
More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy


{kind=link}
Abstract
:1. Introduction
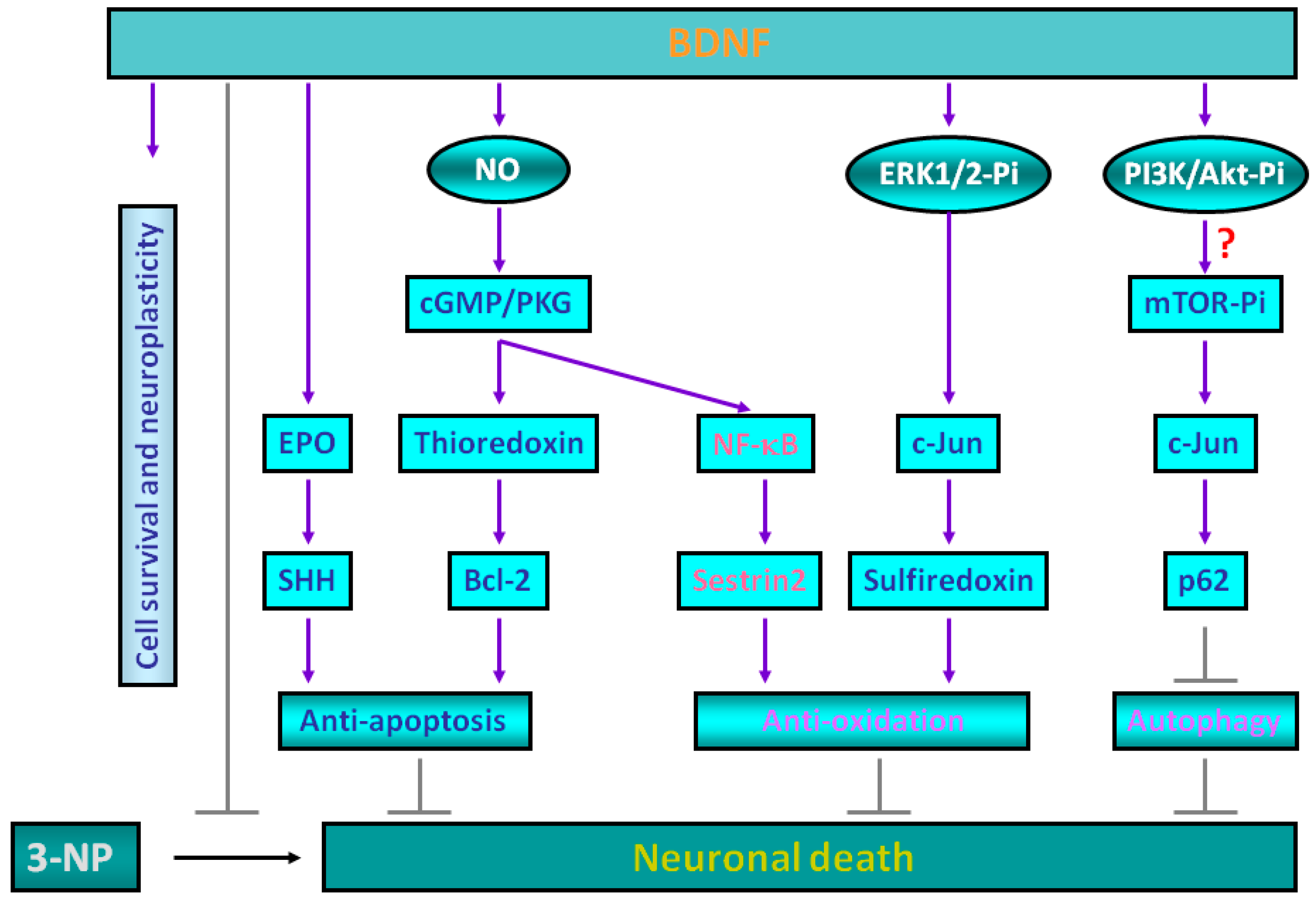
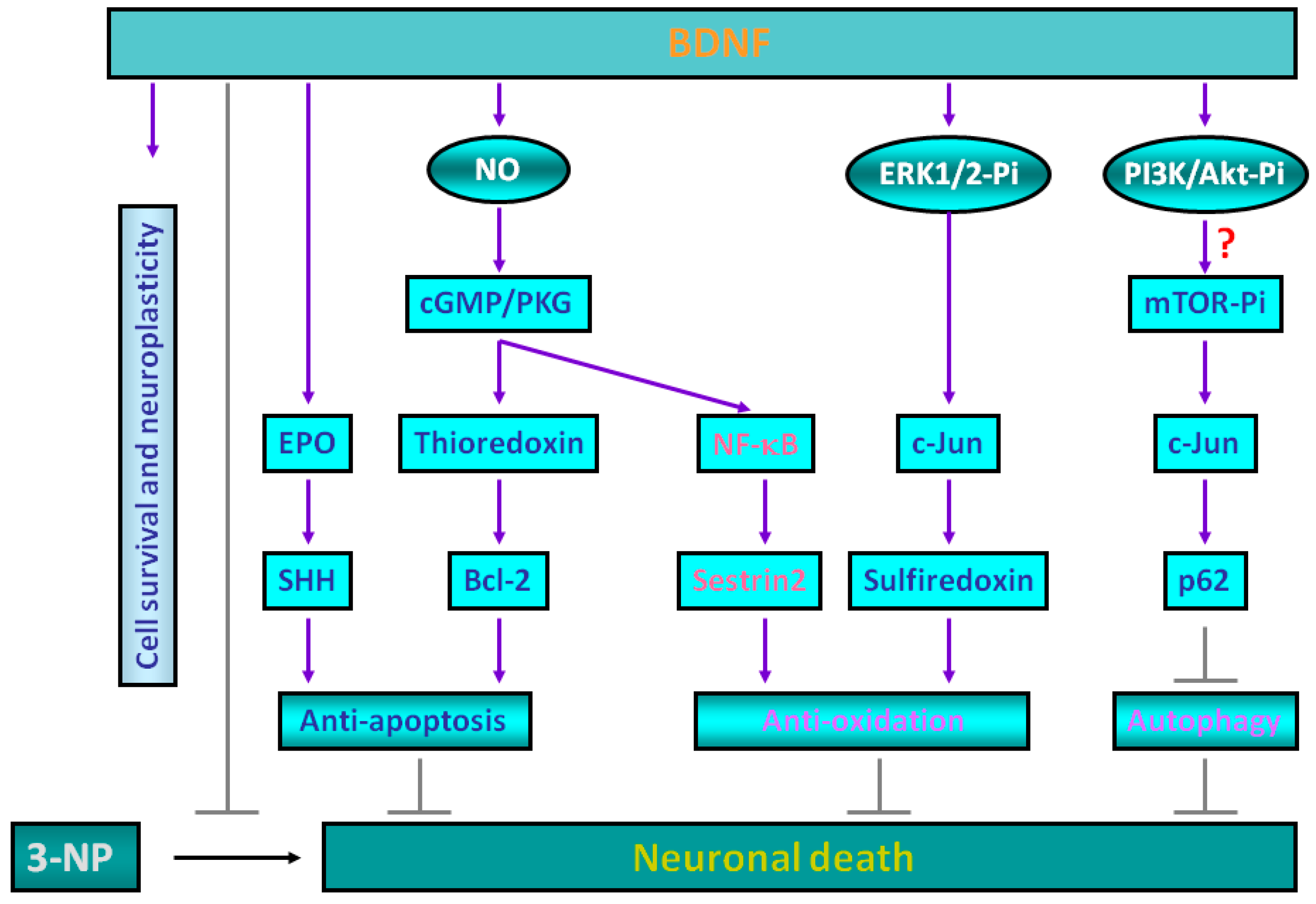
2. Molecular Mechanisms for Brain-Derived Neurotrophic Factor (BDNF)-Induced Protective Effects against 3-Nitropropionic Acid (3-NP) Toxicity
3. Sestrin2 Induction Conducts the Antioxidant Effects of BDNF against Mitochondrial Inhibition
4. Roles of p62 in Mediating BDNF-Dependent Neuroprotection against Mitochondrial Inhibition via Autophagy Suppression
5. Experimental Evidence Supporting BDNF-Mediated Neuroprotection against 3-NP Toxicity in Animal Models
6. Conclusions and Future Prospect
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 3-NP | 3-nitropropionic acid |
| Aβ | amyloid β-peptide |
| AD | Alzheimer’s disease |
| AGC family | protein kinases A, G, and C (PKA, PKG, PKC) |
| AP-1 | activator protein 1 |
| Bcl-2 | B-cell lymphoma 2 |
| BDNF | brain-derived neurotrophic factor |
| cAMP | cyclic adenosine monophosphate |
| cGMP | cyclic guanosine monophosphate |
| ChIP | chromatin immunoprecipitation |
| CREB | cAMP response-element binding protein |
| EPO | erythropoietin |
| ERK1/2 | extracellular signal-regulated kinase-1/2 |
| HD | Huntington’s disease |
| HIF-1 | hypoxia-inducible factor-1 |
| hNSCs | human neural stem cells |
| JNKs | c-Jun N-terminal kinases |
| l-NAME | l-nitroarginine methylester |
| MAPKs | mitogen-activated protein kinases |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MSCs | mesenchymal stem cells |
| mTOR | mammalian target of rapamycin |
| NF-κB | nuclear factor-κB |
| NGF | nerve growth factor |
| NMDAR | N-methyl-d-aspartate receptor |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| Nrf2 | nuclear factor (erythroid-derived 2)-like 2 |
| NT3 | neurotrophin 3 |
| NT-4 | neurotrophin 4 |
| p70S6K | p70 ribosomal protein S6 kinase |
| PD | Parkinson’s disease |
| PI3-K | phosphatidylinositol 3-kinase |
| PKA | cAMP-dependent protein kinase |
| PKG | cGMP-dependent protein kinase |
| PLCγ | phospholipase Cγ |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SHH | sonic hedgehog |
| SODs | superoxide dismutases |
| TrkB | tropomyosin-related kinase B |
| UCP2 | uncoupling protein 2 |
References
- Greenberg, M.E.; Xu, B.; Lu, B.; Hempstead, B.L. New insights in the biology of BDNF synthesis and release: Implications in CNS function. J. Neurosci. 2009, 29, 12764–12767. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; deTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Stephens, B.; Mueller, A.J.; Shering, A.F.; Hood, S.H.; Taggart, P.; Arbuthnott, G.W.; Bell, J.E.; Kilford, L.; Kingsbury, A.E.; Daniel, S.E.; et al. Evidence of a breakdown of corticostriatal connections in Parkinson’s disease. Neuroscience 2005, 132, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Graveland, G.A.; Williams, R.S.; DiFiglia, M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 1985, 227, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Pilato, F.; Profice, P.; Ranieri, F.; Capone, F.; Di Iorio, R.; Florio, L.; Di Lazzaro, V. Synaptic plasticity in neurodegenerative diseases evaluated and modulated by in vivo neurophysiological techniques. Mol. Neurobiol. 