
Epigenetic Regulation of BDNF Gene during Development and Diseases

Abstract
:1. Introduction
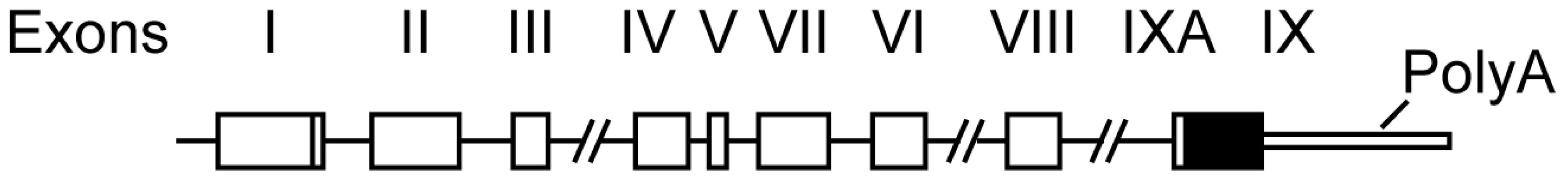
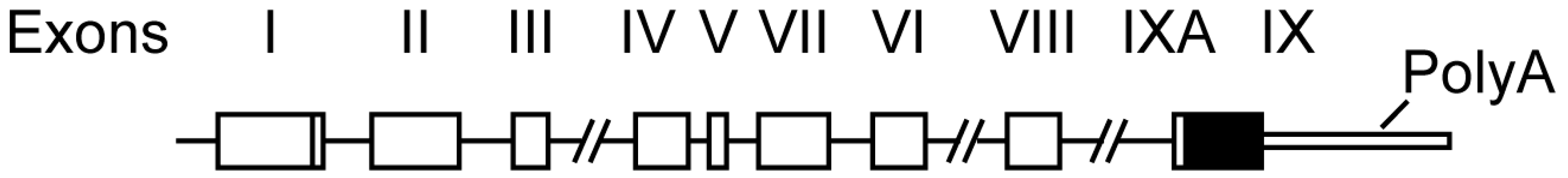
2. Bdnf Gene
3. Data-Mining Using ENCODE
4. Histone Modifications in Gene Regulation
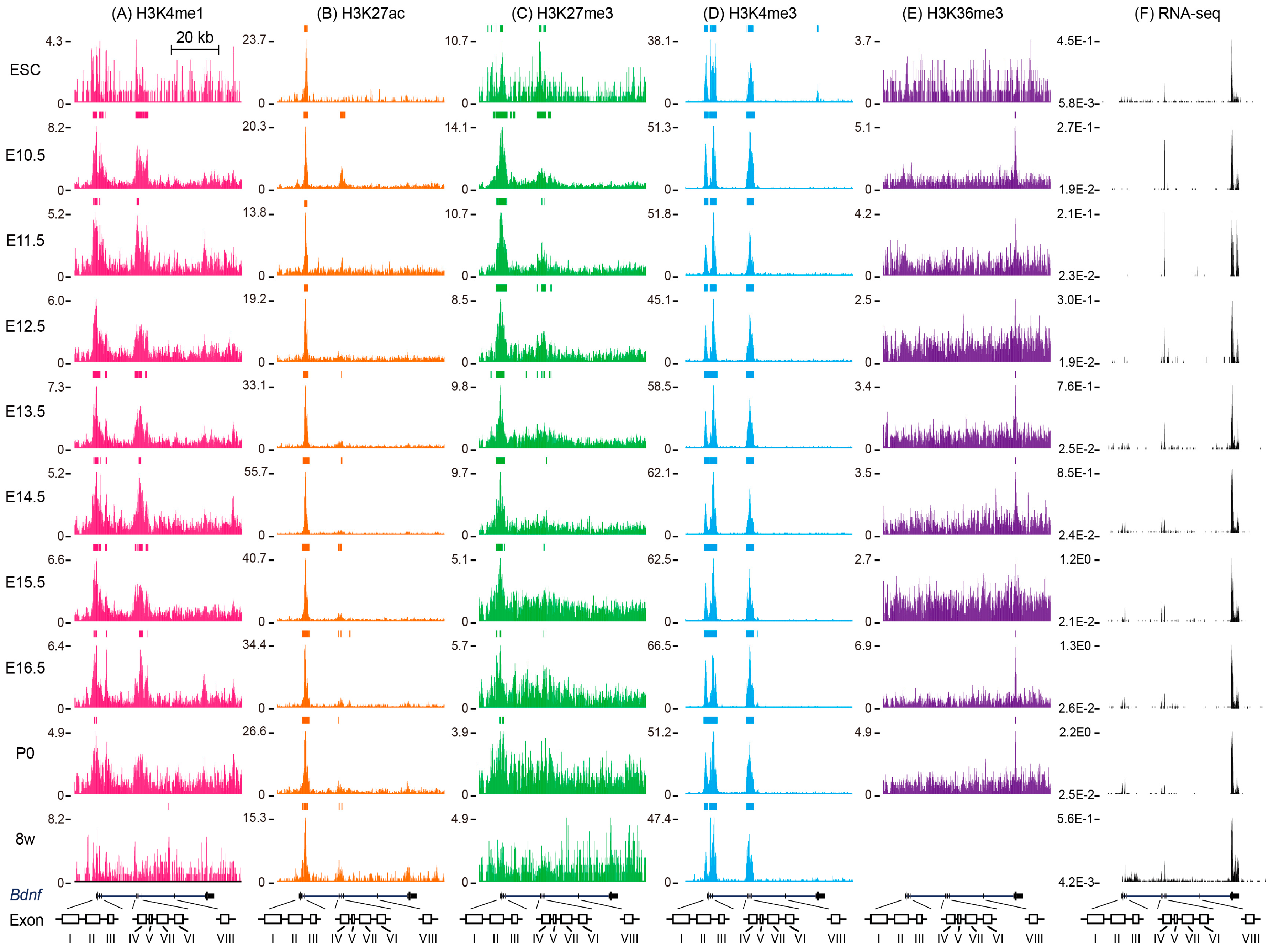
5. Histone Modifications around Mouse Bdnf
5.1. Histone Modifications around Mouse Bdnf in Embryonic Stem Cells
5.2. Histone Modifications around Mouse Bdnf during Brain Development
6. Epigenetics in Neurological Diseases
6.1. Regulation of BDNF by Histone Modifications in Huntington’s Disease
6.2. Regulation of Human BDNF by Histone Modifications in Alzheimer’s Disease
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jones, K.R.; Farinas, I.; Backus, C.; Reichardt, L.F. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 1994, 76, 989–999. [Google Scholar] [CrossRef]
- Schwartz, P.M.; Borghesani, P.R.; Levy, R.L.; Pomeroy, S.L.; Segal, R.A. Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning. Neuron 1997, 19, 269–281. [Google Scholar] [CrossRef]
- Lindsay, R.M. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J. Neurosci. 1988, 8, 2394–2405. [Google Scholar] [PubMed]
- Richner, M.; Ulrichsen, M.; Elmegaard, S.L.; Dieu, R.; Pallesen, L.T.; Vaegter, C.B. Peripheral nerve injury modulates neurotrophin signaling in the peripheral and central nervous system. Mol. Neurobiol. 2014, 50, 945–970. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, R.; Yamashita, T. Concept and molecular basis of axonal regeneration after central nervous system injury. Neurosci. Res. 2014, 78, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, R.M.; Thoenen, H.; Barde, Y.A. Placode and neural crest-derived sensory neurons are responsive at early developmental stages to brain-derived neurotrophic factor. Dev. Boil. 1985, 112, 319–328. [Google Scholar] [CrossRef]
- Kalcheim, C.; Gendreau, M. Brain-derived neurotrophic factor stimulates survival and neuronal differentiation in cultured avian neural crest. Brain Res. 1988, 469, 79–86. [Google Scholar] [CrossRef]
- Ichim, G.; Tauszig-Delamasure, S.; Mehlen, P. Neurotrophins and cell death. Exp. Cell Res. 2012, 318, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Alsina, B.; Vu, T.; Cohen-Cory, S. Visualizing synapse formation in arborizing optic axons in vivo: Dynamics and modulation by BDNF. Nat. Neurosci. 2001, 4, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Nagappan, G.; Ke, Y.; Sacktor, T.C.; Lu, B. BDNF facilitates L-LTP maintenance in the absence of protein synthesis through PKMζ. PLoS ONE 2011, 6, e21568. [Google Scholar] [CrossRef] [PubMed]
- Abidin, I.; Kohler, T.; Weiler, E.; Zoidl, G.; Eysel, U.T.; Lessmann, V.; Mittmann, T. Reduced presynaptic efficiency of excitatory synaptic transmission impairs LTP in the visual cortex of BDNF-heterozygous mice. Eur. J. Neurosci. 2006, 24, 3519–3531. [Google Scholar] [CrossRef] [PubMed]
