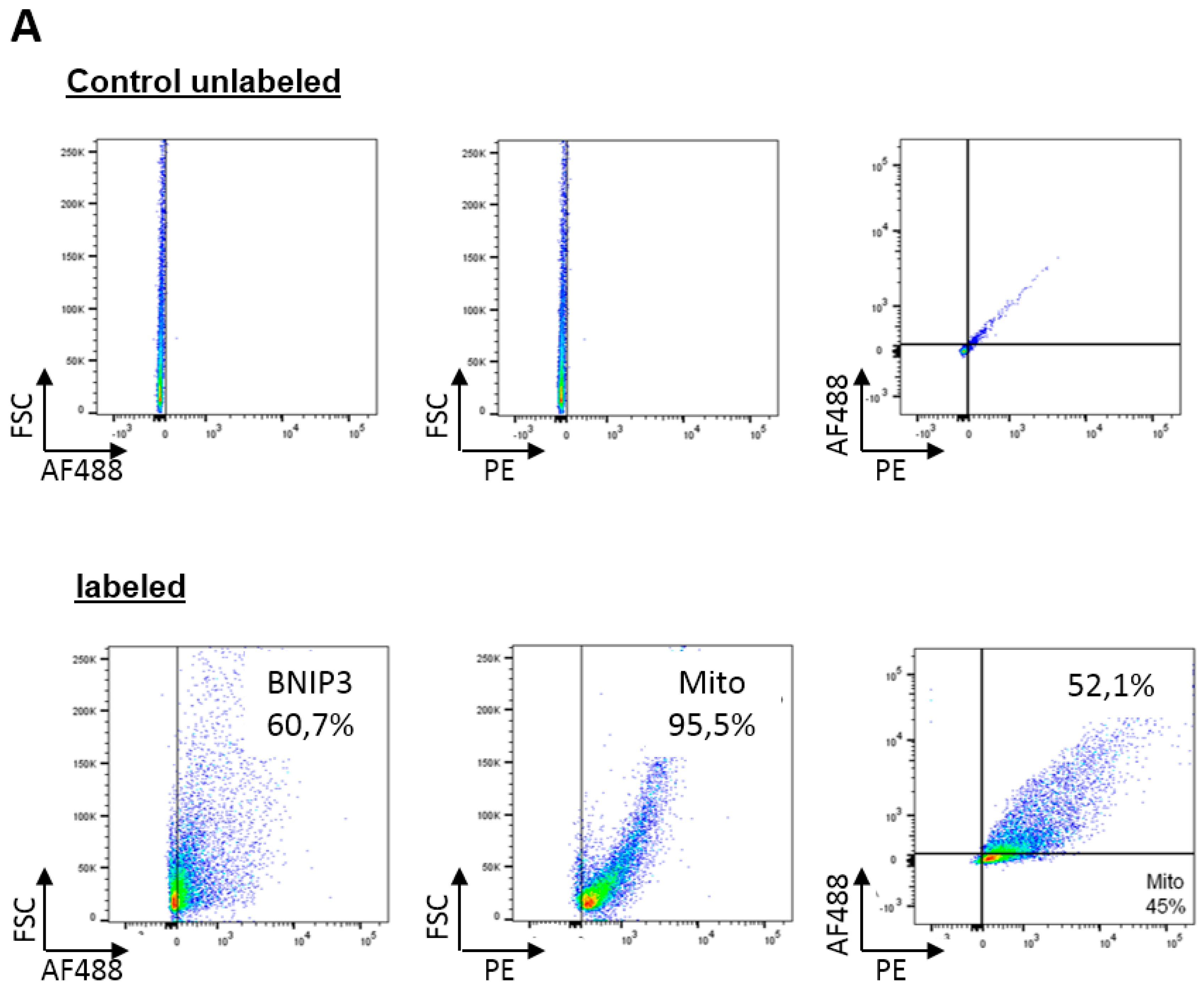
3.4. Preparation of Mouse Heart Mitochondria
3.5. Incubation of rBNIP3 with Mitochondria
3.6. Alkali Extraction
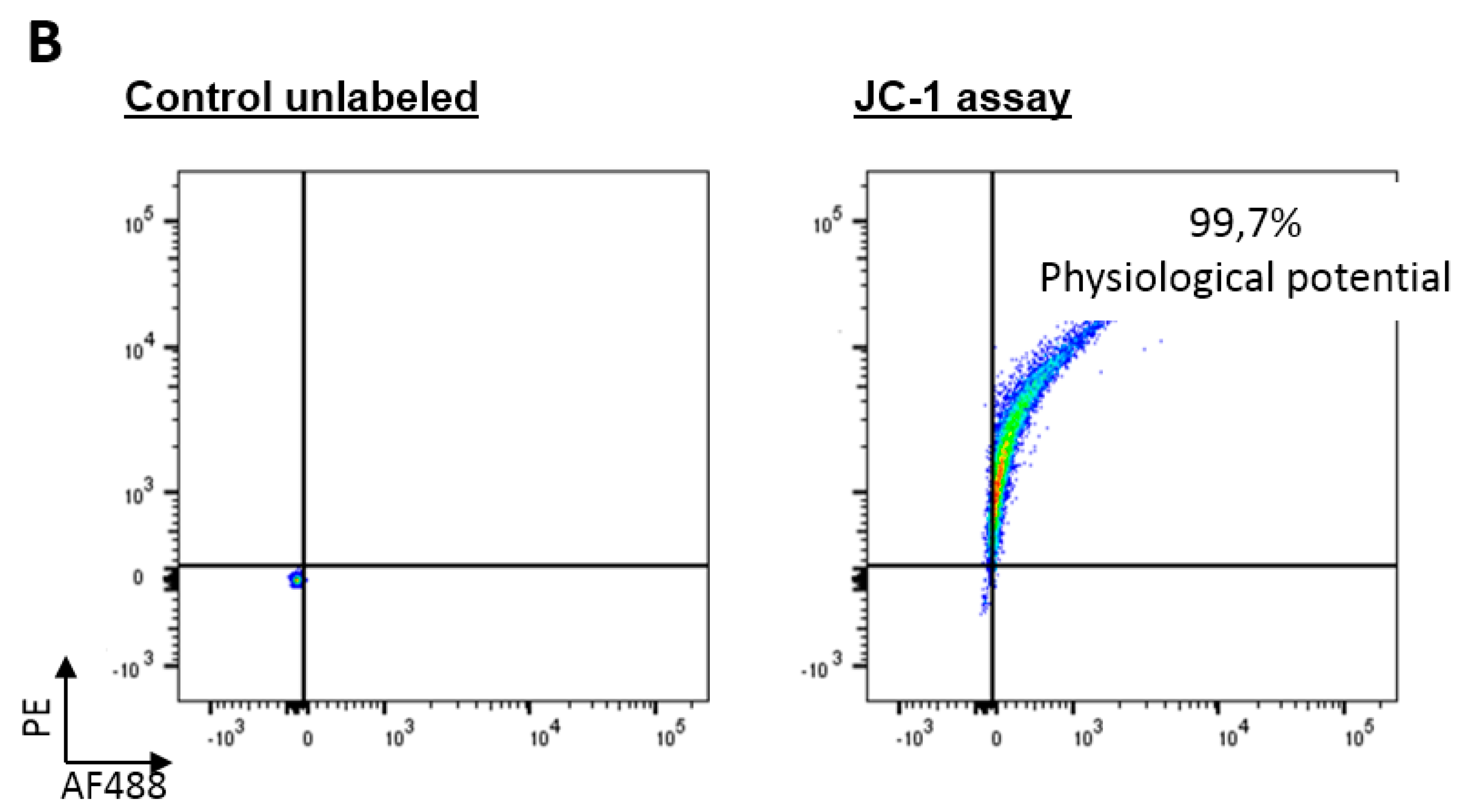
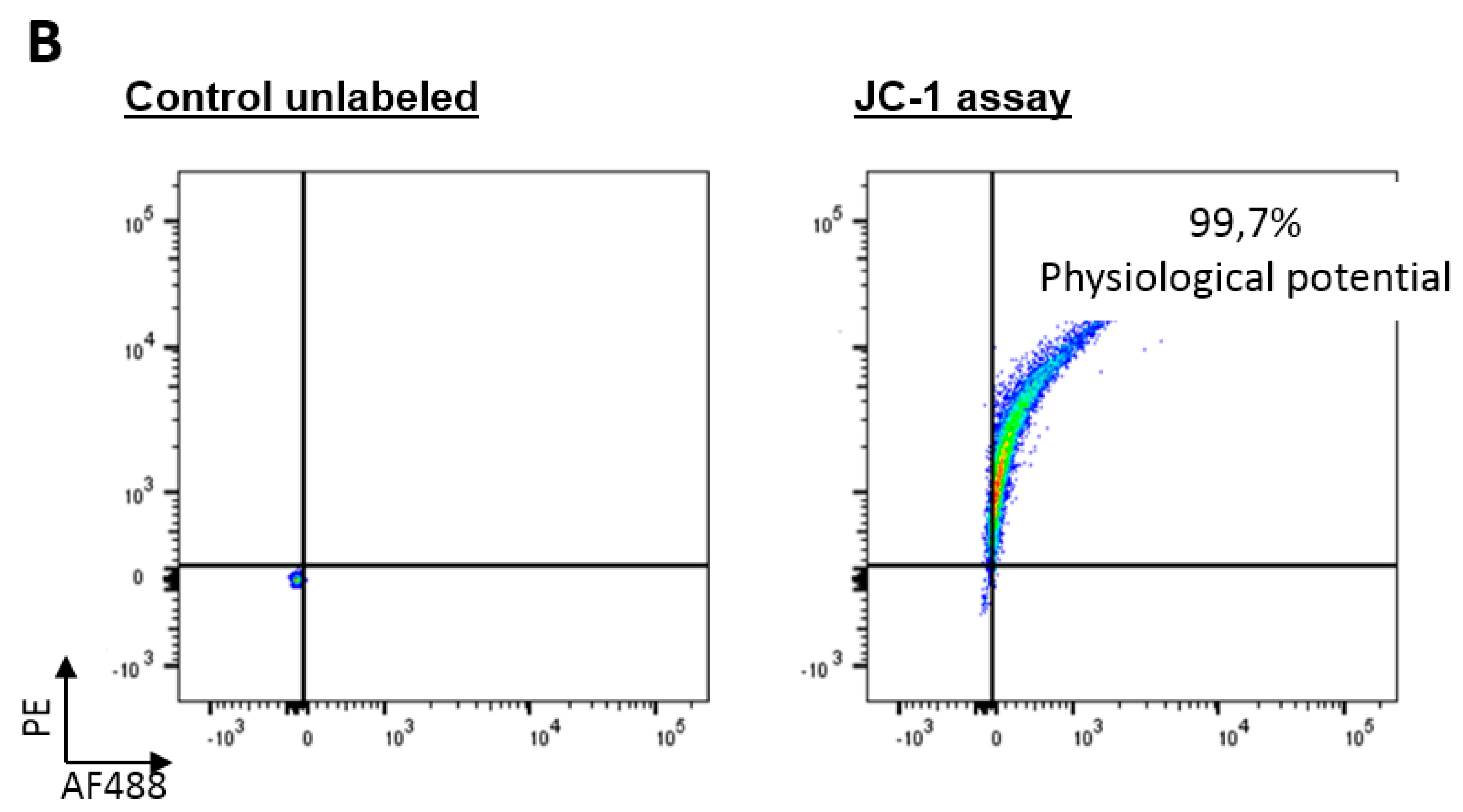
3.7. Flow Cytometry Analysis
3.8. Immunoblotting Analyses
3.9. Co-Immunoprecipitation
3.10. Transmission Electron Microscopy
3.11. Heart Viability Assay
3.12. Spin Labeling of rBNIP3 with miPROXYL
3.13. EPR Spectroscopy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kubli, D.A.; Ycaza, J.E.; Gustafsson, A.B. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem. J. 2007, 405, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Vande Velde, C.; Cizeau, J.; Dubik, D.; Alimonti, J.; Brown, T.; Israels, S.; Hakem, R.; Greenberg, A.H. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol. 2000, 20, 5454–5468. [Google Scholar] [CrossRef] [PubMed]
- Sassone, F.; Margulets, V.; Maraschi, A.; Passafaro, M.; Silani, V.; Ciammola, A.; Kirshenbaum, L.A.; Sassone, J. Bcl-2/adenovirus E1B 19-kDa interacting protein (BNIP3) has a key role in the mitochondrial dysfunction induced by mutant huntingtin. Hum. Mol. Genet. 2015, 24, 6530–6539. [Google Scholar] [CrossRef] [PubMed]
- Kubasiak, L.A.; Hernandez, O.M.; Bishopric, N.H.; Webster, K.A. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc. Natl. Acad. Sci. USA 2002, 99, 12825–12830. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Diwan, A.; Krenz, M.; Syed, F.M.; Wansapura, J.; Ren, X.; Koesters, A.G.; Li, H.; Kirshenbaum, L.A.; Hahn, H.S.; Robbins, J.; et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J. Clin. Investig. 2007, 117, 2825–2833. [Google Scholar] [CrossRef] [PubMed]