2012, 46, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Zundorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Excitotoxic and excitoprotective mechanisms: Abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromol. Med. 2003, 3, 65–94. [Google Scholar] [CrossRef]
- Numakawa, T.; Matsumoto, T.; Numakawa, Y.; Richards, M.; Yamawaki, S.; Kunugi, H. Protective action of neurotrophic factors and estrogen against oxidative stress-mediated neurodegeneration. J. Toxicol. 2011, 2011, 405194. [Google Scholar] [CrossRef] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2015, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef] [PubMed]
- Del Toro, D.; Canals, J.M.; Gines, S.; Kojima, M.; Egea, G.; Alberch, J. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J. Neurosci. 2006, 26, 12748–12757. [Google Scholar] [CrossRef] [PubMed]
- Alston, T.A.; Mela, L.; Bright, H.J. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc. Natl. Acad. Sci. USA 1977, 74, 3767–3771. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Brouillet, E.; Jenkins, B.G.; Ferrante, R.J.; Kowall, N.W.; Miller, J.M.; Storey, E.; Srivastava, R.; Rosen, B.R.; Hyman, B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993, 13, 4181–4192. [Google Scholar] [PubMed]
- Brouillet, E.; Hantraye, P. Effects of chronic MPTP and 3-nitropropionic acid in nonhuman primates. Curr. Opin. Neurol. 1995, 8, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Brouillet, E.; Hantraye, P.; Ferrante, R.J.; Dolan, R.; Leroy-Willig, A.; Kowall, N.W.; Beal, M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. USA 1995, 92, 7105–7109. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Hwang, C.S.; Chen, S.D.; Yin, J.H.; Yang, D.I. Neuroprotective mechanisms of brain-derived neurotrophic factor against 3-nitropropionic acid toxicity: Therapeutic implications for Huntington’s disease. Ann. N. Y. Acad. Sci. 2010, 1201, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Chen, S.D.; Yin, J.H.; Hwang, C.S.; Yang, D.I. Nuclear factor-κB-dependent sestrin2 induction mediates the antioxidant effects of BDNF against mitochondrial inhibition in rat cortical neurons. Mol. Neurobiol. 2016, 53, 4126–4142. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Chen, C.H.; Hwang, C.S.; Chen, S.D.; Hwang, W.C.; Yang, D.I. Roles of p62 in BDNF-dependent autophagy suppression and neuroprotection against mitochondrial dysfunction in rat cortical neurons. J. Neurochem. