
Discriminative Features in Three Autosomal Recessive Cutis Laxa Syndromes: Cutis Laxa IIA, Cutis Laxa IIB, and Geroderma Osteoplastica

 , , ,
, , , 
Abstract
:1. Introduction
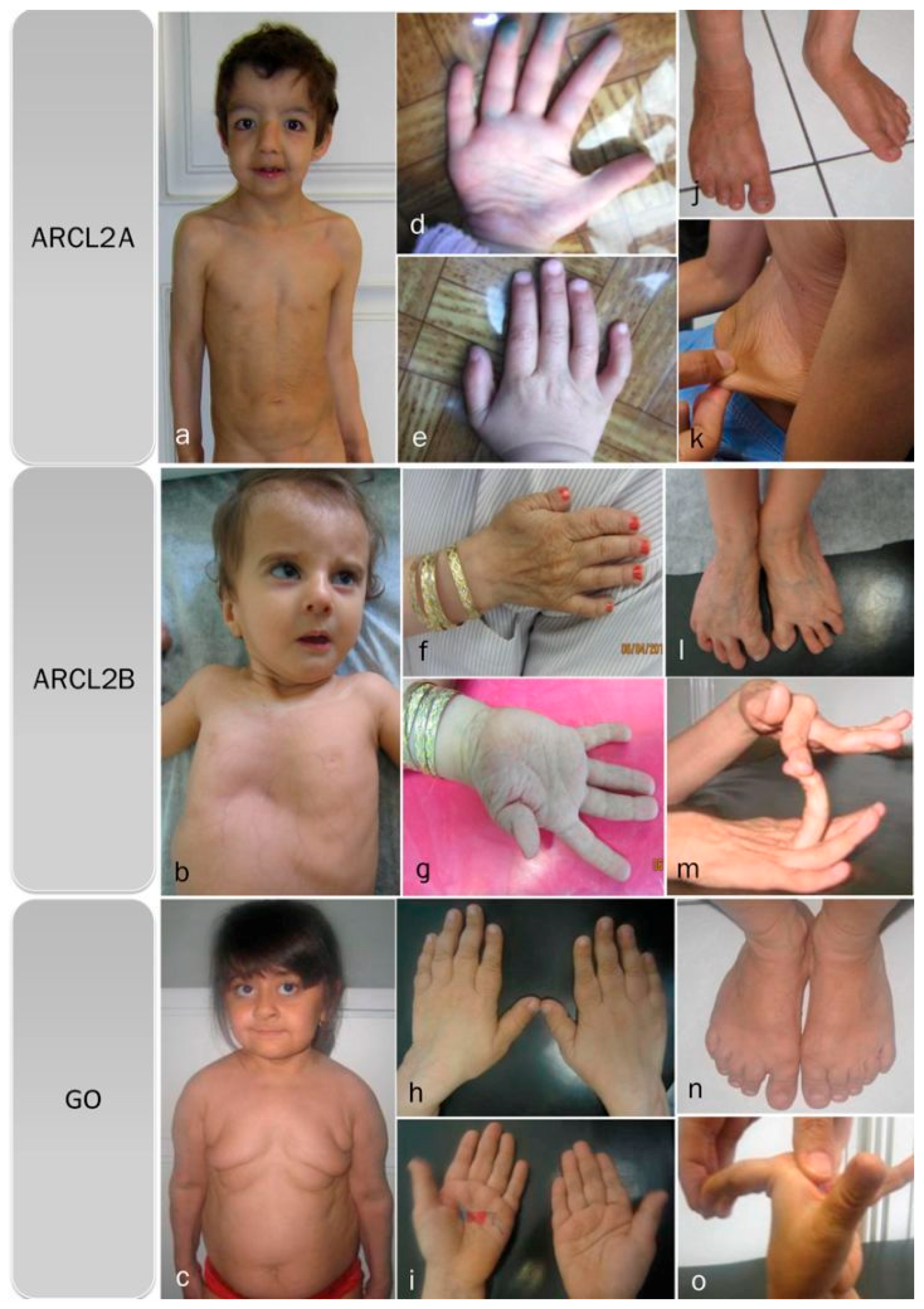
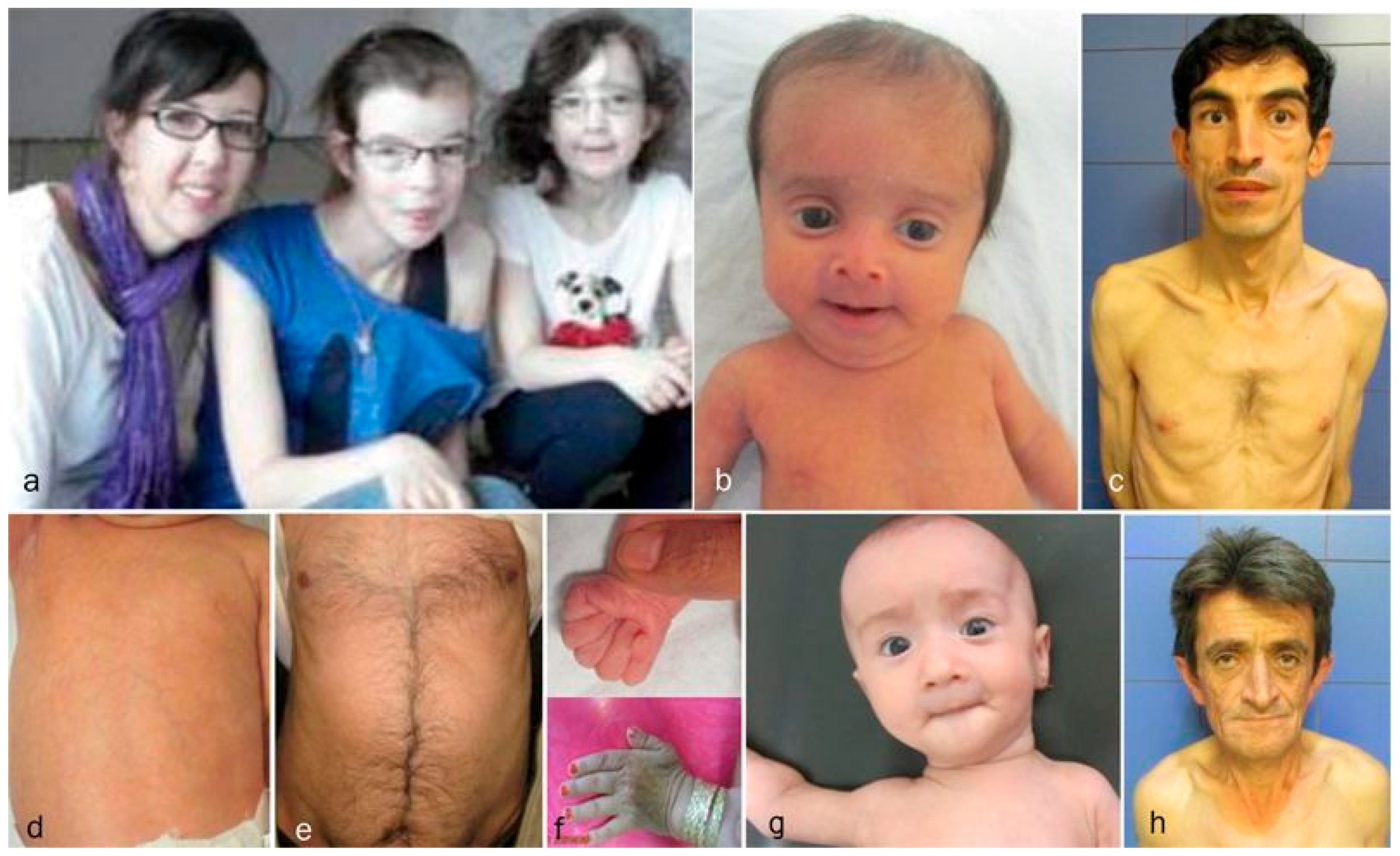
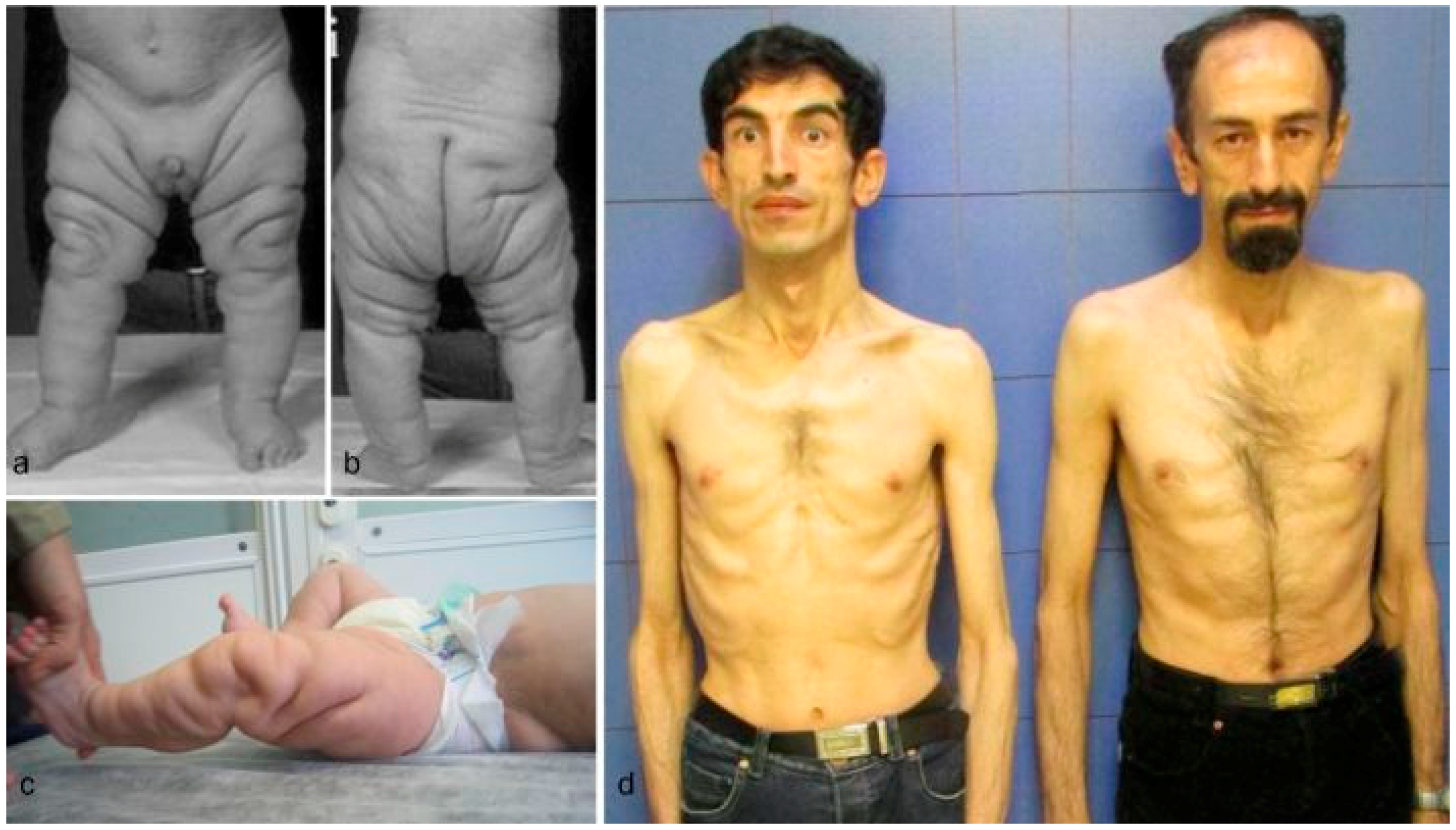
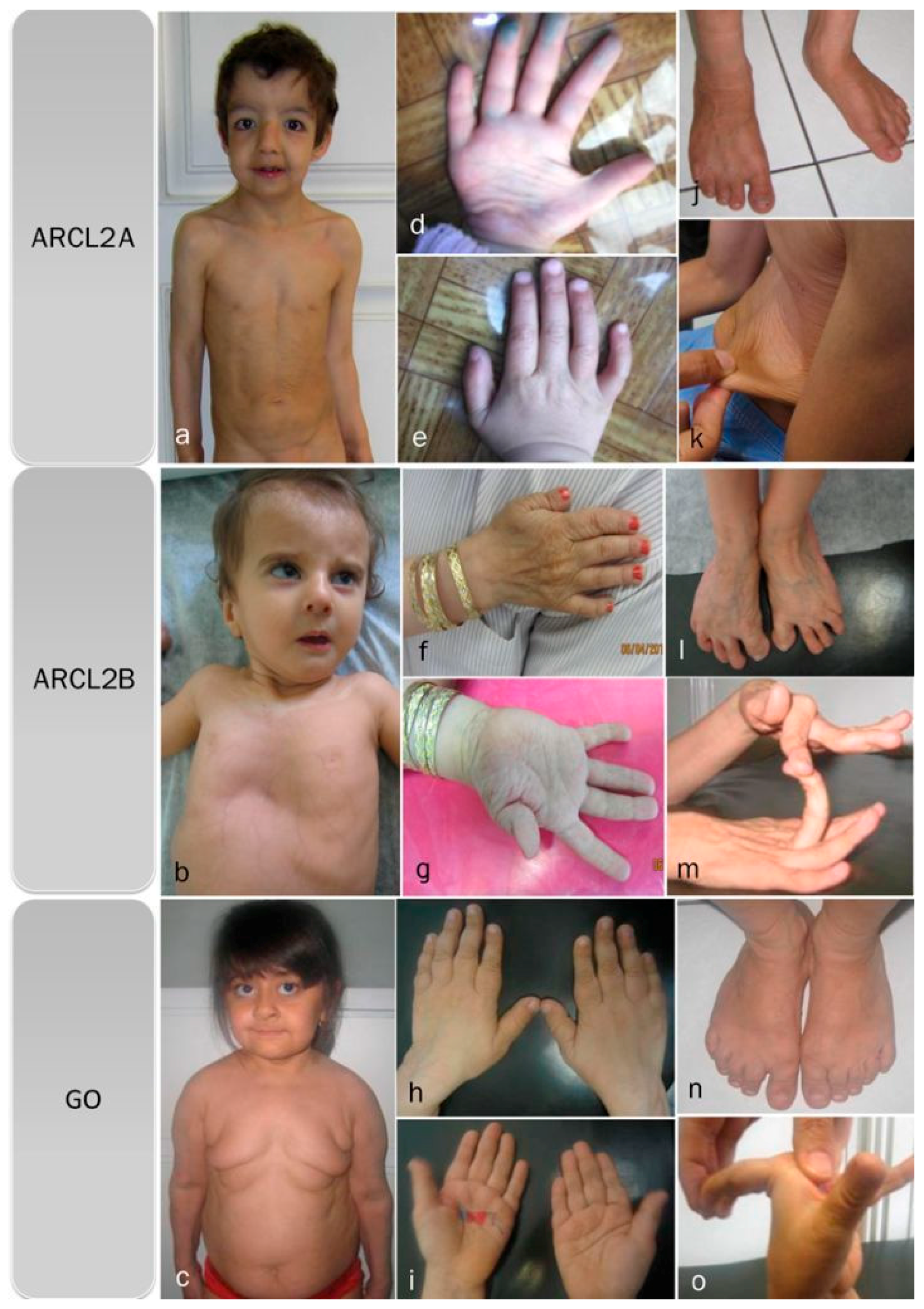
2. Patients
3. Discussion
3.1. Overlapping Features
3.2. Facial Features
3.3. Progeroid Features
3.4. Neurological Features
3.5. Discriminative Features in ARCL2A
3.6. Discriminative Features in ARCL2B
3.7. Discriminative Features in GO
4. Conclusions
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | Morava et al., 2008 [4] | Van Maldergem et al., 2008 [7] | Hucthagowder et al., 2009 [6] | Fischer et al., 2012 [23] | Greally et al., 2014 [24] | Gardeitchik et al. 2014 [22] | Bahena-Bahena et al., 2014 [25] | Ritelli et al., 2014 [26] | Goyal et al., 2015 [27] | Cohen et al., 2016 [28] | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Diagnosis | ARCL2A | ARCL2A | ARCL2A | ARCL2A | ARCL2A | ARCL2A | ARCL2A | ARCL2A | ARCL 2A | ARCL2A | |
| Intra-uterine growth retardation | 2/10 | NE | NE | NE | 3/3 | NE | NE | + | + | 0/2 | 5/7 |
| Postnatal growth delay | NE | NE | 7/17 | NE | 0/3 | NE | 2/2 | - | + | NE | 10/24 |
| Aged appearance | 10/10 | 11/11 | NE | NE | NE | NE | 2/2 | + | + | NE | 4/4 |
| Persistent open fontanel | 10/10 | 11/11 | 16/17 | 10/12 | 3/3 | NE | 2/2 | + | NE | 0/2 | 32/37 |
| Blue sclera | NE | NE | NE | NE | NE | NE | NE | + | + | 0/2 | 2/4 |
| Intellectual disability | 8/10 | 11/11 | 12/17 | 6/11 | 3/3 | NE | 2/2 | - | + | 2/2 | 27/37 |
| Microcephaly | 7/10 | 4/11 | 14/17 | NE | 3/3 | NE | 2/2 | - | + | 2/2 | 23/26 |
| Hypotonia | 8/10 | 11/11 | NE | NE | 1/3 | 6/6 | 2/2 | - | NE | ½ | 10/13 |
| seizures | 3/10 | 5/11 | 1/17 | NE | 1/3 | 2/6 | 0/2 | - | - | 1/2 | 5/30 |
| Athetoid/dystonic movements | NE | NE | NE | NE | 0/3 | 1/6 | 0/2 | - | - | 0/2 | 1/15 |
| Corpus callosum dysgenesis | NE | 1/11 | NE | NE | 1/3 | 0/4 | NE | NE | NE | 0/2 | 1/9 |
| Pachygryria/cobblestone like dysgenesis | 4/10 | 8/11 | 9/15 | 1/1 | 2/3 | 4/4 | NE | NE | NE | 2/2 | 18/25 |
| Strabismus | 7/10 | 0/11 | 13/17 | NE | NE | 2/6 | NE | - | - | 1/2 | 16/27 |
| Cataract/corneal clouding | NE | 0/11 | 0/17 | NE | NE | 1/6 | NE | - | - | 0/2 | 0/27 |
| Osteopenia | NE | NE | NE | NE | NE | NE | NE | NE | - | NE | NE |
| Fractures | NE | 4/11 | 0/17 | 0/12 | 0/3 | 0/6 | 0/2 | + | - | 0/2 | 1/44 |
| Congenital hip dislocation | NE | 8/8 | NE | NE | 0/3 | NE | NE | - | + | 0/2 | 1/7 |
| Wormian bonses | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE |
| Hyperextensibility of joints | 9/10 | NE | 14/17 | 10/12 | 3/3 | NE | 2/2 | NE | NE | 2/2 | 31/36 |
| Dislocations | NE | NE | NE | NE | 0/3 | NE | NE | - | - | 0/2 | 0/7 |
| Adducted thumbs | NE | NE | NE | NE | 0/3 | NE | NE | NE | NE | 0/2 | 0/5 |
| Wrinkled skin | 10/10 | 11/11 | 17/17 | 13/13 | 3/3 | NE | 2/2 | + | + | 2/2 | 39/39 |
| Visible veins on the chest | NE | NE | NE | NE | NE | NE | NE | NE | - | NE | NE |
| hernia | NE | 6/11 | 7/16 | NE | 2/3 | NE | ½ | + | - | 0/2 | 11/25 |
| Flat feet | NE | NE | NE | NE | NE | NE | NE | + | NE | 0/2 | 1/3 |