- Abidin, I.; Eysel, U.T.; Lessmann, V.; Mittmann, T. Impaired gabaergic inhibition in the visual cortex of brain-derived neurotrophic factor heterozygous knockout mice. J. Physiol. 2008, 586, 1885–1901. [Google Scholar] [CrossRef] [PubMed]
- Soliman, F.; Glatt, C.E.; Bath, K.G.; Levita, L.; Jones, R.M.; Pattwell, S.S.; Jing, D.; Tottenham, N.; Amso, D.; Somerville, L.H.; et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science 2010, 327, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Tuszynski, M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2011, 10, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Pruunsild, P.; Kazantseva, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.R.; Lu, L.; Zhu, X.G.; Gong, J.P.; Shaham, Y.; Uhl, G.R. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006, 1067, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.R.; Walther, D.; Drgon, T.; Polesskaya, O.; Lesnick, T.G.; Strain, K.J.; de Andrade, M.; Bower, J.H.; Maraganore, D.M.; Uhl, G.R. Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson’s Disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2005, 134B, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Tognoli, C.; Rossi, F.; Di Cola, F.; Baj, G.; Tongiorgi, E.; Terova, G.; Saroglia, M.; Bernardini, G.; Gornati, R. Acute stress alters transcript expression pattern and reduces processing of proBDNF to mature BDNF in Dicentrarchus labrax. BMC Neurosci. 2010, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Baj, G.; Del Turco, D.; Schlaudraff, J.; Torelli, L.; Deller, T.; Tongiorgi, E. Regulation of the spatial code for bdnf mrna isoforms in the rat hippocampus following pilocarpine-treatment: A systematic analysis using laser microdissection and quantitative real-time PCR. Hippocampus 2013, 23, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Rosenbloom, K.R.; Sloan, C.A.; Malladi, V.S.; Dreszer, T.R.; Learned, K.; Kirkup, V.M.; Wong, M.C.; Maddren, M.; Fang, R.; Heitner, S.G.; et al. Encode data in the UCSC genome browser: Year 5 update. Nucleic Acids Res. 2013, 41, D56–D63. [Google Scholar] [CrossRef] [PubMed]
- Psych, E.C.; Akbarian, S.; Liu, C.; Knowles, J.A.; Vaccarino, F.M.; Farnham, P.J.; Crawford, G.E.; Jaffe, A.E.; Pinto, D.; Dracheva, S.; et al. The psychencode project. Nat. Neurosci. 2015, 18, 1707–1712. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Tesar, P.J.; Scacheri, P.C. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011, 21, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Chantalat, S.; Depaux, A.; Hery, P.; Barral, S.; Thuret, J.Y.; Dimitrov, S.; Gerard, M. Histone H3 trimethylation at lysine 36 is associated with constitutive and facultative heterochromatin. Genome Res. 2011, 21, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Schneider, R.