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Brain-derived neurotrophic factor: Regulation, effects, and potential clinical relevance. Neurology 2015, 84, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Lovell, M.A.; Furukawa, K.; Markesbery, W.R. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J. Neurochem. 1995, 65, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Chan, J.Y.; Hsu, K.S.; Chang, A.Y.; Chan, S.H. Brain-derived neurotrophic factor ameliorates brain stem cardiovascular dysregulation during experimental temporal lobe status epilepticus. PLoS ONE 2012, 7, e33527. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Cao, R.; Choi, Y.S.; Cho, H.Y.; Rhee, A.D.; Hah, C.K.; Hoyt, K.R.; Obrietan, K. The CREB/CRE transcriptional pathway: Protection against oxidative stress-mediated neuronal cell death. J. Neurochem. 2009, 108, 1251–1265. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Wu, C.W.; Chang, A.Y.; Hsu, K.S.; Chan, J.Y. Transcriptional upregulation of brain-derived neurotrophic factor in rostral ventrolateral medulla by angiotensin II: Significance in superoxide homeostasis and neural regulation of arterial pressure. Circ. Res. 2010, 107, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Laco, M.; Cunha-Oliveira, T.; Oliveira, C.R.; Rego, A.C. BDNF regulates BIM expression levels in 3-nitropropionic acid-treated cortical neurons. Neurobiol. Dis. 2009, 35, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Cunha-Oliveira, T.; Laco, M.; Oliveira, C.R.; Rego, A.C. Dysregulation of CREB activation and histone acetylation in 3-nitropropionic acid-treated cortical neurons: Prevention by BDNF and NGF. Neurotox. Res. 2010, 17, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Chen, S.D.; Hwang, C.S.; Yang, D.I. Sonic hedgehog mediates BDNF-induced neuroprotection against mitochondrial inhibitor 3-nitropropionic acid. Biochem. Biophys. Res. Commun. 2009, 385, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Chen, S.D.; Yin, J.H.; Hwang, C.S.; Yang, D.I. Erythropoietin and sonic hedgehog mediate the neuroprotective effects of brain-derived neurotrophic factor against mitochondrial inhibition. Neurobiol. Dis. 2010, 40, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Hwang, C.S.; Yang, D.I. Protective effects of brain-derived neurotrophic factor against neurotoxicity of 3-nitropropionic acid in rat cortical neurons. Neurotoxicology 2009, 30, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Yin, J.H.; Hwang, C.S.; Chen, S.D.; Yang, D.Y.; Yang, D.I. c-Jun-dependent sulfiredoxin induction mediates BDNF protection against mitochondrial inhibition in rat cortical neurons. Neurobiol. Dis. 2012, 46, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Hwang, C.S.; Yin, J.H.; Chen, S.D.; Yang, D.I. Oncostatin M-dependent Mcl-1 induction mediated by JAK1/2-STAT1/3 and CREB contributes to bioenergetic improvements and protective effects against mitochondrial dysfunction in cortical neurons. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2306–2325. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. Erythropoietin: Structure, control of production, and function. Physiol. Rev. 1992, 72, 449–489. [Google Scholar] [PubMed]