| Clinical Features | Reversade et al., 2009 [2] | Guernsey et al., 2009 [3] | Yildirim et al., 2010 [16] | Lin et al., 2011 [29] | Kretz et al., 2011 [15] | Dimopoulou et al., 2013 [17] | Nouri et al., 2013 [30] | Scherrer et al., 2013 [31] | Gardeitchik et al. 2014 [22] | Rahmati et al., 2015 [32] | Goyal et al., 2015 [27] | Alazami et al., 2016 [33] | Vahidnezhed et al., 2016 [34] | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diagnosis | ARCL2B | ARCL2B | ARCL2B | ARCL2B | ARCL2B | ARCL2B | ARCL2B | ARCL2B | ARCL2B | ARCL2B | ARCL2B | ARCL2B | ARCL2B | |
| Intra-uterine growth retardation | 29/31 | 5/5 | 4/4 | 2/2 | 6/6 | 23/25 | - | 1/1 | NE | + | + | + | 5/5 | 69/83 |
| Postnatal growth delay | 4/13 | 5/5 | 0/4 | 1/2 | 6/6 | 21/28 | NE | 3/3 | NE | + | + | + | NE | 43/64 |
| Aged appearance | 31/31 | 5/5 | 4/4 | 2/2 | 6/6 | 29/30 | + | 3/3 | NE | + | + | + | 5/5 | 86/87 |
| Persistent open fontanel | 3/6 | 3/3 | 2/3 | 1/2 | NE | 14/19 | NE | 3/3 | NE | - | + | + | 2/2 | 31/40 |
| Blue sclera | NE | 4/5 | NE | 1/2 | 1/6 | 13/30 | NE | NE | NE | + | + | - | 2/2 | 23/47 |
| Intellectual disability | 34/35 | 5/5 | 4/4 | 2/2 | 3/6 | 27/29 | + | 3/3 | NE | NE | + | + | 5/5 | 86/92 |
| Microcephaly | NE | 4/5 | 2/4 | 2/2 | 4/5 | 19/27 | NE | 0/3 | NE | - | + | - | NE | 32/47 |
| Hypotonia | NE | 0/3 | NE | 0/2 | NE | 25/30 | NE | 2/3 | 5/5 | + | - | - | NE | 33/44 |
| seizures | NE | NE | 2/4 | 0/2 | 0/6 | 3/33 | + | 0/3 | 5/5 | - | - | + | 2/5 | 14/61 |
| Athetoid /dystonic movements | 4/35 | 0/5 | 0/4 | ½ | NE | 9/28 | NE | 0/3 | 3/5 | - | - | - | NE | 17/85 |
| Corpus callosum dysgenesis | 15/21 | 2/3 | 1/4 | 0/2 | NE | 4/13 | + | NE | 2/5 | - | NE | + | 0/2 | 26/53 |
| pachygryria | 0/21 | 0/3 | 0/4 | 0/2 | NE | 0/13 | - | NE | 0/5 | - | NE | - | 0/2 | 0/53 |
| Strabismus | NE | NE | NE | ½ | 1/6 | 12/27 | NE | 0/3 | 2/5 | - | - | - | ½ | 17/43 |
| Cataract/corneal clouding | 4/35 | 0/5 | NE | 0/2 | 0/6 | 4/27 | NE | 0/3 | 4/5 | - | - | + | 0/2 | 12/88 |
| Osteopenia | 12/18 | 0/1 | 4/4 | 0/2 | 3/3 | 14/16 | NE | 3/3 | NE | - | - | - | 2/2 | 38/52 |
| Fractures | NE | 0/3 | 0/4 | 0/2 | 1/6 | NE | NE | 2/3 | NE | - | - | - | 0/2 | 3/23 |
| Congenital hip dislocation | 19/34 | 2/4 | ¼ | ½ | 4/6 | 15/25 | NE | 3/3 | NE | + | + | + | 5/5 | 43/86 |
| Wormian bonses | 6/9 | 1/2 | 4/4 | 0/2 | 1/1 | 8/12 | NE | NE | NE | - | NE | - | NE | 20/32 |
| Hyperextensibility of joints | 35/35 | NE | 4/4 | 2/2 | 4/6 | 24/26 | NE | 3/3 | NE | NE | NE | + | 2/2 | 75/79 |
| Dislocations | NE | NE | NE | 0/2 | NE | NE | NE | 0/3 | NE | NE | NE | - | 2/2 | 2/8 |
| Adducted thumbs | NE | 3/3 | 4/4 | ½ | 4/6 | 13/26 | NE | 2/3 | NE | NE | NE | + | NE | 24/45 |
| Wrinkled skin | 35/35 | 3/3 | 4/4 | 2/2 | 6/6 | 31/31 | + | 3/3 | NE | + | + | + | 5/5 | 93/93 |
| Visible veins on the chest | NE | 5/5 | 4/4 | 2/2 | 6/6 | 24/31 | NE | NE | NE | + | + | + | 2/2 | 46/53 |
| hernia | 17/35 | 2/3 | 1/4 | 0/2 | 0/6 | 10/28 | NE | 3/3 | NE | - | - | - | NE | 33/81 |
| Flat feet | NE | NE | NE | NE | 5/6 | NE | NE | NE | NE | NE | NE | - | NE | 5/7 |
| Clinical Features | Newman et al., 2008 [35] | Al-Dosari et al., 2009 [19] | Gardeitchik et al., 2014 [22] | Alazami et al., 2016 [33] | Total |
|---|---|---|---|---|---|
| Diagnosis | GO | GO | GO | GO | GO |
| Intra-uterine growth retardation | NE | NE | NE | NE | NE |
| Postnatal growth delay | 5/9 | 1/7 | NE | 2/27 | 8/43 |
