
Strategies for Overcoming Resistance in Tumours Harboring BRAF Mutations

Abstract
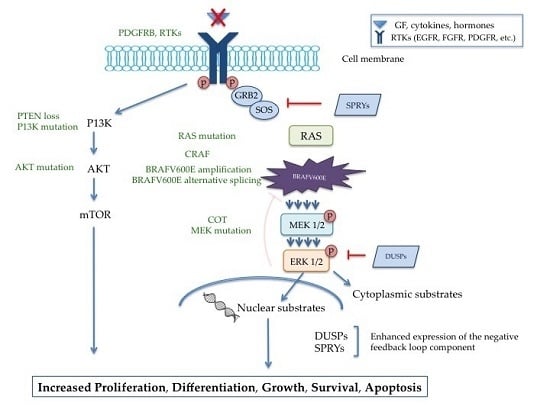
:
1. Introduction
2. Involvement of BRAF in the MAPK Pathway
Regulation of the MAPK Pathway
3. Conferred Resistance Mechanisms in BRAFV600E Tumours
3.1. Resistance Through MAPK Pathway Reactivation
3.2. Resistance Involving Insensitivity to MAPK Regulators
3.3. Other Mechanisms of Resistance
4. Challenges Encountered by Colorectal Cancer (CRC) Patients with BRAFV600E Mutation
Evidence of Specific Resistance Mechanisms in BRAFV600E Mutated CRC
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | protein kinase B |
| ATP | adenosine triphosphate |
| BRAF | v-RAF murine sarcoma viral oncogene homolog B |
| CRC | colorectal cancer |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular signal-regulated kinase |
| HSP90 | heat shock protein 90 |
| LKB1 | liver kinase B1 |
| MAPK | mitogen activated protein kinase |
| MEK | mitogen-activated protein kinase kinase |
| mTOR | mammalian target of rapamycin |
| PCR | polymerase chain reaction |
| PTEN | phosphatase and tensin homolog |
| RTK | receptor tyrosine kinase |
| TSC2 | tuberous sclerosis complex 2 |
References
- Niault, T.S.; Baccarini, M. Targets of RAF in tumorigenesis. Carcinogenesis 2010, 31, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Perrimon, N. The torso receptor protein-tyrosine kinase signaling pathway: An endless story. Cell 1993, 74, 219–222. [Google Scholar] [CrossRef]
- Ikawa, S.; Fukui, M.; Ueyama, Y.; Tamaoki, N.; Yamamoto, T.; Toyoshima, K. BRAF, a new member of the RAF family, is activated by DNA rearrangement. Mol. Cell. Biol. 1988, 8, 2651–2654. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [PubMed]
- Cohen, Y.; Xing, M.; Mambo, E.; Guo, Z.; Wu, G.; Trink, B.; Beller, U.; Westra, W.H.; Ladenson, P.W.; Sidransky, D. BRAF mutation in papillary thyroid carcinoma. J. Natl. Cancer Inst. 2003, 95, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Singer, G.; Oldt, R.; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Ie, M.S. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nakayama, N.; Kurman, R.J.; Cope, L.; Pohl, G.; Samuels, Y.; Velculescu, V.E.; Wang, T.; Shih, I. Sequence mutations and amplification of Pik3ca and Akt2 genes in purified ovarian serous neoplasms. Cancer Biol. Ther. 2006, 5, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Volpe, P.; Feldman, M.; Kumar, M.; Rishi, I.; Gerrero, R.; Einhorn, E.; Herlyn, M.; Minna, J.; Nicholson, A. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002, 62, 6997–7000. [Google Scholar] [PubMed]
- Yuen, S.T.; Davies, H.; Chan, T.L.; Ho, J.W.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Tsui, W.W.; Chan, A.S. Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res. 2002, 62, 6451–6455. [Google Scholar] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J. High frequency of BRAF mutations in nevi. Nat. Genet. 2002, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.S.; Palmedo, G.; Flaig, M.J.; Puchta, U.; Reckwerth, A.; Rütten, A.; Mentzel, T.; Hügel, H.; Hantschke, M.; Schmid-Wendtner, M. Mutations of the BRAF gene in benign and malignant melanocytic lesions. J. Investig. Dermatol. 2003, 121, 1160–1162. [Google Scholar] [CrossRef] [PubMed]