- Marti, H.H. Erythropoietin and the hypoxic brain. J. Exp. Biol. 2004, 207, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Siren, A.L.; Fratelli, M.; Brines, M.; Goemans, C.; Casagrande, S.; Lewczuk, P.; Keenan, S.; Gleiter, C.; Pasquali, C.; Capobianco, A.; et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc. Natl. Acad. Sci. USA 2001, 98, 4044–4049. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Z.G.; Gregg, S.R.; Zhang, R.L.; Jiao, Z.; LeTourneau, Y.; Liu, X.; Feng, Y.; Gerwien, J.; Torup, L.; et al. The Sonic hedgehog pathway mediates carbamylated erythropoietin-enhanced proliferation and differentiation of adult neural progenitor cells. J. Biol. Chem. 2007, 282, 32462–32470. [Google Scholar] [CrossRef] [PubMed]
- Marosi, K.; Mattson, M.P. BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol. Metab. 2014, 25, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Kobori, N.; Clifton, G.L.; Dash, P. Altered expression of novel genes in the cerebral cortex following experimental brain injury. Brain Res. Mol. Brain Res. 2002, 104, 148–158. [Google Scholar] [CrossRef]
- Truettner, J.; Busto, R.; Zhao, W.; Ginsberg, M.D.; Perez-Pinzon, M.A. Effect of ischemic preconditioning on the expression of putative neuroprotective genes in the rat brain. Brain Res. Mol. Brain Res. 2002, 103, 106–115. [Google Scholar] [CrossRef]
- Kim, H.; Jung, Y.; Shin, B.S.; Song, H.; Bae, S.H.; Rhee, S.G.; Jeong, W. Redox regulation of lipopolysaccharide-mediated sulfiredoxin induction, which depends on both AP-1 and Nrf2. J. Biol. Chem. 2010, 285, 34419–34428. [Google Scholar] [CrossRef] [PubMed]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Shoshani, T.; Faerman, A.; Zelin, E.; Kamer, I.; Kalinski, H.; Gorodin, S.; Fishman, A.; Chajut, A.; Einat, P.; et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 2002, 21, 6017–6031. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid. Redox Signal. 2011, 15, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhan, C.; Zhong, Q.; Li, S. Upregulation of sestrin-2 expression via p53 protects against 1-methyl-4-phenylpyridinium (MPP+) neurotoxicity. J. Mol. Neurosci. 2013, 51, 967–975. [Google Scholar] [PubMed]
- Chen, Y.S.; Chen, S.D.; Wu, C.L.; Huang, S.S.; Yang, D.I. Induction of sestrin2 as an endogenous protective mechanism against amyloid β-peptide neurotoxicity in primary cortical culture. Exp. Neurol. 2014, 253, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Yang, J.L.; Yang, D.I.; Lin, T.K.; Liou, C.W.; Chen, S.D. Roles of Sestrin2 and ribosomal protein S6 in transient global ischemia-induced hippocampal neuronal Injury. Int. J. Mol. Sci. 2015, 16, 26406–26416. [Google Scholar] [CrossRef] [PubMed]