| Aged appearance | 10/10 | 7/7 | NE | 23/27 | 40/44 |
| Persistent open fontanel | NE | 1/7 | NE | NE | 1/7 |
| Blue sclera | 1/10 | NE | NE | NE | 1/10 |
| Intellectual disability | 0/10 | 0/7 | NE | 0/27 | 0/44 |
| Microcephaly | 0/10 | 0/7 | NE | 0/27 | 0/44 |
| Hypotonia | 8/8 | NE | 3/3 | 2/27 | 13/38 |
| seizures | 0/10 | NE | 1/3 | 0/27 | 1/40 |
| Athetoid/dystonic movements | 0/10 | NE | 0/3 | 0/27 | 0/40 |
| Corpus callosum dysgenesis | NE | NE | 0/3 | NE | 0/3 |
| pachygryria | NE | NE | 0/3 | NE | 0/3 |
| Strabismus | NE | NE | 0/3 | NE | 0/3 |
| Cataract/corneal clouding | NE | NE | 0/3 | NE | 0/3 |
| Osteopenia | 10/10 | 6/7 | NE | 26/27 | 42/44 |
| Fractures | 9/10 | 5/7 | NE | 17/27 | 31/44 |
| Congenital hip dislocation | 4/10 | 2/7 | NE | 25/27 | 31/44 |
| Wormian bonses | NE | NE | NE | 15/27 | 15/27 |
| Hyperextensibility of joints | 10/10 | NE | NE | 26/27 | 36/37 |
| Dislocations | NE | NE | NE | NE | NE |
| Adducted thumbs | NE | NE | NE | NE | NE |
| Wrinkled skin | 10/10 | 7/7 | NE | 27/27 | 44/44 |
| Visible veins on the chest | 0/10 | NE | NE | 0/27 | 0/34 |
| hernia | 0/10 | NE | NE | 0/27 | 0/34 |
| Flat feet | NE | NE | NE | NE | NE |
References
- Kornak, U.; Reynders, E.; Dimopoulou, A.; van Reeuwijk, J.; Fischer, B.; Rajab, A.; Budde, B.; Nürnberg, P.; Foulquier, F.; Lefeber, D.; et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat. Genet. 2008, 40, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Reversade, B.; Escande-Beillard, N.; Dimopoulou, A.; Fischer, B.; Chng, S.C.; Li, Y.; Shboul, M.; Tham, P.Y.; Kayserili, H.; Al-Gazali, L.; et al. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat. Genet. 2009, 41, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Guernsey, D.L.; Jiang, H.; Evans, S.C.; Ferguson, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.L.; Provost, S.; Bedard, K.; Orr, A.; et al. Mutation in pyrroline-5-carboxylate reductase 1 gene in families with cutis laxa type 2. Am. J. Hum. Genet. 2009, 85, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Morava, E.; Lefeber, D.J.; Urban, Z.; de Meileir, L.; Meinecke, P.; Gillessen Kaesbach, G.; Sykut-Cegielska, J.; Adamowicz, M.; Salafsky, I.; Ranells, J.; et al. Defining the phenotype in an autosomal recessive cutis laxa syndrome with a combined congenital defect of glycosylation. Eur. J. Hum. Genet. 2008, 16, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Morava, E.; Wevers, R.A.; Willemsen, M.A.; Lefeber, D. Cobblestone-like brain dysgenesis and altered glycosylation in congenital cutis laxa, Debré type. Neurology 2009, 73, 1164. [Google Scholar] [CrossRef] [PubMed]
- Hucthagowder, V.; Morava, E.; Kornak, U.; Lefeber, D.J.; Fischer, B.; Dimopoulou, A.; Aldinger, A.; Choi, J.; Davis, E.C.; Abuelo, D.N.; et al. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum. Mol. Genet. 2009, 18, 2149–2165. [Google Scholar] [CrossRef] [PubMed]