- Braig, M.; Schmitt, C.A. Oncogene-induced senescence: Putting the brakes on tumor development. Cancer Res. 2006, 66, 2881–2884. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Tufano, R.P.; Tufaro, A.P.; Basaria, S.; Ewertz, M.; Rosenbaum, E.; Byrne, P.J.; Wang, J.; Sidransky, D.; Ladenson, P.W. Detection of BRAF mutation on fine needle aspiration biopsy specimens: A new diagnostic tool for papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2004, 89, 2867–2872. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [PubMed]
- Mercer, K.E.; Pritchard, C.A. RAF proteins and cancer: BRAF is identified as a mutational target. Biochim. Biophys. Acta Rev. Cancer 2003, 1653, 25–40. [Google Scholar] [CrossRef]
- Houben, R.; Becker, J.C.; Kappel, A.; Terheyden, P.; Bröcker, E.; Goetz, R.; Rapp, U.R. Constitutive Activation of the RAS-RAF signaling pathway in metastatic melanoma is associated with poor prognosis. J. Carcinog. 2004, 3, 6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ogino, S.; Shima, K.; Meyerhardt, J.A.; McCleary, N.J.; Ng, K.; Hollis, D.; Saltz, L.B.; Mayer, R.J.; Schaefer, P.; Whittom, R. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: Results from intergroup trial CALGB 89803. Clin. Cancer Res. 2012, 18, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Alzahrani, A.S.; Carson, K.A.; Viola, D.; Elisei, R.; Bendlova, B.; Yip, L.; Mian, C.; Vianello, F.; Tuttle, R.M. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA 2013, 309, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.J.; Cobb, M.H. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 1997, 9, 180–186. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. In MAP Kinase Signaling Protocols; Springer: New York, NY, USA, 2010; pp. 3–38. [Google Scholar]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W. Meaningful relationships: The regulation of the RAS/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000, 351, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Troppmair, J.; Bruder, J.T.; App, H.; Cai, H.; Liptak, L.; Szeberenyi, J.; Cooper, G.M.; Rapp, U.R. RAS controls coupling of growth factor receptors and protein kinase C in the membrane to RAF-1 and BRAF protein serine kinases in the cytosol. Oncogene 1992, 7, 1867–1873. [Google Scholar] [PubMed]
- Chen, R.H.; Sarnecki, C.; Blenis, J. Nuclear localization and regulation of ERK- and RSK-encoded protein kinases. Mol. Cell. Biol. 1992, 12, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Weinberg, R.A. RAS oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Eblen, S.T.; Slack-Davis, J.K.; Tarcsafalvi, A.; Parsons, J.T.; Weber, M.J.; Catling, A.D. Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol. Cell. Biol. 2004, 24, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.G.; Li, P.; Marsden, L.; Williams, N.; Roberts, T.M.; Sturgill, T.W. RAF-1 is a potential substrate for mitogen-activated protein kinase in vivo. Biochem. J. 1991, 277, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, M.K.; Müller, J.; Ritt, D.A.; Zhou, M.; Zhou, X.Z.; Copeland, T.D.; Conrads, T.P.; Veenstra, T.D.; Lu, K.P.; Morrison, D.K. Regulation of RAF-1 by direct feedback phosphorylation. Mol. Cell 2005, 17, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Brummer, T.; Naegele, H.; Reth, M.; Misawa, Y. Identification of novel ERK-mediated feedback phosphorylation sites at the C-terminus of BRAF. Oncogene 2003, 22, 8823–8834. [Google Scholar] [CrossRef] [PubMed]
