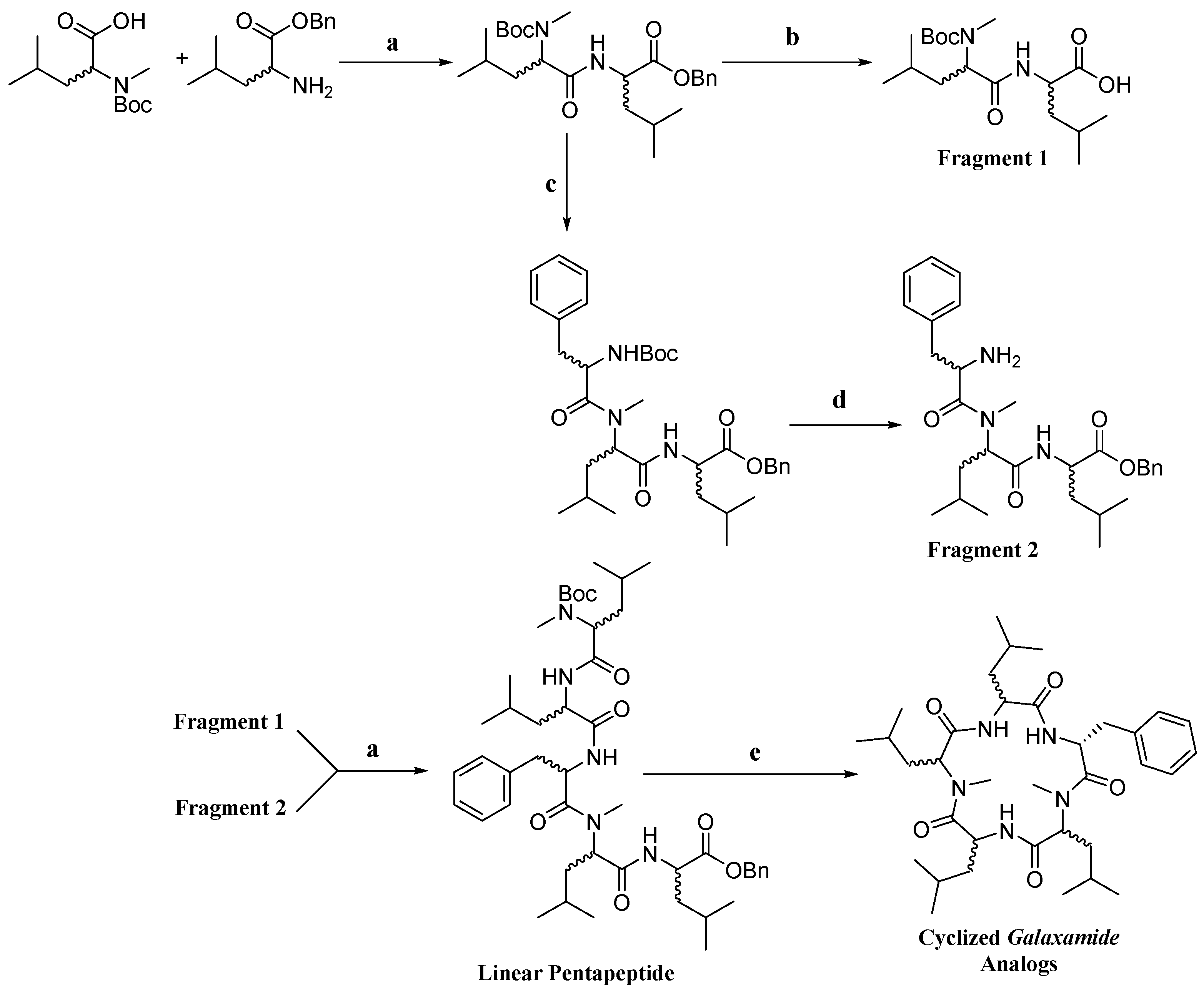
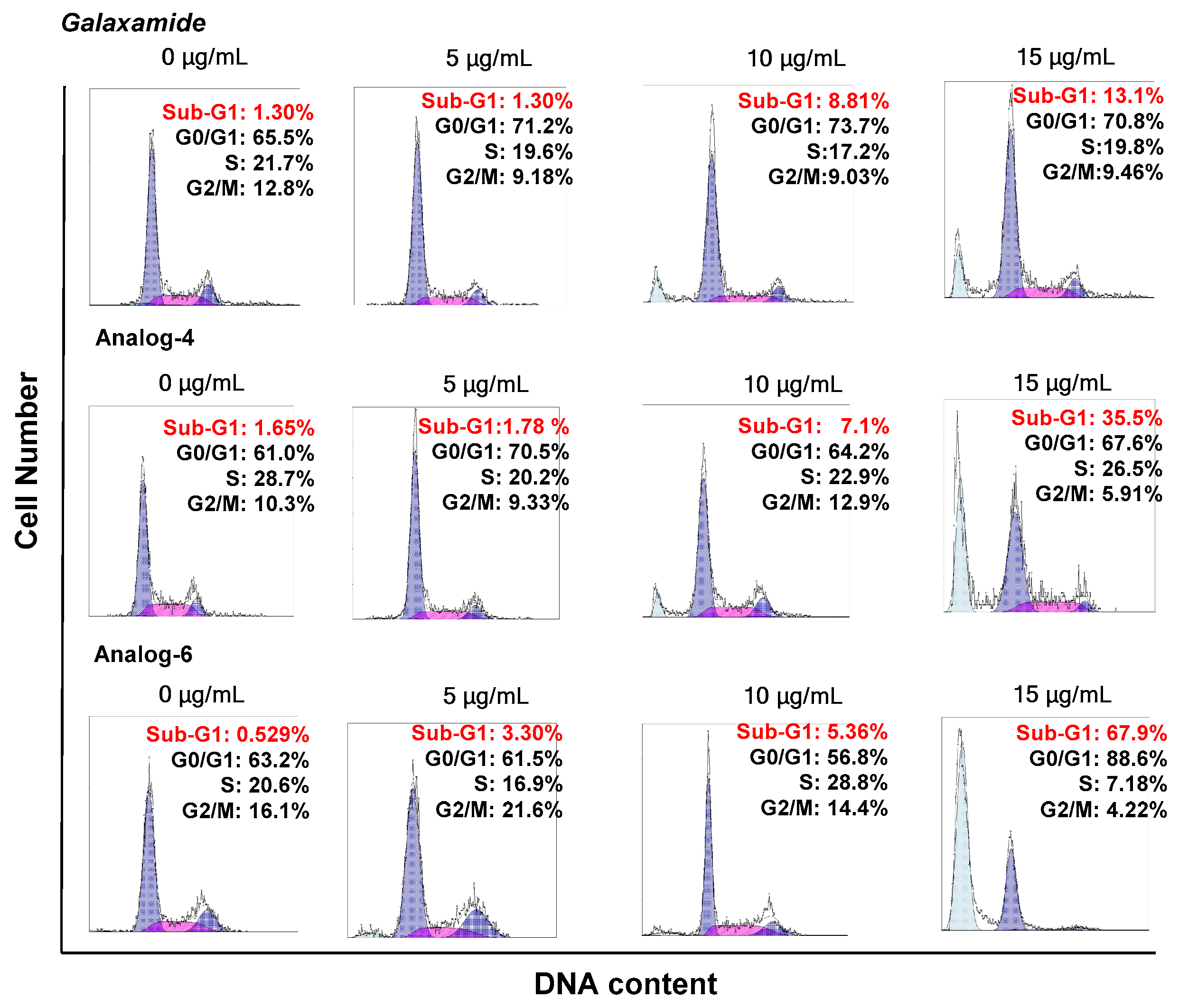
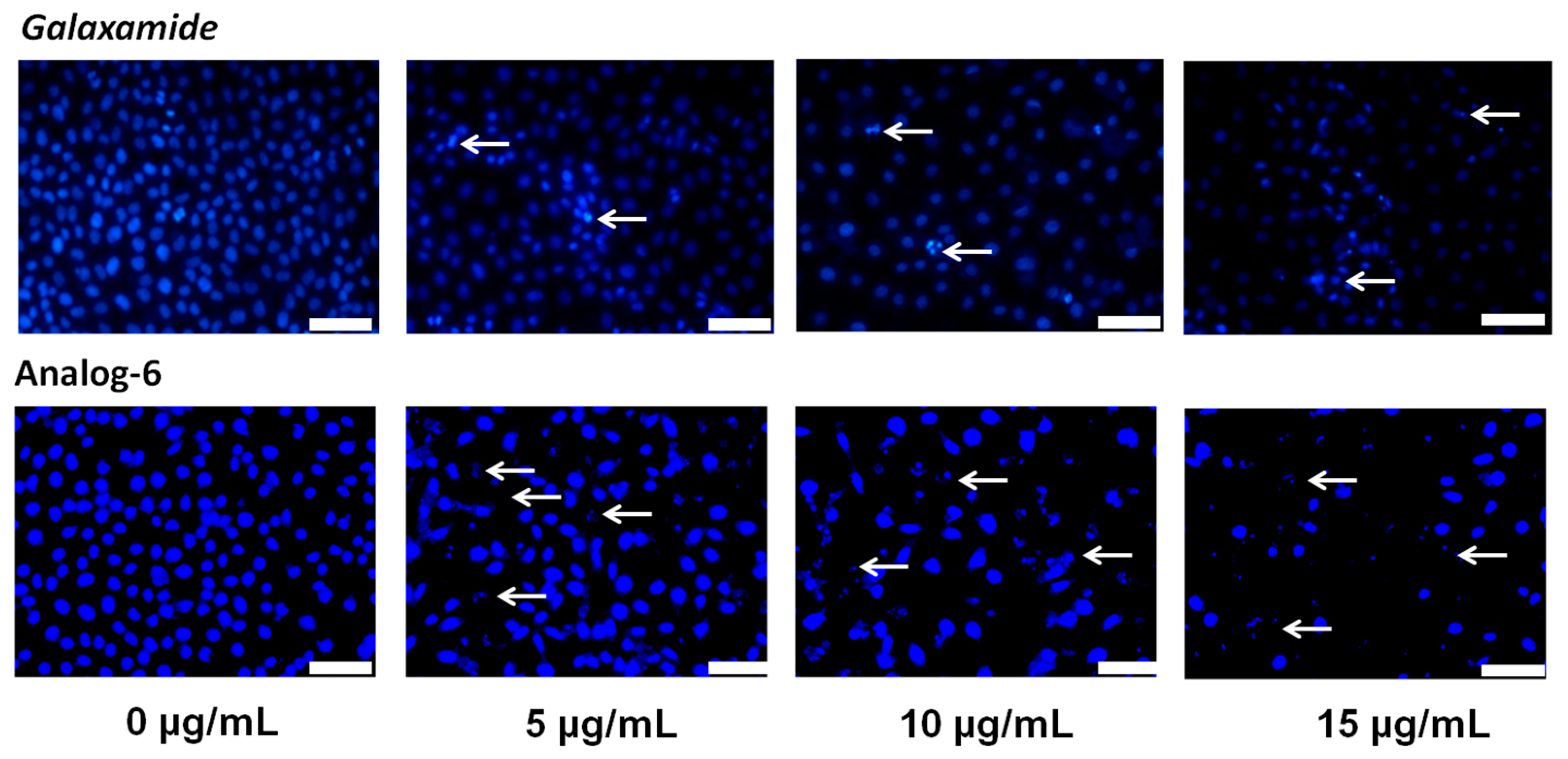
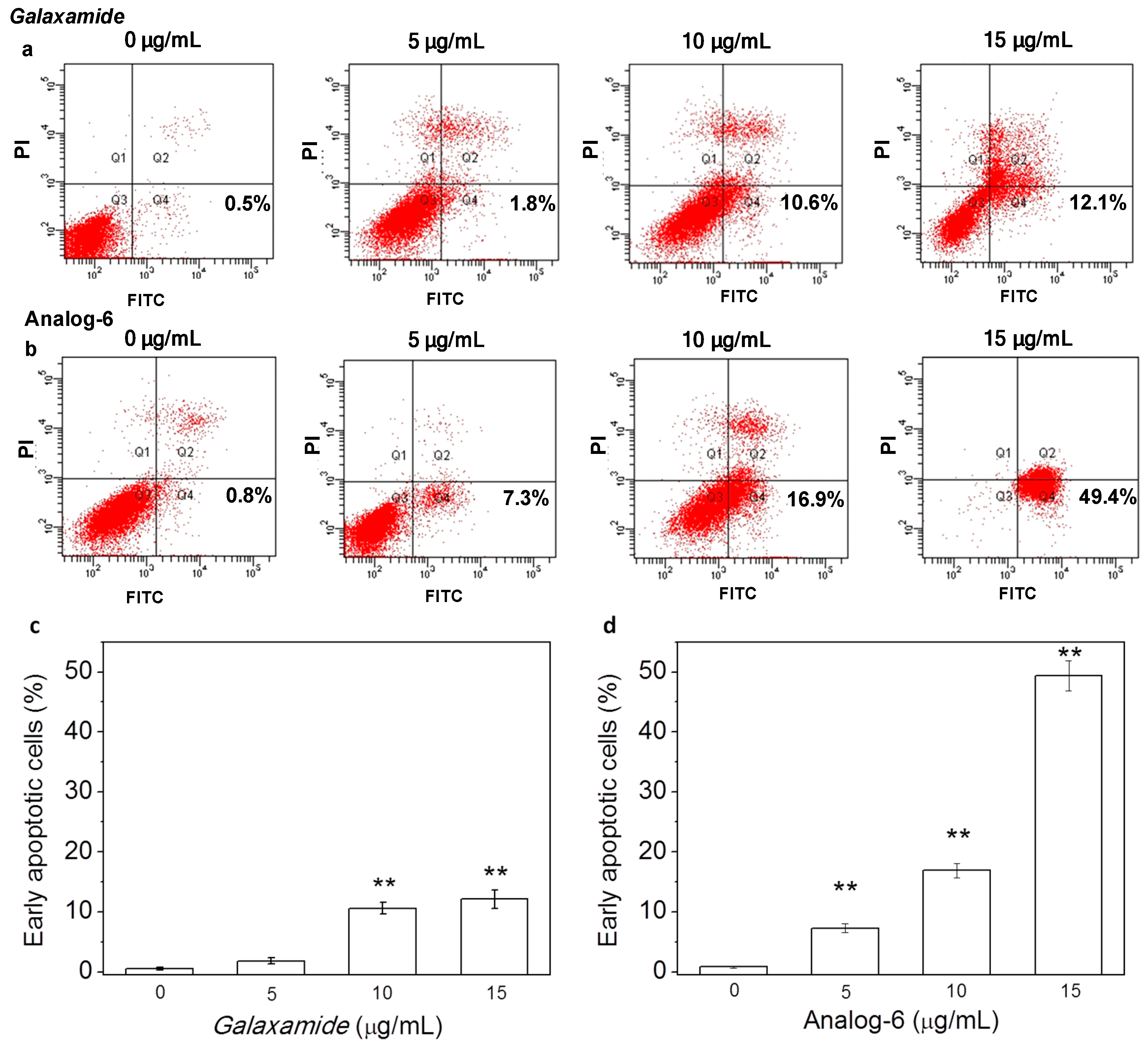
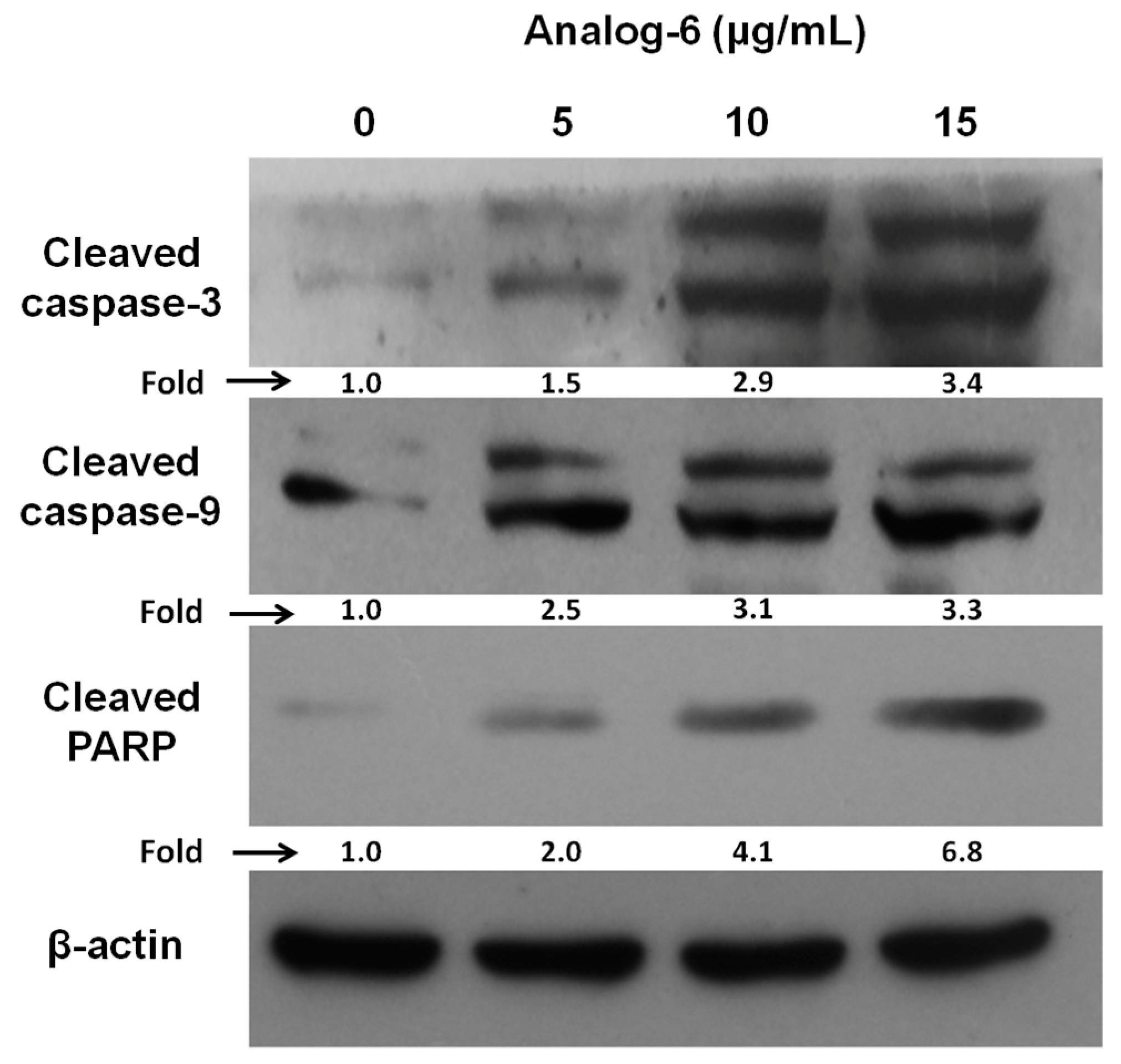
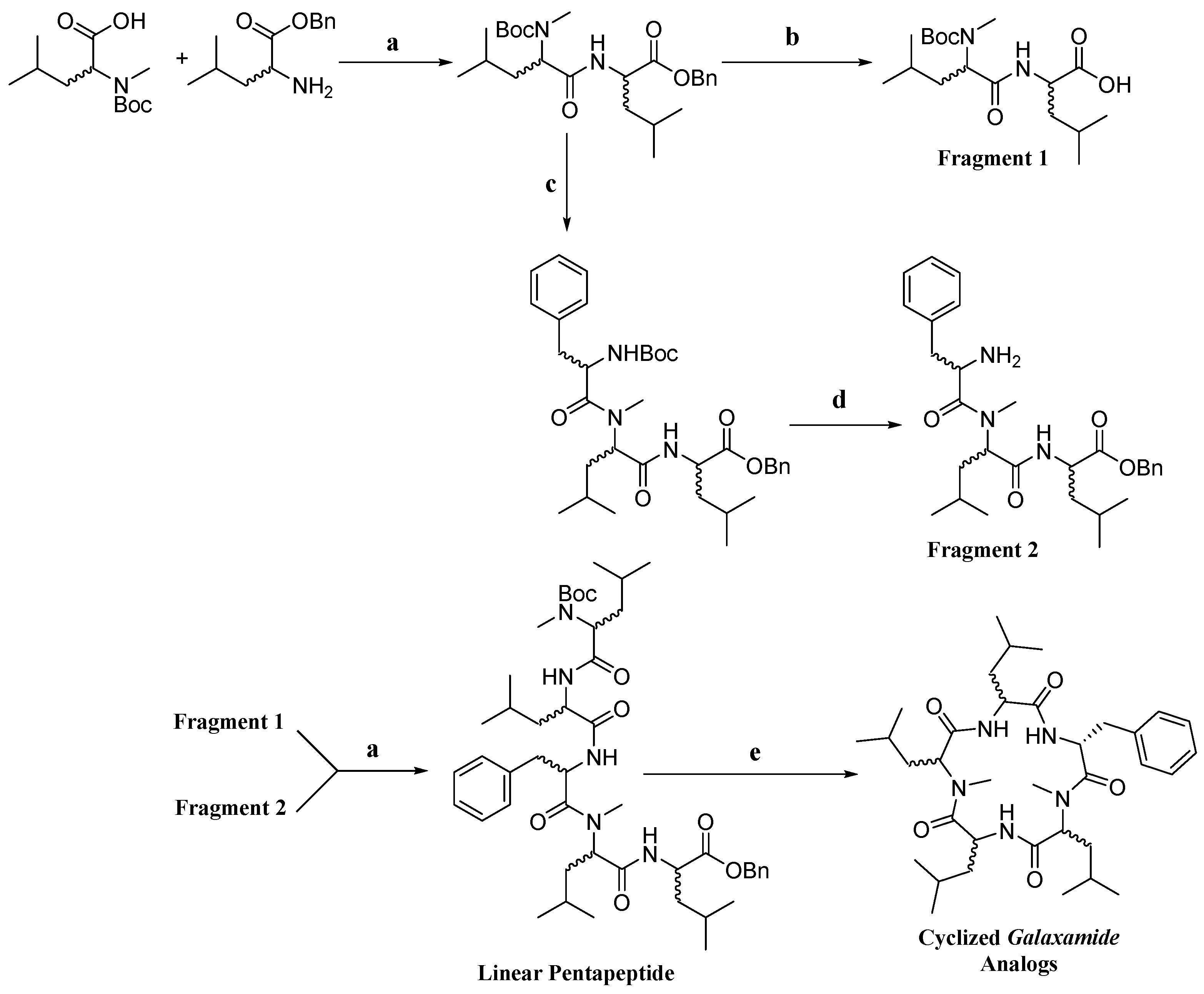
4.1. Synthesis of Galaxamide Analogs
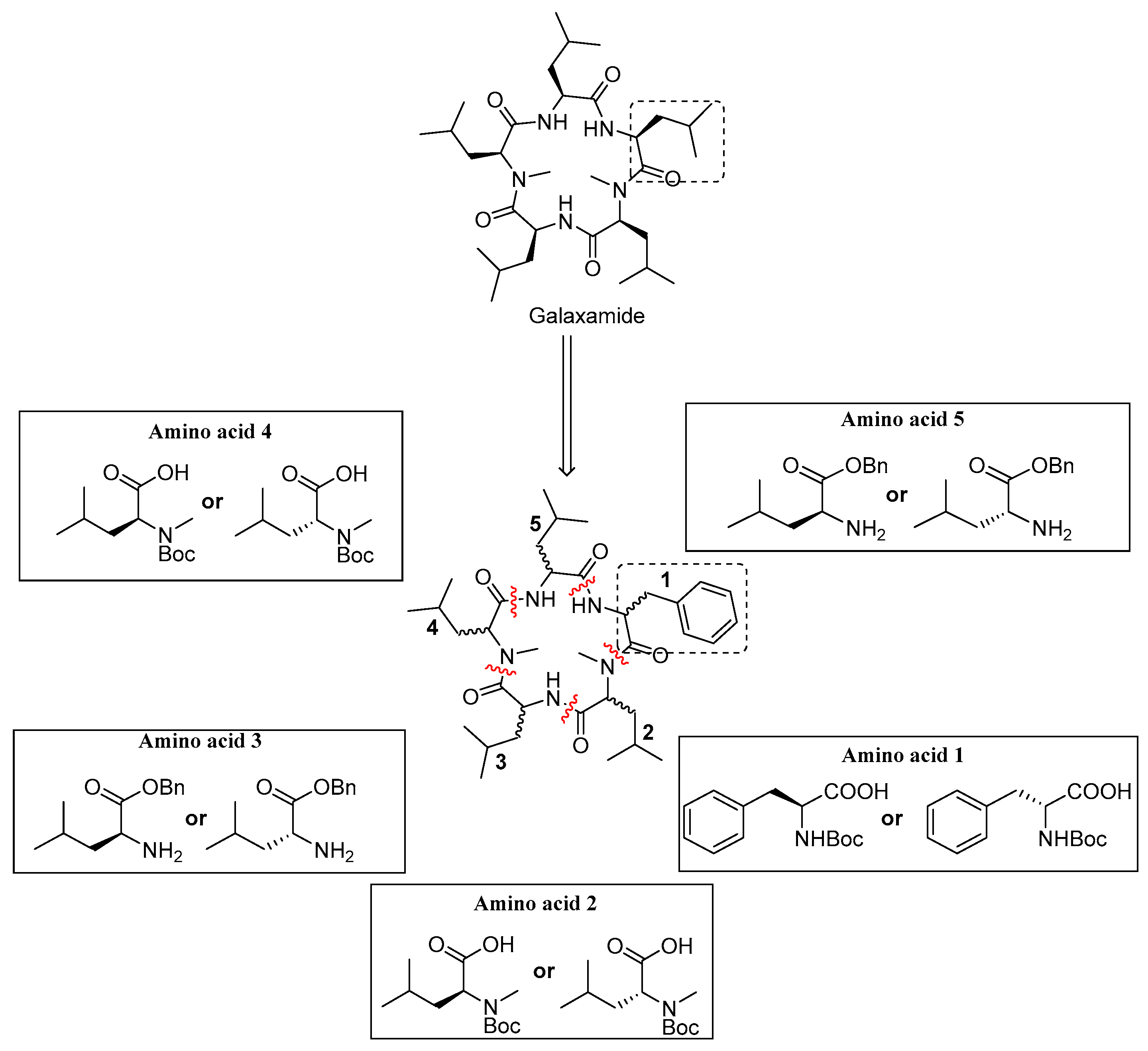