- Van Maldergem, L.; Yuksel-Apak, M.; Kayserili, H.; Seemanova, E.; Giurgea, S.; Basel-Vanagaite, L.; Leao-Teles, E.; Vigneron, J.; Foulon, M.; Greally, M.; et al. Cobblestone-like brain dysgenesis and altered glycosylation in congenital cutis laxa, Debre type. Neurology 2008, 71, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, K.; Kurosawa, K.; Makita, Y.; Masuno, M.; Kuroki, Y. Male with type II autosomal recessive cutis laxa. Clin. Genet. 1994, 45, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Morava, E.; Wopereis, S.; Coucke, P.; Gillessen-Kaesbach, G.; Voit, T.; Smeitink, J.; Wevers, R.; Grunewald, S. Defective protein glycosylation in patients with cutis laxa syndrome. Eur. J. Hum. Genet. 2005, 13, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Wopereis, S.; Morava, E.; Grünewald, S.; Mills, P.B.; Winchester, B.G.; Clayton, P.; Coucke, P.; Huijben, K.M.; Wevers, R.A. A combined defect in the biosynthesis of N- and O-glycans in patients with cutis laxa and neurological involvement: The biochemical characteristics. Biochim. Biophys. Acta 2005, 1741, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Al-Gazali, L.I.; Sztriha, L.; Skaff, F.; Haas, D. Gerodermia osteodysplastica and wrinkly skin syndrome: Are they the same? Am. J. Med. Genet. 2001, 101, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Hamamy, H.; Masri, A.; Ajlouni, K. Wrinkly skin syndrome. Clin. Exp. Dermatol. 2005, 30, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Nanda, A.; Alsaleh, Q.A.; Al-Sabah, H.; Marzouk, E.E.; Salam, A.M.; Nanda, M.; Anim, J.T. Gerodermia osteodysplastica/wrinkly skin syndrome: Report of three patients and brief review of the literature. Pediatr. Dermatol. 2008, 25, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Rajab, A.; Kornak, U.; Budde, B.S.; Hoffmann, K.; Jaeken, J.; Nurnberg, P.; Mundlos, S. Geroderma osteodysplasticum hereditaria and wrinkly skin syndrome in 22 patients from Oman. Am. J. Med. Genet. 2008, 146, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Kretz, R.; Bozorgmehr, B.; Kariminejad, M.H.; Rohrbach, M.; Hausser, I.; Baumer, A.; Baumgartner, M.; Giunta, C.; Kariminejad, A.; Häberle, J. Defect in proline synthesis: Pyrroline-5-carboxylate reductase 1 deficiency leads to a complex clinical phenotype with collagen and elastin abnormalities. J. Inherit Metab. Dis. 2011, 34, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, Y.; Tolun, A.; Tüysüz, B. The phenotype caused by PYCR1 mutations corresponds to geroderma osteodysplasticum rather than autosomal recessive cutis laxa type 2. Am. J. Med. Genet. A 2011, 155A, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulou, A.; Fischer, B.; Gardeitchik, T.; Schroter, P.; Kayserili, H.; Schlack, C.; Li, Y.; Brum, J.M.; Barisic, I.; Castori, M.; et al. Genotype-phenotype spectrum of PYCR1-related autosomal recessive cutis laxa. Mol. Genet. Metab. 2013, 110, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Bamatter, F.; Franceschetti, A.; Klein, D.; Sierro, A. Gerodermie osteodysplastique hereditaire. Ann. Pediatr. 1950, 174, 126–127. [Google Scholar]
- Al-Dosari, M.; Alkuraya, F.S. A novel missense mutation in SCYL1BP1 produces geroderma osteodysplastica phenotype indistinguishable from that caused by nullimorphic mutations. Am. J. Med. Genet. A 2009, 149A, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.G.; Martsolf, J.T.; Baker, C.G.; Reed, M.H. Geroderma osteodysplastica. A report of two affected families. Hum. Genet. 1978, 40, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Hennies, H.C.; Kornak, U.; Zhang, H.; Egerer, J.; Zhang, X.; Seifert, W.; Kuhnisch, J.; Budde, B.; Natebus, M.; Brancati, F.; et al. Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nat. Genet. 2008, 40, 1410–1412. [Google Scholar] [CrossRef] [PubMed]