- Sturgill, T.W.; Ray, L.B.; Erikson, E.; Maller, J.L. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature 1988, 334, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, N.K.; Price, D.J.; Kyriakis, J.M.; Pelech, S.; Sanghera, J.; Avruch, J. An array of insulin-activated, proline-directed serine/threonine protein kinases phosphorylate the P70 S6 kinase. J. Biol. Chem. 1992, 267, 3325–3335. [Google Scholar] [PubMed]
- Wang, L.; Gout, I.; Proud, C.G. Cross-talk between the ERK and P70 S6 kinase (S6k) signaling pathways. MEK-dependent activation of S6k2 in cardiomyocytes. J. Biol. Chem. 2001, 276, 32670–32677. [Google Scholar] [CrossRef] [PubMed]
- Camps, M.; Nichols, A.; Gillieron, C.; Antonsson, B.; Muda, M.; Chabert, C.; Boschert, U.; Arkinstall, S. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science 1998, 280, 1262–1265. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, S.; Gimond, C.; Chambard, J.; Touboul, T.; Roux, D.; Pouysségur, J.; Pagès, G. Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol. Cell. Biol. 2005, 25, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Shima, H.; Katagiri, C.; Kikuchi, K. Activation of ERK induces phosphorylation of MAPK phosphatase-7, a JNK specific phosphatase, at Ser-446. J. Biol. Chem. 2003, 278, 32448–32456. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Miyazaki, S.; Tanimura, S.; Kohno, M. Efficient suppression of FGF-2-induced ERK activation by the cooperative interaction among mammalian sprouty isoforms. J. Cell Sci. 2005, 118, 5861–5871. [Google Scholar] [CrossRef] [PubMed]
- Hacohen, N.; Kramer, S.; Sutherland, D.; Hiromi, Y.; Krasnow, M.A. Sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the drosophila airways. Cell 1998, 92, 253–263. [Google Scholar] [CrossRef]
- Alvarez, E.; Northwood, I.C.; Gonzalez, F.A.; Latour, D.A.; Seth, A.; Abate, C.; Curran, T.; Davis, R.J. Pro-Leu-Ser/Thr-Pro is a consensus primary sequence for substrate protein phosphorylation. Characterization of the phosphorylation of c-Myc and c-Jun proteins by an epidermal growth factor receptor threonine 669 protein kinase. J. Biol. Chem. 1991, 266, 15277–15285. [Google Scholar] [PubMed]
- Arnaud, M.; Crouin, C.; Deon, C.; Loyaux, D.; Bertoglio, J. Phosphorylation of GRB2-associated binder 2 on serine 623 by ERK/MAPK regulates its association with the phosphatase SHP-2 and decreases STAT5 activation. J. Immunol. 2004, 173, 3962–3971. [Google Scholar] [CrossRef]
- Langlois, W.J.; Sasaoka, T.; Saltiel, A.R.; Olefsky, J.M. Negative feedback regulation and desensitization of insulin-and epidermal growth factor-stimulated P21RAS activation. J. Biol. Chem. 1995, 270, 25320–25323. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, F.; D’Alessandris, C.; Federici, M.; Laratta, E.; del Guerra, S.; del Prato, S.; Marchetti, P.; Lauro, R.; Perticone, F.; Sesti, G. Activation of the hexosamine pathway leads to phosphorylation of insulin receptor substrate-1 on Ser307 and Ser612 and impairs the phosphatidylinositol 3-Kinase/Akt/mammalian target of rapamycin insulin biosynthetic pathway in rin pancreatic Β-Cells. Endocrinology 2004, 145, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Ito, H.; Kamei, K.; Inaguma, Y.; Iwamoto, I.; Saga, S. Phosphorylation of Αb-crystallin in mitotic cells and identification of enzymatic activities responsible for phosphorylation. J. Biol. Chem. 1998, 273, 28346–28354. [Google Scholar] [CrossRef] [PubMed]