- Chaaniene, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness and endoplasmic reticulum-mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ. Heart Fail. 2014, 6, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, X.; Zhang, S.; Ma, X.; Kong, J. BNIP3 upregulation and EndoG translocation in delayed neuronal death in stroke and in hypoxia. Stroke 2007, 38, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Burton, T.R.; Gibson, S.B. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 2009, 16, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, Q.; Yang, D.; Bollag, W.B.; Robertson, K.; Wu, P.; Liu, K. Verticillin A overcomes apoptosis resistance in human colon carcinoma through DNA methylation-dependent upregulation of BNIP3. Cancer Res. 2011, 71, 6807–6816. [Google Scholar] [CrossRef] [PubMed]
- Ern, Y.T.; Campo, L.; Han, C.; Turley, H.; Pezzella, F.; Gatter, K.C.; Harris, A.L.; Fox, S.B. BNIP3 as a progression marker in primary human breast cancer opposing functions in in situ versus invasive cancer. Clin. Cancer Res. 2007, 13, 467–474. [Google Scholar]
- Kubli, D.A.; Quinsay, M.N.; Huang, C.; Lee, Y.; Gustafsson, A.B. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2025–H2031. [Google Scholar] [CrossRef] [PubMed]
- Quinsay, M.N.; Lee, Y.; Rikka, S.; Sayen, M.R.; Molkentin, J.D.; Gottlieb, R.A.; Gustafsson, A.B. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J. Mol. Cell. Cardiol. 2010, 48, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bian, X.; Kong, J. The proapoptotic protein BNIP3 interacts with VDAC to induce mitochondrial release of endonuclease G. PLoS ONE 2014, 3, e113642. [Google Scholar] [CrossRef] [PubMed]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ray, R.; Dubik, D.; Shi, L.; Cizeau, J.; Bleackley, C.R.; Saxena, S.; Gietz, D.R.; Greenberg, A.H. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J. Exp. Med. 1997, 186, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Chen, G.; van de Velde, C.; Cizeau, J.; Park, J.; Reed, J.; Gietz, R.; Greenberg, A. BNIP3 heterodimerizes with Bcl-2/Bcl-XL and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J. Biol. Chem. 2000, 275, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Imazu, T.; Shimizu, S.; Tagami, S.; Matsushima, M.; Nakamura, Y.; Miki, T.; Okuyama, A.; Tsujimoto, Y. Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with Bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene 1999, 18, 4523–4529. [Google Scholar] [CrossRef] [PubMed]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnauné-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Klare, J.P.; Steinhoff, H. Spin labeling EPR. Photosynth. Res. 2009, 102, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.M.; Thompson, J.W.; Wei, J.; Bishopric, N.H.; Webster, K.A. Regulation of Bnip3 death pathways by calcium, phosphorylation, and hypoxia-reoxygenation. Antioxid. Redox Signal. 2007, 9, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Pannier, M.; Veit, S.; Godt, G.; Jeschke, G.; Spiess, H.W. Dead-time free measurement of dipole-dipole interactions between electron spins. J. Magn. Reson. 2011, 213, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G.; Polyhach, Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 2007, 9, 1895–1910. [Google Scholar] [CrossRef] [PubMed]
- Hinds, M.G.; Smits, C.; Risk, J.M.; Bailey, M.; Huang, D.C.S.; Day, C.L. Bim, Bad and Bmf: Intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007, 14, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, T.; Follis, A.V.; Kriwacki, R.W.; Green, D.R. Many players in BCL-2 family affairs. Trends Biochem. Sci. 2015, 39, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 3263, 26–59. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E.; Pines, N.T. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets the roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Hendgen-Cotta, U.B.; Merx, M.W.; Shiva, S.; Schmitz, J.; Becher, S.; Klare, J.P.; Steinhoff, H.-J.; Goedecke, A.; Schrader, J.; Gladwin, M.T.; et al. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2008, 105, 10256–10261. [Google Scholar] [CrossRef] [PubMed]