- Gardeitchik, T.; Mohamed, M.; Fischer, B.; Lammens, M.; Lefeber, D.; Lace, B.; Parker, M.; Kim, K.J.; Lim, B.C.; Häberle, J.; et al. Clinical and biochemical features guiding the diagnostics in neurometabolic cutis laxa. Eur. J. Hum. Genet. 2014, 22, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.; Dimopoulou, A.; Egerer, J.; Gardeitchik, T.; Kidd, A.; Jost, D.; Kayserili, H.; Alanay, Y.; Tantcheva-Poor, I.; Mangold, E.; et al. Further characterization of ATP6V0A2-related autosomal recessive cutis laxa. Hum. Genet. 2012, 131, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- Greally, M.T.; Kalis, N.N.; Agab, W.; Ardati, K.; Giurgea, S.; Kornak, U.; van Maldergem, L. Autosomal recessive cutis laxa type 2A (ARCL2A) mimicking Ehlers-Danlos syndrome by its dermatological manifestations: Report of three affected patients. Am. J. Med. Genet. A 2014, 164, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Bahena-Bahena, D.; López-Valdez, J.; Raymond, K.; Salinas-Marín, R.; Ortega-García, A.; Ng, B.G.; Freeze, H.H.; Ruíz-García, M.; Martínez-Duncker, I. ATP6V0A2 mutations present in two Mexican Mestizo children with an autosomal recessive cutis laxa syndrome type IIA. Mol. Genet. Metab. Rep. 2014, 1, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Chiarelli, N.; Quinzani, S.; Dordoni, C.; Venturini, M.; Pezzani, L.; Calzavara-Pinton, P.; Colombi, M. Identification of two novel ATP6V0A2 mutations in an infant with cutis laxa by exome sequencing. J. Dermatol. Sci. 2014, 75, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.; Singh, A.; Kornak, U.; Kapoor, S. The diagnostic dilemma of cutis laxa: A report of two cases with genotypic dissimilarity. Indian J. Dermatol. 2015, 60, 521. [Google Scholar] [PubMed]
- Cohen, R.; Halevy, A.; Aharoni, S.; Kraus, D.; Konen, O.; Basel-Vanagaite, L.; Goldberg-Stern, H.; Straussberg, R. Polymicrogyria and myoclonic epilepsy in autosomal recessive cutis laxa type 2A. Neurogenetics 2016, 17, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.S.; Yeung, C.Y.; Liu, H.L.; Ho, C.S.; Shu, C.H.; Chuang, C.K.; Huang, Y.W.; Wu, T.Y.; Huang, Z.D.; Jian, Y.R.; et al. A novel mutation in PYCR1 causes an autosomal recessive cutis laxa with premature aging features in a family. Am. J. Med. Genet. A 2011, 155A, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Nouri, N.; Aryani, O.; Nouri, N.; Kamalidehghan B Houshmand, M. Cutis laxa type II with mutation in the pyrroline-5-carboxylate reductase 1 gene. Pediatr. Dermatol. 2013, 30, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, D.Z.; Baptista, M.B.; Matos, A.H.; Maurer-Morelli, C.V.; Steiner, C.E. Mutations in PYCR1 gene in three families with autosomal recessive cutis laxa, type 2. Eur. J. Med. Genet. 2013, 56, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, M.; Yazdanparast, M.; Jahanshahi, K.; Zakeri, M. Congenital cutis laxa type 2 associated with recurrent aspiration pneumonia and growth delay: Case report. Electron. Physician 2015, 7, 1391–1393. [Google Scholar] [PubMed]