- Kallenborn-Gerhardt, W.; Lu, R.; Syhr, K.M.; Heidler, J.; von Melchner, H.; Geisslinger, G.; Bangsow, T.; Schmidtko, A. Antioxidant activity of sestrin2 controls neuropathic pain after peripheral nerve injury. Antioxid. Redox Signal. 2013, 19, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Koesling, D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ. Res. 2003, 93, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F.; Feil, R.; Kleppisch, T.; Schlossmann, J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev. 2006, 86, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Essler, S.; Dehne, N.; Brune, B. Role of sestrin2 in peroxide signaling in macrophages. FEBS Lett. 2009, 583, 3531–3535. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Kim, K.M.; Kim, M.G.; Seo, K.H.; Han, J.Y.; Ka, S.O.; Park, B.H.; Shin, S.M.; Ku, S.K.; Cho, I.J.; Ki, S.H. Role of sestrin2 in the regulation of proinflammatory signaling in macrophages. Free Radic. Biol. Med. 2015, 78, 156–167. [Google Scholar]
- Shin, B.Y.; Jin, S.H.; Cho, I.J.; Ki, S.H. Nrf2-ARE pathway regulates induction of Sestrin-2 expression. Free Radic. Biol. Med. 2012, 53, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Singh, S.; De Haro, S.; Master, S.; Ponpuak, M.; Dinkins, C.; Ornatowski, W.; Vergne, I.; Deretic, V. Autophagy and pattern recognition receptors in innate immunity. Immunol. Rev. 2009, 227, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Cecconi, F. mTOR, AMBRA1, and autophagy: An intricate relationship. Cell Cycle 2013, 12, 2524–2525. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Peter, M.; Hofmann, K. Selective autophagy: Ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 2010, 12, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Into, T.; Inomata, M.; Takayama, E.; Takigawa, T. Autophagy in regulation of Toll-like receptor signaling. Cell. Signal. 2012, 24, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Ichimura, Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010, 584, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Wang, Y.; Wu, J.C.; Lin, F.; Han, R.; Han, F.; Fukunaga, K.; Qin, Z.H. Down-regulation of Bcl-2 enhances autophagy activation and cell death induced by mitochondrial dysfunction in rat striatum. J. Neurosci. Res. 2009, 87, 3600–3610. [Google Scholar] [CrossRef] [PubMed]