- Luedike, P.; Hendgen-Cotta, U.B.; Sobierajski, J.; Totzeck, M.; Reeh, M.; Dewor, M.; Lue, H.; Krisp, C.; Wolters, D.; Kelm, M.; et al. Cardioprotection through S-nitros(yl)ation of macrophage migration inhibitory factor. Circulation 2012, 125, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Rassaf, T.; Totzeck, M.; Hendgen-Cotta, U.B.; Shiva, S.; Heusch, G.; Kelm, M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ. Res. 2014, 114, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Hendgen-Cotta, U.B.; Luedike, P.; Totzeck, M.; Kropp, M.; Schicho, A.; Stock, P.; Rammos, C.; Niessen, M.; Heiss, C.; Lundberg, J.O.; et al. Dietary nitrate supplementation improves revascularization in chronic ischemia. Circulation 2012, 126, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Totzeck, M.; Schicho, A.; Stock, P.; Kelm, M.; Rassaf, T.; Hendgen-Cotta, U.B. Nitrite circumvents canonical cGMP signaling to enhance proliferation of myocyte precursor cells. Mol. Cell. Biochem. 2015, 401, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Totzeck, M.; Hendgen-Cotta, U.B.; Luedike, P.; Berenbrink, M.; Klare, J.P.; Steinhoff, H.-J.; Semmler, D.; Shiva, S.; Williams, D.; Kipar, A.; et al. Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation 2012, 126, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Pohl, J.; Hendgen-Cotta, U.B.; Rammos, C.; Luedike, P.; Mull, E.; Stoppe, C.; Jülicher, K.; Lue, H.; Merx, M.W.; Kelm, M.; et al. Targeted intracellular accumulation of macrophage migration inhibitory factor in the reperfused heart mediates cardioprotection. Thromb. Haemost. 2016, 115, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G.; Chechik, V.; Ionita, P.; Godt, A.; Zimmermann, H.; Banham, J.; Timmel, C.R.; Hilger, D.; Jung, H. DeerAnalysis2006-A comprehensive software package for analyzing pulsed ELDOR data. App. Magn. Reson. 2006, 30, 473–498. [Google Scholar]