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Boil. Chem. 2005, 280, 17732–17736. [Google Scholar] [CrossRef] [PubMed]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Amador-Arjona, A.; Cimadamore, F.; Huang, C.T.; Wright, R.; Lewis, S.; Gage, F.H.; Terskikh, A.V. SOX2 primes the epigenetic landscape in neural precursors enabling proper gene activation during hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E1936–E1945. [Google Scholar] [CrossRef] [PubMed]
- Baj, G.; D’Alessandro, V.; Musazzi, L.; Mallei, A.; Sartori, C.R.; Sciancalepore, M.; Tardito, D.; Langone, F.; Popoli, M.; Tongiorgi, E. Physical exercise and antidepressants enhance BDNF targeting in hippocampal CA3 dendrites: Further evidence of a spatial code for bdnf splice variants. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2012, 37, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Carretero, J.; Benvegnu, S.; Dotti, C.G.; Martin, M.G. Neuronal activity controls Bdnf expression via polycomb de-repression and CREB/CBP/JMJD3 activation in mature neurons. Nat. Commun. 2016, 7, 11081. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Martin-Segura, A.; Baliyan, S.; Ahmed, T.; Balschun, D.; Venero, C.; Martin, M.G.; Dotti, C.G. Aging triggers a repressive chromatin state at Bdnf promoters in hippocampal neurons. Cell Rep. 2016, 16, 2889–2900. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, N.; Fukuchi, M.; Hirai, A.; Chiba, Y.; Tamura, T.; Takahashi, N.; Tabuchi, A.; Tsuda, M.; Shiraishi, M. Differential epigenetic regulation of BDNF and NT-3 genes by trichostatin a and 5-aza-2′-deoxycytidine in Neuro-2a cells. Biochem. Biophys. Res. Commun. 2010, 394, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Koppel, I.; Timmusk, T. Differential regulation of bdnf expression in cortical neurons by class-selective histone deacetylase inhibitors. Neuropharmacology 2013, 75, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Cremins, J.E.; Sauria, M.E.; Sanyal, A.; Gerasimova, T.I.; Lajoie, B.R.; Bell, J.S.; Ong, C.T.; Hookway, T.A.; Guo, C.; Sun, Y.; et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 2013, 153, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Sams, D.S.; Nardone, S.; Getselter, D.; Raz, D.; Tal, M.; Rayi, P.R.; Kaphzan, H.; Hakim, O.; Elliott, E. Neuronal CTCF is necessary for basal and experience-dependent gene regulation, memory formation, and genomic structure of BDNF and Arc. Cell Rep. 2016, 17, 2418–2430. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent Bdnf gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Zheleznyakova, G.Y.; Cao, H.; Schioth, H.B. BDNF DNA methylation changes as a biomarker of psychiatric disorders: Literature review and open access database analysis. Behav. Brain Funct. BBF 2016, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- You, H.J.; Park, J.H.; Pareja-Galeano, H.; Lucia, A.; Shin, J.I. Targeting microRNAs involved in the BDNF signaling impairment in neurodegenerative diseases. NeuroMol. Med. 2016, 18, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Marullo, M.; Conforti, P.; MacDonald, M.E.; Tartari, M.; Cattaneo, E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. 