- Solesio, M.E.; Saez-Atienzar, S.; Jordan, J.; Galindo, M.F. 3-Nitropropionic acid induces autophagy by forming mitochondrial permeability transition pores rather than activating the mitochondrial fission pathway. Br. J. Pharmacol. 2013, 168, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Puerta, E.; Hervias, I.; Barros-Minones, L.; Jordan, J.; Ricobaraza, A.; Cuadrado-Tejedor, M.; Garcia-Osta, A.; Aguirre, N. Sildenafil protects against 3-nitropropionic acid neurotoxicity through the modulation of calpain, CREB, and BDNF. Neurobiol. Dis. 2010, 38, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.J.; Kim, G.W. Differential caspase activity in the cortex and striatum with chronic infusion of 3-nitropropionic acid. Biochem. Biophys. Res. Commun. 2015, 465, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Yang, L.; Zhou, S.M.; Guan, S.Y.; Yang, L.K.; Shi, Q.X.; Zhao, M.G.; Yang, Q. Effect of Praeruptorin C on 3-nitropropionic acid induced Huntington’s disease-like symptoms in mice. Biomed. Pharmacother. 2017, 86, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Ranju, V.; Sathiya, S.; Kalaivani, P.; Priya, R.J.; Saravana Babu, C. Memantine exerts functional recovery by improving BDNF and GDNF expression in 3-nitropropionic acid intoxicated mice. Neurosci. Lett. 2015, 586, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Valdeolivas, S.; Navarrete, C.; Cantarero, I.; Bellido, M.L.; Munoz, E.; Sagredo, O. Neuroprotective properties of cannabigerol in Huntington’s disease: Studies in R6/2 mice and 3-nitropropionate-lesioned mice. Neurotherapeutics 2015, 12, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Kim, J.; Cho, S.J.; Hatori, K.; Nagai, A.; Choi, H.B.; Lee, M.C.; McLarnon, J.G.; Kim, S.U. Proactive transplantation of human neural stem cells prevents degeneration of striatal neurons in a rat model of Huntington disease. Neurobiol. Dis. 2004, 16, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.; Boyer, C.; Leveque, X.; Fink, K.D.; Thinard, R.; Blanchard, F.; Dunbar, G.L.; Lescaudron, L. Mesenchymal stem cell transplantation and DMEM administration in a 3-NP rat model of Huntington’s disease: Morphological and behavioral outcomes. Behav. Brain Res. 2011, 217, 369–378. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-D.; Wu, C.-L.; Hwang, W.-C.; Yang, D.-I. More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy. Int. J. Mol. Sci. 2017, 18, 545. https://doi.org/10.3390/ijms18030545
Chen S-D, Wu C-L, Hwang W-C, Yang D-I. More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy. International Journal of Molecular Sciences. 2017; 18(3):545. https://doi.org/10.3390/ijms18030545
Chicago/Turabian StyleChen, Shang-Der, Chia-Lin Wu, Wei-Chao Hwang, and Ding-I Yang. 2017. "More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy" International Journal of Molecular Sciences 18, no. 3: 545. https://doi.org/10.3390/ijms18030545
APA StyleChen, S.-D., Wu, C.-L., Hwang, W.-C., & Yang, D.-I. (2017). More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy. International Journal of Molecular Sciences, 18(3), 545. https://doi.org/10.3390/ijms18030545