- Ding, J.; Zhang, Z.; Roberts, G.J.; Falcone, M.; Miao, Y.; Shao, Y.; Zhang, X.C.; Andrews, D.W.; Lin, J. Bcl-2 and Bax interact via the BH1-3 groove-BH3 motif interface and a novel interface involving the BH4 motif. J. Biol. Chem. 2010, 285, 28749–28763. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, Å.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Meusburger, S.; Hawkins, C.J.; Riglar, D.T.; Lee, E.F.; Fairlie, W.D.; Huang, D.C.S.; Adams, J.M. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc. Natl. Acad. Sci. USA 2008, 105, 18081–18087. [Google Scholar] [CrossRef] [PubMed]
- Zhai, D.; Jin, C.; Huang, Z.; Satterthwait, A.C.; Reed, J.C. Differential regulation of Bax and Bak by anti-apoptotic Bcl-2 family proteins Bcl-B and Mcl-1. J. Biol. Chem. 2008, 283, 9580–9586. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendgen-Cotta, U.B.; Esfeld, S.; Rudi, K.; Miinalainen, I.; Klare, J.P.; Rassaf, T. Cytosolic BNIP3 Dimer Interacts with Mitochondrial BAX Forming Heterodimers in the Mitochondrial Outer Membrane under Basal Conditions. Int. J. Mol. Sci. 2017, 18, 687. https://doi.org/10.3390/ijms18040687
Hendgen-Cotta UB, Esfeld S, Rudi K, Miinalainen I, Klare JP, Rassaf T. Cytosolic BNIP3 Dimer Interacts with Mitochondrial BAX Forming Heterodimers in the Mitochondrial Outer Membrane under Basal Conditions. International Journal of Molecular Sciences. 2017; 18(4):687. https://doi.org/10.3390/ijms18040687
Chicago/Turabian StyleHendgen-Cotta, Ulrike B., Sonja Esfeld, Katharina Rudi, Ilkka Miinalainen, Johann P. Klare, and Tienush Rassaf. 2017. "Cytosolic BNIP3 Dimer Interacts with Mitochondrial BAX Forming Heterodimers in the Mitochondrial Outer Membrane under Basal Conditions" International Journal of Molecular Sciences 18, no. 4: 687. https://doi.org/10.3390/ijms18040687
APA StyleHendgen-Cotta, U. B., Esfeld, S., Rudi, K., Miinalainen, I., Klare, J. P., & Rassaf, T. (2017). Cytosolic BNIP3 Dimer Interacts with Mitochondrial BAX Forming Heterodimers in the Mitochondrial Outer Membrane under Basal Conditions. International Journal of Molecular Sciences, 18(4), 687. https://doi.org/10.3390/ijms18040687