2008, 18, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Gharami, K.; Xie, Y.; An, J.J.; Tonegawa, S.; Xu, B. Brain-derived neurotrophic factor over-expression in the forebrain ameliorates huntington’s disease phenotypes in mice. J. Neurochem. 2008, 105, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hayden, M.R.; Xu, B. BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J. Neurosci. 2010, 30, 14708–14718. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Vashishtha, M.; Ng, C.W.; Yildirim, F.; Gipson, T.A.; Kratter, I.H.; Bodai, L.; Song, W.; Lau, A.; Labadorf, A.; Vogel-Ciernia, A.; et al. Targeting H3K4 trimethylation in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, E3027–E3036. [Google Scholar] [CrossRef] [PubMed]
- Wilczek, C.; Chitta, R.; Woo, E.; Shabanowitz, J.; Chait, B.T.; Hunt, D.F.; Shechter, D. Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2a and H4 and the histone chaperone nucleoplasmin in Xenopus laevis eggs. J. Boil. Chem. 2011, 286, 42221–42231. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, T.; Arbez, N.; Stewart, J.C.; Chighladze, E.; Ross, C.A. PRMT5-mediated symmetric arginine dimethylation is attenuated by mutant huntingtin and is impaired in Huntington’s disease (HD). Cell Cycle 2015, 14, 1716–1729. [Google Scholar] [CrossRef] [PubMed]
- Girardot, M.; Hirasawa, R.; Kacem, S.; Fritsch, L.; Pontis, J.; Kota, S.K.; Filipponi, D.; Fabbrizio, E.; Sardet, C.; Lohmann, F.; et al. PRMT5-mediated histone H4 arginine-3 symmetrical dimethylation marks chromatin at G + C-rich regions of the mouse genome. Nucleic Acids Res. 2014, 42, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Belyaev, N.; Conforti, P.; Ooi, L.; Tartari, M.; Papadimou, E.; MacDonald, M.; Fossale, E.; Zeitlin, S.; Buckley, N.; et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci. 2007, 27, 6972–6983. [Google Scholar] [CrossRef] [PubMed]
- Naruse, Y.; Aoki, T.; Kojima, T.; Mori, N. Neural restrictive silencer factor recruits mSin3 and histone deacetylase complex to repress neuron-specific target genes. Proc. Natl. Acad. Sci. USA 1999, 96, 13691–13696. [Google Scholar] [CrossRef] [PubMed]
- Roopra, A.; Sharling, L.; Wood, I.C.; Briggs, T.; Bachfischer, U.; Paquette, A.J.; Buckley, N.J. Transcriptional repression by neuron-restrictive silencer factor is mediated via the Sin3-histone deacetylase complex. Mol. Cell. Boil. 2000, 20, 2147–2157. [Google Scholar] [CrossRef]
- Grimes, J.A.; Nielsen, S.J.; Battaglioli, E.; Miska, E.A.; Speh, J.C.; Berry, D.L.; Atouf, F.; Holdener, B.C.; Mandel, G.; Kouzarides, T. The co-repressor mSin3A is a functional component of the REST-coREST repressor complex. J. Boil. Chem. 2000, 275, 9461–9467. [Google Scholar] [CrossRef]