- Alazami, A.M.; Al-Qattan, S.M.; Faqeih, E.; Alhashem, A.; Alshammari, M.; Alzahrani, F.; Al-Dosari, M.S.; Patel, N.; Alsagheir, A.; Binabbas, B.; et al. Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum. Genet. 2016, 135, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Vahidnezhad, H.; Karamzadeh, R.; Saeidian, A.H.; Youssefian, L.; Sotoudeh, S.; Zeinali, S.; Vasei, M.; Golnabi, F.; Baghdadi, T.; Uitto, J. Molecular dynamics simulation of the consequences of a PYCR1 mutation (p.Ala189Val) in patients with complex connective tissue disorder and severe intellectual disability. J. Investig. Dermatol. 2017, 137, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Newman, W.G.; Clayton-Smith, J.; Metcalfe, K.; Cole, R.; Tartaglia, M.; Brancati, F.; Morara, S.; Novelli, A.; Liu, X.; Siminovitch, K.A.; et al. Geroderma osteodysplastica maps to a 4 Mb locus on chromosome 1q24. Am. J. Med. Genet. A 2008, 146A, 3034–3037. [Google Scholar] [CrossRef] [PubMed]
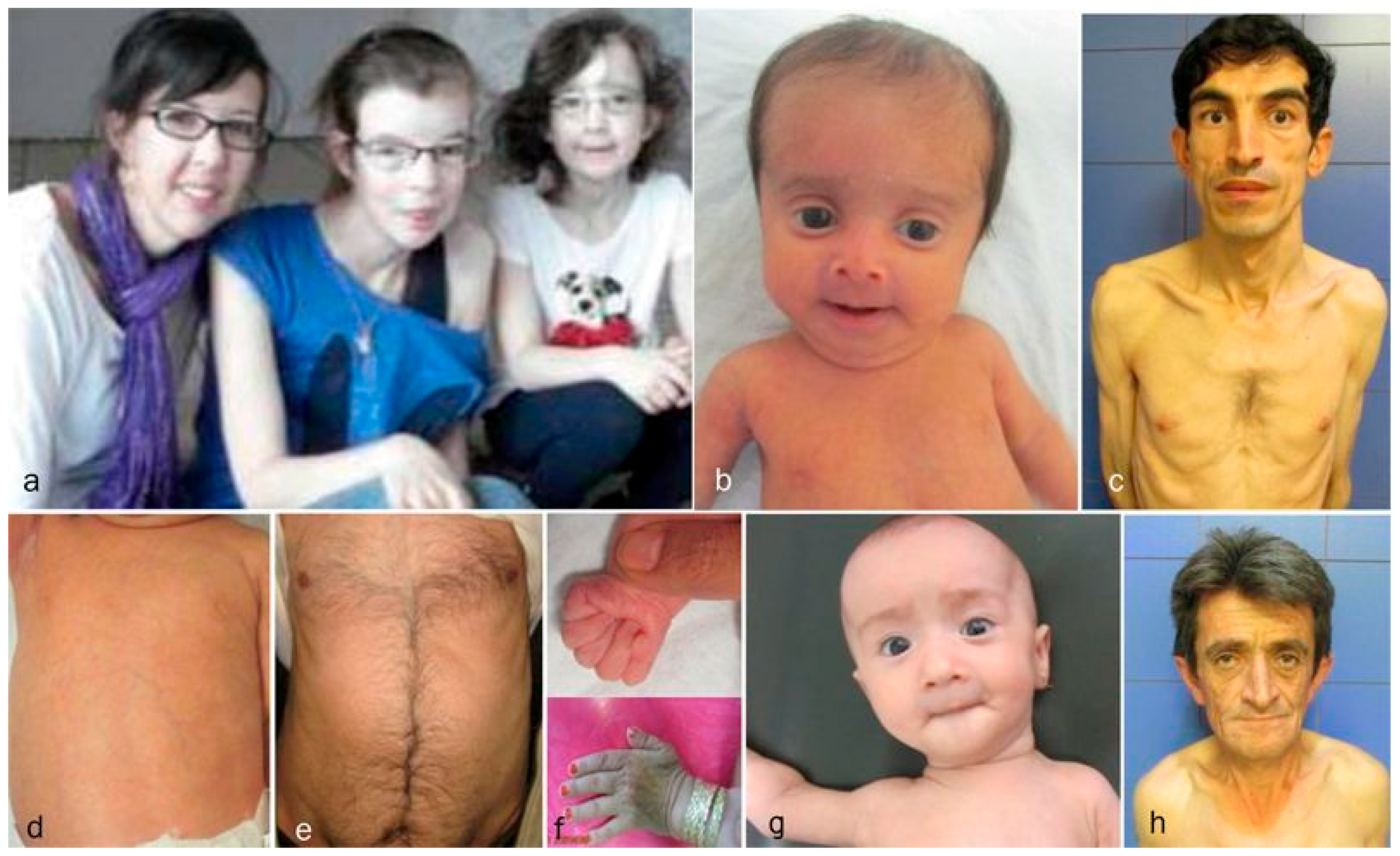
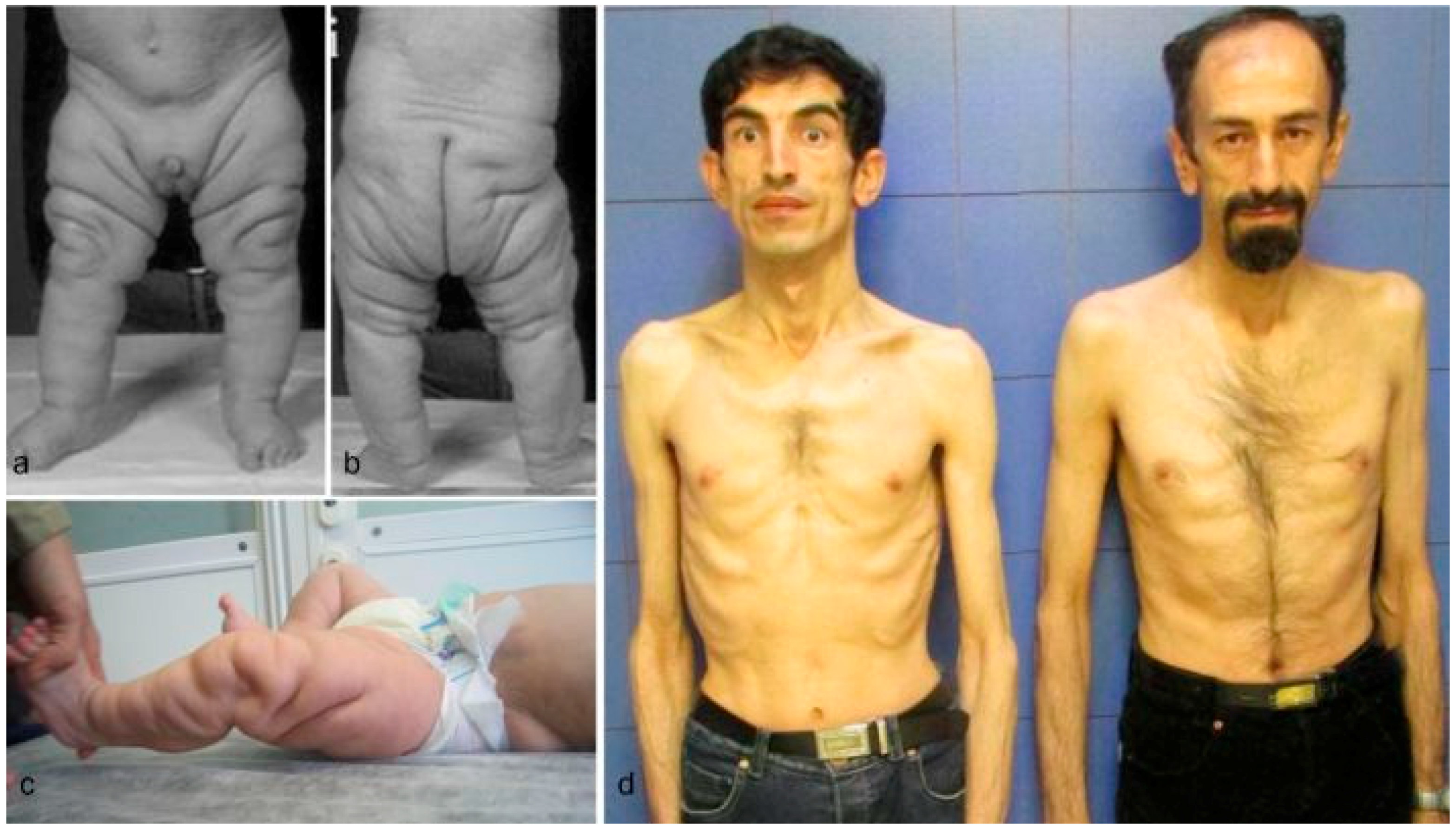

| Diagnosis | Cutis laxa IIA | Family I Case 1 | Family II Case 2 * | Cutis laxa IIB | Family III Case 3 | FamilyIV Case 4 | Family IV Case 5 | Family V Case 6 | GO | Family VI Case 7 | Family VII Case 8 | Family VIII Case 9 | Family IX Case 10 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ARCL2A | ARCL 2A | ARCL2B | ARCL2B | ARCL2B | ARCL2B | GO | GO | GO | GO | ||||
| Age at examination | 4 years | 12 years | 14 months | 35 years | 36 years | 1 year | 9 years | 3 years | 6 months | 40 years | |||
| Sex | F | M | F | M | M | F | F | M | F | M | |||
| Intra-uterine growth retardation | + | + | + | + | + | + | + | + | - | - | - | + | NE |
| height | 108 cm 95th centile | 140 cm 10th centile | 75 cm <50th centile | 173 cm <50th centile | 175 cm <50th centile | 68 cm <25th centile | 115 cm <3rd centile | 69 cm <3rd centile | 65 cm 50th centile | 158 cm <3rd centile | |||
| weight | 12 kg <3rd centile | 27 Kg <3rd centile | 6.8 kg <3rd centile | 40 kg <3rd centile | 45 <3rd centile | 6.2 kg <3rd centile | 24 kg <25th centile | 7.4 kg <3rd centile | 6.4 kg <50th centile | 50 kg <3rd centile | |||
| Head Circumference | 47.5 cm <50th centile | 51 cm <50th centile | 42 cm <2nd centile | 53 cm <50th centile | 54.5 <50th centile | 40 cm <2nd centile | 50 cm <50th centile | 42 cm <2nd centile | 41 cm <25th centile | 57.5 cm <98th centile | |||
| Facial features | |||||||||||||
| Aged appearance | + | + | +/− | + | + | + | + | + | + | + | + | + | + |
| Persistent open fontanel | + | NE | + | + | - | NE | NE | + | − | NE | − | − | NE |
| Bossing forehead/broad forehead | + | − | − | + | + | − | − | + | − | − | + | − | − |
| Downslanting palpebral fissures | + | + | + | − | − | − | − | − | − | − | − | − | − |
| Reverse V eyebrows | + | + | + | − | − | − | − | − | − | − | − | − | − |
| Long philtrum | + | + | + | − | − | − | − | − | − | − | − | − | − |
| Anteverted nares | + | + | + | − | − | − | − | − | − | − | − | − | − |
| Blue sclera | + | − | − | + | + | − | − | + | − | − | − | − | − |
| Triangular face | − | − | + | + | − | + | + | − | − | − | − | − | |
| Thin nose | − | − | + | + | + | + | + | − | − | − | − | − | |
| Long face | − | − | + ** | − | + | + | − | − | − | − | − | − | |
| prognathism | − | − | + ** | − | + | + | − | − | − | − | − | − | |
| Maxillary hypoplasia | − | − | − | − | − | − | − | + | + | + | − | + | |
| Oblique furrowing extending from the outer canthus to the lateral border of the supraorbital ridge | − | − | − | − | − | − | − | + | − | + | + | − | |