- Saal, S.K.G.; Parsons, R. Is the small heat shock protein Αb-crystallin an oncogene? J. Clin. Investig. 2006, 116, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Cruzalegui, F.H.; Cano, E.; Treisman, R. ERK activation induces phosphorylation of ELK-1 at multiple S/TP motifs to high stoichiometry. Oncogene 1999, 18, 7948–7957. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.O.; MacKeigan, J.P.; Blenis, J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol. Cell. Biol. 2004, 24, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.; Davis, R.J.; McLaren, A.; Cohen, P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003, 22, 3876–3886. [Google Scholar] [CrossRef] [PubMed]
- Milne, D.M.; Campbell, D.G.; Caudwell, F.B.; Meek, D.W. Phosphorylation of the tumor suppressor protein P53 by mitogen-activated protein kinases. J. Biol. Chem. 1994, 269, 9253–9260. [Google Scholar] [PubMed]
- Yeh, P.Y.; Chuang, S.; Yeh, K.; Song, Y.C.; Chang, L.L.; Cheng, A. Phosphorylation of P53 on Thr55 by ERK2 is necessary for doxorubicin-induced P53 activation and cell death. Oncogene 2004, 23, 3580–3588. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.C.; Greene, L.A. Nerve growth factor (NGF) down-regulates the Bcl-2 homology 3 (BH3) domain-only protein BIM and suppresses its proapoptotic activity by phosphorylation. J. Biol. Chem. 2002, 277, 49511–49516. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.A.; Morrice, N.; Brady, S.; Magee, G.; Pathak, S.; Clarke, P.R. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK/MAPK. Nat. Cell Biol. 2003, 5, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Scheid, M.P.; Schubert, K.M.; Duronio, V. Regulation of bad phosphorylation and association with Bcl-Xl by the MAPK/ERK kinase. J. Biol. Chem. 1999, 274, 31108–31113. [Google Scholar] [CrossRef] [PubMed]
- Garnovskaya, M.N.; Mukhin, Y.V.; Vlasova, T.M.; Grewal, J.S.; Ullian, M.E.; Tholanikunnel, B.G.; Raymond, J.R. Mitogen-induced rapid phosphorylation of serine 795 of the retinoblastoma gene product in vascular smooth muscle cells involves ERK activation. J. Biol. Chem. 2004, 279, 24899–24905. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Gabuzda, D. Mitogen-activated protein kinase phosphorylates and regulates the HIV-1 VIF protein. J. Biol. Chem. 1998, 273, 29879–29887. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, O.; Pagès, G.; Gimond, C. The dual-specificity MAP kinase phosphatases: Critical roles in development and cancer. Am. J. Physiol. Cell Physiol. 2010, 299, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.; Litvak, V.; Medvedovsky, H.; Zwang, Y.; Lev, S.; Yarden, Y. Sprouty fine-tunes EGF signaling through interlinked positive and negative feedback loops. Curr. Biol. 2003, 13, 297–307. [Google Scholar] [CrossRef]
- Therrien, M.; Chang, H.C.; Solomon, N.M.; Karim, F.D.; Wassarman, D.A.; Rubin, G.M. KSR, a novel protein kinase required for RAS signal transduction. Cell 1995, 83, 879–888. [Google Scholar] [CrossRef]
- Keller, E.T.; Fu, Z.; Brennan, M. The role of RAF kinase inhibitor protein (RKIP) in health and disease. Biochem. Pharmacol. 2004, 68, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Yeung, M.L.; Beach, S.; Shields, J.M.; Yeung, K.C. RKIP downregulates BRAF kinase activity in melanoma cancer cells. Oncogene 2005, 24, 3535–3540. [Google Scholar] [CrossRef] [PubMed]