- Huang, Y.; Myers, S.J.; Dingledine, R. Transcriptional repression by REST: Recruitment of Sin3A and histone deacetylase to neuronal genes. Nat. Neurosci. 1999, 2, 867–872. [Google Scholar] [PubMed]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Lunyak, V.V.; Burgess, R.; Prefontaine, G.G.; Nelson, C.; Sze, S.H.; Chenoweth, J.; Schwartz, P.; Pevzner, P.A.; Glass, C.; Mandel, G.; et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science 2002, 298, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- McFarland, K.N.; Huizenga, M.N.; Darnell, S.B.; Sangrey, G.R.; Berezovska, O.; Cha, J.H.; Outeiro, T.F.; Sadri-Vakili, G. MeCP2: A novel huntingtin interactor. Hum. Mol. Genet. 2014, 23, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991, 7, 695–702. [Google Scholar] [CrossRef]
- Holsinger, R.M.; Schnarr, J.; Henry, P.; Castelo, V.T.; Fahnestock, M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: Decreased levels in Alzheimer’s disease. Brain Res. Mol. Brain Res. 2000, 76, 347–354. [Google Scholar] [CrossRef]
- Wang, B.Y.; Zhong, Y.; Zhao, Z.; Miao, Y. Epigenetic suppression of hippocampal BDNF mediates the memory deficiency induced by amyloid fibrils. Pharmacol. Biochem. Behav. 2014, 126, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, A.; Pierrot, N.; Tasiaux, B.; Schakman, O.; Kienlen-Campard, P.; De Smet, C.; Octave, J.N. Epigenetic regulations of immediate early genes expression involved in memory formation by the amyloid precursor protein of Alzheimer disease. PLoS ONE 2014, 9, e99467. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Aβ oligomers reduce BDNF expression via HDAC nuclear translocation. J. Neurosci. 2015, 35, 7538–7551. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Modification | Functional Association |
|---|---|
| H3K4me1 | Active enhancer |
| H3K27ac | Active enhancer and promoter |
| H3K27me3 | Inactive chromatin |
| H3K4me3 | Active promoter |
| H3K36me3 | Active or inactive gene body |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.-W.; Chen, L. Epigenetic Regulation of BDNF Gene during Development and Diseases. Int. J. Mol. Sci. 2017, 18, 571. https://doi.org/10.3390/ijms18030571
Chen K-W, Chen L. Epigenetic Regulation of BDNF Gene during Development and Diseases. International Journal of Molecular Sciences. 2017; 18(3):571. https://doi.org/10.3390/ijms18030571
Chicago/Turabian StyleChen, Kuan-Wei, and Linyi Chen. 2017. "Epigenetic Regulation of BDNF Gene during Development and Diseases" International Journal of Molecular Sciences 18, no. 3: 571. https://doi.org/10.3390/ijms18030571
APA StyleChen, K.-W., & Chen, L. (2017). Epigenetic Regulation of BDNF Gene during Development and Diseases. International Journal of Molecular Sciences, 18(3), 571. https://doi.org/10.3390/ijms18030571