| Drooping/sagging cheeks | + | + | − | − | − | − | − | − | + | + | + | +/− | − |
| Neurological abnormalities | |||||||||||||
| Developmental delay | + | + | + | + | + | + | + | + | − | − | − | − | − |
| Intellectual disability | + | + | Very mild | + | + | + | + | + | − | − | − | − | − |
| Microcephaly | − | − | + | + | − | − | + | − | − | − | − | − | |
| Hypotonia | + | + | + at birth | + | + | − | − | + | − | − | − | − | − |
| Eye abnormalities | − | ||||||||||||
| Myopia | + | + | − | − | NE | − | − | − | − | − | − | − | − |
| Strabismus | + | − | + | − | + | − | − | − | − | − | − | − | − |
| Skeleton | |||||||||||||
| Pectus excavatum | + | − | + | + | − | − | − | + | − | − | − | − | − |
| Osteopenia | + | + | − | + | + | NE | NE | NE | + | + | + | + | + |
| Fractures | − | − | +/− | − | − | − | − | + | + | − | − | + | |
| Congenital hip dislocation | + | + | − | + | − | − | − | + | +/− | − | − | + | − |
| Skin & joints | |||||||||||||
| Hyperextensibility of joints | + | + | + | + | + | +/− | +/− | + | + | + | + | + | − |
| Dislocations | − | − | + | - | − | + | − | − | − | − | − | ||
| Adducted thumbs | − | − | + | + | + | + | + | − | + | − | − | − | |
| Clenched hands | − | − | + | + | − | − | + | − | + | − | − | − | |
| Increased palmar creases | + | + | + | + | + | + | + | + | + | + | |||
| Wrinkled skin | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Severe wrinkling of skin on dorsum of hands and feet | − | − | + | + | + | + | + | − | + | − | − | − | |
| Visible veins on the chest | − | − | + | + | + | + | + | − | − | + | − | − | |
| Inguinal hernia | + | − | + | + | − | − | − | − | − | − | + | − | − |
| Umbilical hernia | − | − | + | − | − | − | + | − | − | − | + | − | |
| Lipodystrophy | − | − | + | − | + | + | + | − | − | − | − | − | |
| Flat feet | + | − | + | + | + | + | + | + | − | − | − | + | |
| Causative gene Mutation cDNA Mutation protein | ATP6V0A2 | c.2255C>T homozygous mutation in ATP6V0A2 novel | c.754delT homozygous mutation in ATP6V0A2 22 | c.797 G>A homozygous mutation in PYCR1 gene 2,3 | c. 355C>T homozygous mutation in PYCR1 gene 2 | c. 355C>T homozygous mutation in PYCR1 gene 2 | c.572G>A homozygous mutation in PYCR1 gene 15 | c.-1_1GA>CT homozygous mutation in GORAB gene 21 | c.-1_1GA>CT homozygous mutation in GORAB gene | c.391C>T homozygous mutation in GORAB gene novel | c.75_76delinsCT homozygous mutation in GORAB gene 21 | ||
| other | Glycosylation abnormalitites | Glycosylation abnoramalities | Cleft palate | Small ASD | |||||||||
| Clinical Features | ARCL2A Total Cases from Literature | ARCL2A Present Cases | ARCL2B Total Cases from the Literature | ARCL2B Present Cases | GO Total Cases from the Literature | GO Present Cases | |||
|---|---|---|---|---|---|---|---|---|---|
| Number of Positive Cases from Number of Evaluated Cases | Frequency (%) | Number of Positive Cases from Number of Evaluated Cases | Frequency (%) | Number of Positive Cases From Number of Evaluated Cases | Frequency (%) | ||||
| Intra-Uterine Growth Retardation | 7/17 | 41% | 2/2 | 78/83 | 94% | 4/4 | 0/9 | 0% | 1/3 |
| Postnatal Growth Delay | 10/24 | 42% | 0/2 | 43/64 | 67% | 4/4 | 8/52 | 15% | 3/4 |
| Aged Appearance | 25/25 | 100% | 2/2 | 89/87 | 98% | 4/4 | 49/53 | 92% | 4/4 |
| Persistent open Fontanel | 53/58 | 91% | 1/1 | 30/40 | 75% | 1/2 | 2/12 | 17% | 0/2 |