- Brummer, T.; Martin, P.; Herzog, S.; Misawa, Y.; Daly, R.J.; Reth, M. Functional analysis of the regulatory requirements of BRAF and the BRAF V600E oncoprotein. Oncogene 2006, 25, 6262–6276. [Google Scholar] [CrossRef] [PubMed]
- Tsavachidou, D.; Coleman, M.L.; Athanasiadis, G.; Li, S.; Licht, J.D.; Olson, M.F.; Weber, B.L. Spry2 is an inhibitor of the RAS/extracellular signal-regulated kinase pathway in melanocytes and melanoma cells with wild-type BRAF but not with the V599e mutant. Cancer Res. 2004, 64, 5556–5559. [Google Scholar] [CrossRef] [PubMed]
- Pratilas, C.A.; Solit, D.B. Targeting the mitogen-activated protein kinase pathway: Physiological feedback and drug response. Clin. Cancer Res. 2010, 16, 3329–3334. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600e mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Messina, J.L.; Fedorenko, I.V.; Sondak, V.K.; Smalley, K.S. Paradoxical oncogenesis—The long-term effects of braf inhibition in melanoma. Nat. Rev. Clin. Oncol. 2013, 10, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Infante, J.R.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kwak, E.L.; Ryan, D.; Kurzrock, R. Phase 1–2 trial of the BRAF inhibitor dabrafenib (D) plus MEK inhibitor trametinib (T) in BRAF V600 mutant colorectal cancer (CRC): Updated efficacy and biomarker analysis. In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 30 May–3 June 2014.
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Oberholzer, P.A.; Kee, D.; Dziunycz, P.; Sucker, A.; Kamsukom, N.; Jones, R.; Roden, C.; Chalk, C.J.; Ardlie, K.; Palescandolo, E. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J. Clin. Oncol. 2012, 30, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Alcala, A.M.; Flaherty, K.T. BRAF inhibitors for the treatment of metastatic melanoma: Clinical trials and mechanisms of resistance. Clin. Cancer Res. 2012, 18, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. melanomas acquire resistance to BRAF V600E inhibition by RTK or NRAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; Macconaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraat, M.; Hooijdonk, C.G.G.; Ubink, I.; Besselink, N.J.; Pieterse, M.; Veldhuis, W.; van Stralen, M.; Meijer, E.F.; Willems, S.M.; Hadders, M.A.; et al. Detailed imaging and genetic analysis reveal a secondary BRAF L505h resistance mutation and extensive intrapatient heterogeneity in metastatic BRAF mutant melanoma patients treated with vemurafenib. Pigment Cell Melanoma Res. 2015, 28, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Wagenaar, T.R.; Ma, L.; Roscoe, B.; Park, S.M.; Bolon, D.N.; Green, M.R. Resistance to vemurafenib resulting from a novel mutation in the BRAF V600E kinase domain. Pigment Cell Melanoma Res. 2014, 27, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF V600E. Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.I.; Widmer, D.S.; Narechania, A.; Eichhoff, O.; Freiberger, S.N.; Wenzina, J.; Cheng, P.F.; Mihic-Probst, D.; Desalle, R.; Dummer, R.; et al. Co-existence of BRAF and NRAS driver mutations in the same melanoma cells results in heterogeneity of targeted therapy resistance. Oncotarget 2016, 7, 77163–77174. [Google Scholar] [CrossRef] [PubMed]
- Danysh, B.P.; Rieger, E.Y.; Sinha, D.K.; Evers, C.V.; Cote, G.J.; Cabanillas, M.E.; Hofmann, M.C. Long-term vemurafenib treatment drives inhibitor resistance through a spontaneous KRAS G12D mutation in a BRAF V600E papillary thyroid carcinoma model. Oncotarget 2016, 7, 30907–30923. [Google Scholar] [CrossRef] [PubMed]
- Ahronian, L.G.; Sennott, E.M.; van Allen, E.M.; Wagle, N.; Kwak, E.L.; Faris, J.E.; Godfrey, J.T.; Nishimura, K.; Lynch, K.D.; Mermel, C.H.; et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 2015, 5, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Sharma, S.V.; Shioda, T.; McDermott, U.; Ulman, M.; Ulkus, L.E.; Dias-Santagata, D.; Stubbs, H.; Lee, D.Y.; Singh, A.; et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008, 68, 4853–4861. [Google Scholar] [CrossRef] [PubMed]
- Grbovic, O.M.; Basso, A.D.; Sawai, A.; Ye, Q.; Friedlander, P.; Solit, D.; Rosen, N. V600e BRAF requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha Dias, S.; Friedlos, F.; Light, Y.; Springer, C.; Workman, P.; Marais, R. Activated BRAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005, 65, 10686–10691. [Google Scholar] [CrossRef] [PubMed]
- Maloney, A.; Clarke, P.A.; Workman, P. Genes and proteins governing the cellular sensitivity to Hsp90 inhibitors: A mechanistic perspective. Curr. Cancer Drug Targets 2003, 3, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, J.; Smith, D.L.; Jimenez, J.P.; Zhang, C.; Sequeira, M.; He, S.; Sang, J.; Bates, R.C.; Proia, D.A. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol. Cancer Ther. 2014, 13, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.; Haarberg, H.E.; Wood, E.; Rebecca, V.W.; Chen, Y.A.; Xiang, Y.; Ribas, A.; Lo, R.S.; Weber, J.S.; Sondak, V.K.; et al. The Hsp90 inhibitor Xl888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin. Cancer Res. 2012, 18, 2502–2514. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, M.; Yao, M.; Zhu, W. Effects of treatment with an Hsp90 inhibitor in tumors based on 15 phase II clinical trials. Mol. Clin. Oncol. 2016, 5, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Osman, I.; Polsky, D.; Panageas, K.S.; Daud, A.; Goydos, J.S.; Teitcher, J.; Wolchok, J.D.; Germino, F.J.; Krown, S.E.; et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin. Cancer Res. 2008, 14, 8302–8307. [Google Scholar] [CrossRef] [PubMed]
- Pacey, S.; Gore, M.; Chao, D.; Banerji, U.; Larkin, J.; Sarker, S.; Owen, K.; Asad, Y.; Raynaud, F.; Walton, M.; et al. A phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-Aag, Tanespimycin) in patients with metastatic melanoma. Investig. New Drugs 2012, 30, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. BRAF V600e is associated with disabled feedback inhibition of RAF/MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, F.; Ghiorzo, P.; Queirolo, P. Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget 2014, 5, 10206–10221. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Grammer, T.C.; Lemon, K.P.; Kazlauskas, A.; Blenis, J. PDGF- and insulin-dependent PP70S6K activation mediated by phosphatidylinositol-3-OH kinase. Nature 1994, 370, 71–75. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF V600e inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kong, X.; Ribas, A.; Lo, R.S. Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to BRAF V600e inhibition. Cancer Res. 2011, 71, 5067–5074. [Google Scholar] [CrossRef]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Aguissa-Toure, A.H.; Li, G. Genetic alterations of PTEN in human melanoma. Cell Mol. Life Sci. 2012, 69, 1475–1491. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Furney, S.J.; Stamp, G.; Rana, S.; Ricken, G.; Oduko, Y.; Saturno, G.; Springer, C.; Hayes, A.; Gore, M.; et al. Whole-genome sequencing reveals complex mechanisms of intrinsic resistance to BRAF inhibition. Ann. Oncol. 2014, 25, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Alicea, G.M.; Paraiso, K.H.; Lawrence, H.; Gibney, G.T.; Smalley, K.S. Vertical inhibition of the MAPK pathway enhances therapeutic responses in NRAS-mutant melanoma. Pigment Cell Melanoma Res. 2014, 27, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef] [PubMed]
- Marampon, F.; Ciccarelli, C.; Zani, B.M. Down-regulation of C-Myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol. Cancer 2006, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Zawistowski, J.S.; Bevill, S.M.; Goulet, D.R.; Stuhlmiller, T.J.; Beltran, A.S.; Olivares-Quintero, J.F.; Singh, D.; Sciaky, N.; Parker, J.S.; Rashid, N.U.; et al. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacological targeting of the P-TEFB complex. Cancer Discov. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Mancini, R.; Acunzo, M.; Romano, G.; Lagana, A.; Pisanu, M.E.; Malpicci, D.; Madonna, G.; Mallardo, D.; Capone, M.; et al. Mir-579-3p controls melanoma progression and resistance to target therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 5005–5013. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.N.; Kopetz, E.S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: Clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 2015, 6, 660–667. [Google Scholar] [PubMed]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies BRAF V600e amplification-mediated acquired BRAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF V600e inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; He, G.; Nelman-Gonzalez, M.; Ashorn, C.L.; Gallick, G.E.; Stukenberg, P.T.; Kirschner, M.W.; Kuang, J. Regulation of Cdc25C by ERK/MAP kinases during the G2/M transition. Cell 2007, 128, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Edwin, F.; Anderson, K.; Ying, C.; Patel, T.B. Intermolecular interactions of sprouty proteins and their implications in development and disease. Mol. Pharmacol. 2009, 76, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R.; Dayyani, F.; Gopal, Y.N.V.; Jiang, Z.; Wistuba, I.I.; Tang, X.M. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin. Cancer Res. 2013, 19, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Cercek, A.; O’Reilly, E.M.; Reidy, D.L.; Kemeny, N.; Wolinsky, T.; Capanu, M.; Gollub, M.J.; Rosen, N.; Berger, M.F.; et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin. Cancer Res. 2015, 21, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.; O’dwyer, P.; Lee, R.; Nolop, K.; Saltz, L. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. In Proceedings of the ASCO Annual Meeting, Hollywood, FL, USA, 18–20 October 2010; p. 3534.