| Blue sclera | 2/4 | 50% | 1/2 | 23/48 | 48% | 2/2 | 1/19 | 5% | 0/4 |
| Intellectual Disability | 45/58 | 78% | 2/2 | 86/92 | 93% | 4/4 | 3/53 | 6% | 0/4 |
| Microcephaly | 33/47 | 70% | 0/2 | 32/49 | 65% | 2/4 | 1/53 | 2% | 1/4 |
| Hypotonia | 29/35 | 83% | 2/2 | 33/46 | 72% | 2/4 | 13/38 | 34% | 0/4 |
| Seizures | 13/53 | 25% | 0/2 | 14/62 | 23% | 0/4 | 1/49 | 2% | 0/4 |
| Athetoid/Dystonic Movements | 1/15 | 6% | 0/2 | 17/85 | 20% | 0/4 | 0/49 | 0% | 0/4 |
| Corpus Callosum Dysgenesis | 2/20 | 10% | NE | 26/53 | 49% | NE | 0/3 | 0% | NE |
| Pachygryria | 30/46 | 65% | NE | 0/53 | 0% | NE | 0/3 | 0% | NE |
| Strabismus | 21/48 | 44% | 1/2 | 17/48 | 35% | 1/4 | 0/3 | 0% | 0/4 |
| Cataract/corneal clouding | 1/38 | 3% | 0/2 | 13/88 | 15% | 0/4 | 0/3 | 0% | 0/4 |
| Osteopenia | NE | NE | 1/2 | 38/52 | 73% | 1/1 | 50/52 | 96% | 4/4 |
| Fractures | 5/55 | 9% | 0/2 | 3/23 | 13% | 0/4 | 39/53 | 74% | 2/4 |
| Congenital hip dislocation | 9/15 | 60% | 1/2 | 53/86 | 62% | 1/4 | 36/53 | 68% | 1/4 |
| Wormian bonses | NE | NE | 0/2 | 20/32 | 63% | NE | 15/36 | 42% | NE |
| Hyperextensibility of joints | 40/46 | 87% | 2/2 | 75/79 | 95% | 4/4 | 45/46 | 98% | 3/4 |
| Dislocations | 0/7 | 0% | 0/2 | 2/8 | 25% | 2/4 | NE | NE | 0/4 |
| Adducted thumbs | 0/5 | 0% | 0/2 | 28/45 | 62% | 4/4 | NE | NE | 1/4 |
| Wrinkled skin | 60/60 | 100% | 2/2 | 93/93 | 100% | 4/4 | 53/53 | 100% | 4/4 |
| Visible veins on the chest | NE | NE | 0/2 | 46/53 | 87% | 4/4 | 9/43 | 21% | 0/4 |
| Hernia | 17/36 | 47% | 1/2 | 33/84 | 39% | 2/4 | 1/43 | 2% | 2/4 |
| Flat feet | 1/3 | 33% | 1/2 | 5/7 | 71% | 3/4 | NE | NE | 1/4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kariminejad, A.; Afroozan, F.; Bozorgmehr, B.; Ghanadan, A.; Akbaroghli, S.; Khorram Khorshid, H.R.; Mojahedi, F.; Setoodeh, A.; Loh, A.; Tan, Y.X.; et al. Discriminative Features in Three Autosomal Recessive Cutis Laxa Syndromes: Cutis Laxa IIA, Cutis Laxa IIB, and Geroderma Osteoplastica. Int. J. Mol. Sci. 2017, 18, 635. https://doi.org/10.3390/ijms18030635
Kariminejad A, Afroozan F, Bozorgmehr B, Ghanadan A, Akbaroghli S, Khorram Khorshid HR, Mojahedi F, Setoodeh A, Loh A, Tan YX, et al. Discriminative Features in Three Autosomal Recessive Cutis Laxa Syndromes: Cutis Laxa IIA, Cutis Laxa IIB, and Geroderma Osteoplastica. International Journal of Molecular Sciences. 2017; 18(3):635. https://doi.org/10.3390/ijms18030635
Chicago/Turabian StyleKariminejad, Ariana, Fariba Afroozan, Bita Bozorgmehr, Alireza Ghanadan, Susan Akbaroghli, Hamid Reza Khorram Khorshid, Faezeh Mojahedi, Aria Setoodeh, Abigail Loh, Yu Xuan Tan, and et al. 2017. "Discriminative Features in Three Autosomal Recessive Cutis Laxa Syndromes: Cutis Laxa IIA, Cutis Laxa IIB, and Geroderma Osteoplastica" International Journal of Molecular Sciences 18, no. 3: 635. https://doi.org/10.3390/ijms18030635
APA StyleKariminejad, A., Afroozan, F., Bozorgmehr, B., Ghanadan, A., Akbaroghli, S., Khorram Khorshid, H. R., Mojahedi, F., Setoodeh, A., Loh, A., Tan, Y. X., Escande-Beillard, N., Malfait, F., Reversade, B., Gardeitchik, T., & Morava, E. (2017). Discriminative Features in Three Autosomal Recessive Cutis Laxa Syndromes: Cutis Laxa IIA, Cutis Laxa IIB, and Geroderma Osteoplastica. International Journal of Molecular Sciences, 18(3), 635. https://doi.org/10.3390/ijms18030635