- Samowitz, W.S.; Sweeney, C.; Herrick, J.; Albertsen, H.; Levin, T.R.; Murtaugh, M.A.; Wolff, R.K.; Slattery, M.L. Poor survival associated with the BRAF V600e mutation in microsatellite-stable colon cancers. Cancer Res. 2005, 65, 6063–6069. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Ura, T.; Shibata, N.; Takahari, D.; Shitara, K.; Nomura, M.; Kondo, C.; Mizota, A.; Utsunomiya, S.; Muro, K. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br. J. Cancer 2011, 104, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Heynen, G.J.; Germano, G.; Willems, S.M.; Evers, B.; Vecchione, L.; Gambino, V.; Lieftink, C.; Beijersbergen, R.L.; di Nicolantonio, F.; et al. PTPN11 is a central node in intrinsic and acquired resistance to targeted cancer drugs. Cell Rep. 2015, 12, 1978–1985. [Google Scholar] [CrossRef]
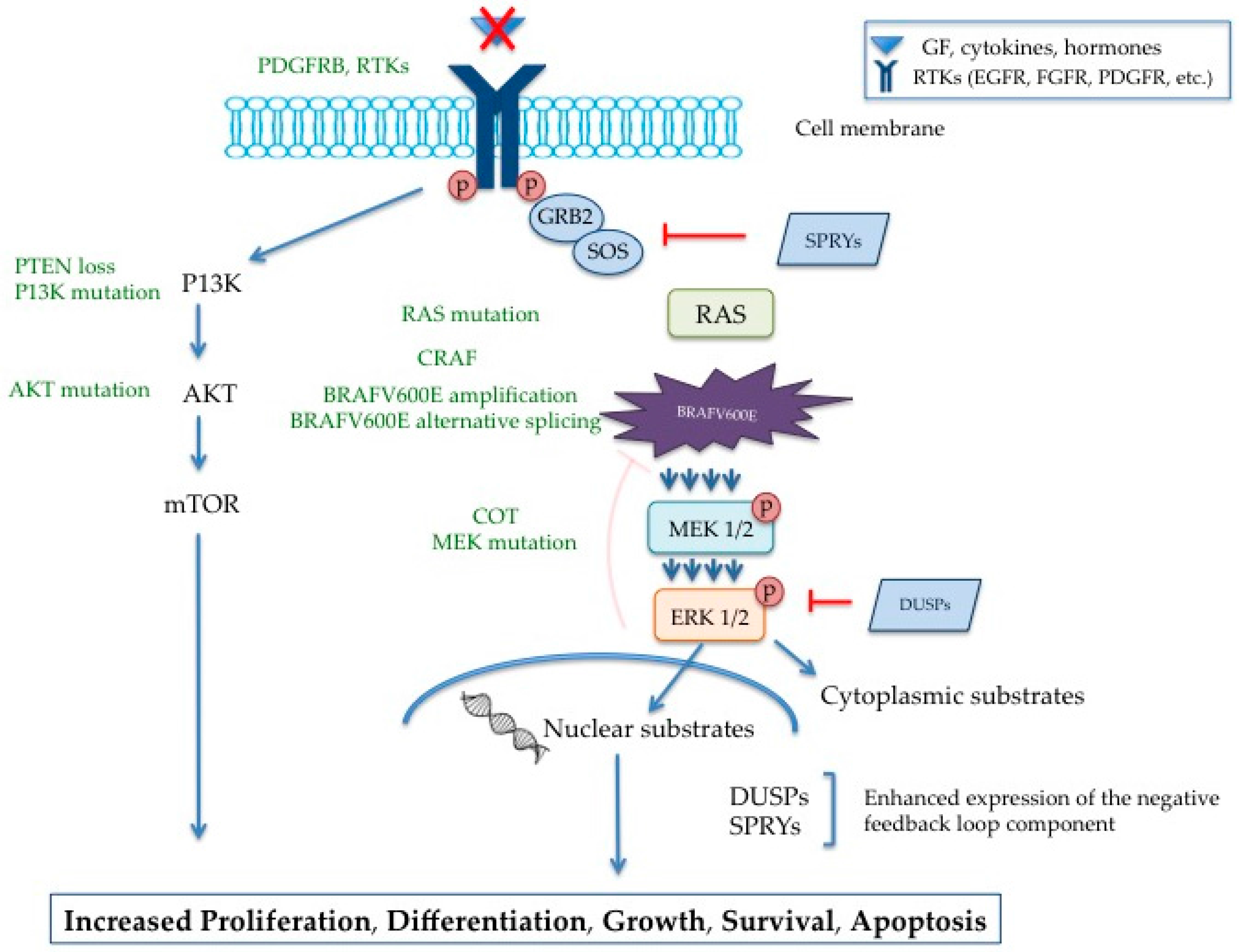

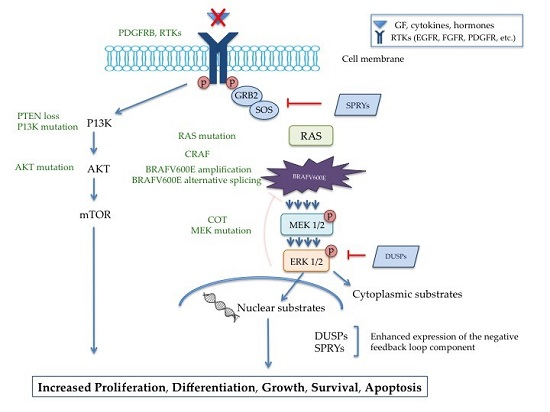{kind=link}
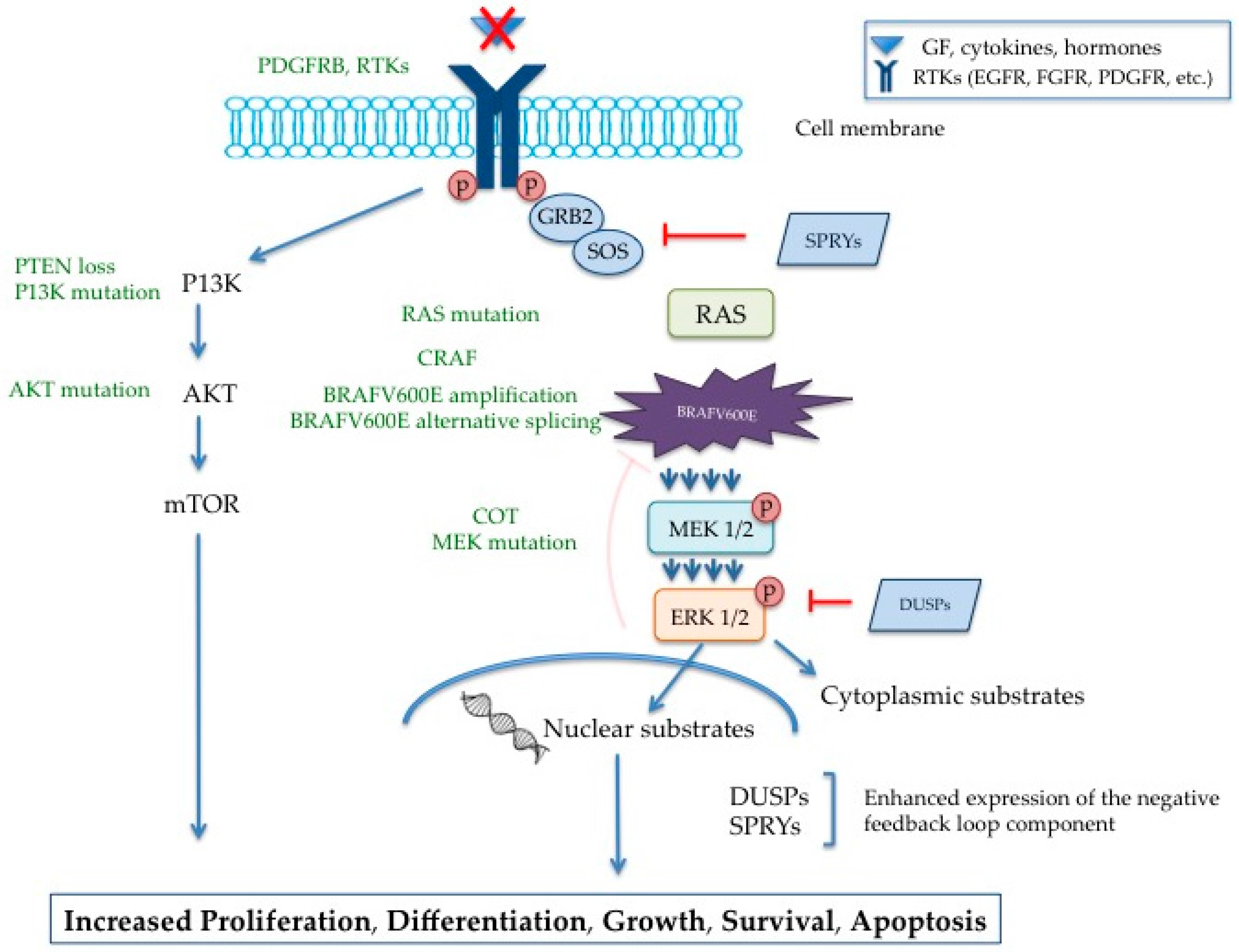{kind=link}
| Category | Protein | Effect of ERK Phosphorylation on Its Functions | Reference |
|---|---|---|---|
| Kinases and phosphatases | MEK1/2 | Either enhances its activity or reduces it depending on the phosphorylation site | [29] |
| CRAF | Inhibits its activity | [30,31] | |
| BRAF | Inhibits its activity | [32] | |
| RSK | Activation and further signal transduction | [33] | |
| S6K | Activation | [34,35] | |
| DUSPs | Negative feedback loop-indirectly via dephosphorylating ERK1/2 | [36,37,38] | |
| SPRYs | Negative feedback loop-directly inactivating upstream | [39,40] | |
| Signalling proteins | EGFR | Downregulation of the MAPK pathway | [41] |
| Gab2 * | Reduces its activation | [42] | |
| SOS * | Negative feedback mechanism via preventing its association with Gab2 | [43] | |
| IRS1 * | Impaired its downstream signalling | [44] | |
| TSC2 | Weakens its ability to pair with TSC1, therefore Impairs its ability to inhibit mTOR signalling | [45] | |
| Cytoskeletal proteins | Crystalline α | Anti-apoptotic protection | [46,47] |
| Transcription Factors | ELK * | Transcription of c-Fos | [47,48] |
| c-Fos * | Acts as a sensor for ERKs’ signal duration | [49] | |
| c-Jun * | Transcription of c-Jun | [50] | |
| p53 | Tumour suppressor protein, plays a role in cell cycle | [51,52] | |
| c-Myc * | Transcription | [41] | |
| Apoptotic proteins | BIM * | Inhibit its pro-apoptotic function | [53] |
| Caspase9 | Reduce its pro-apoptotic function | [54] | |
| Bad * | Inhibit its pro-apoptotic function | [55] | |
| Other proteins | RB * | Cell cycle progression | [56] |
| Vif * | Activates HIV-1 replication | [57] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obaid, N.M.; Bedard, K.; Huang, W.-Y. Strategies for Overcoming Resistance in Tumours Harboring BRAF Mutations. Int. J. Mol. Sci. 2017, 18, 585. https://doi.org/10.3390/ijms18030585
Obaid NM, Bedard K, Huang W-Y. Strategies for Overcoming Resistance in Tumours Harboring BRAF Mutations. International Journal of Molecular Sciences. 2017; 18(3):585. https://doi.org/10.3390/ijms18030585
Chicago/Turabian StyleObaid, Nourah Mohammad, Karen Bedard, and Weei-Yuarn Huang. 2017. "Strategies for Overcoming Resistance in Tumours Harboring BRAF Mutations" International Journal of Molecular Sciences 18, no. 3: 585. https://doi.org/10.3390/ijms18030585
APA StyleObaid, N. M., Bedard, K., & Huang, W.-Y. (2017). Strategies for Overcoming Resistance in Tumours Harboring BRAF Mutations. International Journal of Molecular Sciences, 18(3), 585. https://doi.org/10.3390/ijms18030585