Galaxamide analogues were synthesized following our reported protocols with some modification [
6,
8]. Briefly, a classic “3 + 2” synthetic strategy was used in the synthesis, and the whole process was performed in a liquid phase with
N-Boc-
l(
d)-phenylalanine,
N-Boc-
l(
d)-leucine and
l(
d)-Leu-OBn·TosOH as the starting materials. After sequential experimental steps of amine protection and deprotection, acid deprotection, methylation and condensation over amino acids, cyclization was finished using a combination of condensation reagents DEPBT, HATU and TBTU, leading to the product of cyclic pentapeptides.
4.1.1. Synthesis of N-Boc-Leu-OH
In a 500-mL round-bottom flask under N2 atmosphere, 1 M NaOH solution (63 mL) in distilled water and 3.93 g l-Leu-OH (30 mmol) were added in sequence. The system was then immerged into the ice bath for cooling for 15 min. After the solution of l-Leu-OH become clear, 7.86 g Boc2O dissolved in THF (84 mL, 36 mmol) were added and stirred for 10 min; the pH was adjusted to around 10.5. The reaction temperature was then slowly increased to room temperature, and the solution was stirred overnight. After 24 h, the solvent was removed by vacuum distillation, and the rest was extracted twice using 200 mL ethyl ether. The resultant aqueous phase was cooled in the ice bath, acidified by adding an appropriate amount of 1 M HCl solution to increase the pH to 2. Then, the aqueous phase was further extracted three times with ethyl acetate (150 mL), washed twice with saturated NaCl solution (150 mL). Subsequently, the organic layer was dried by anhydrous sodium sulfate and filtered. After complete distillation of the organic solvent, 6.94 g white powder were obtained with a yield of 98.4%. 1H-NMR (500 MHz, CDCl3), δH 4.87 (m, 1H), 4.31 (m, 1H), 1.75 (m, 2H), 1.54 (m, 1H), 1.45 (s, 9H), 0.95 (d, 6H, J = 6.4 Hz); MS (ESI) m/z: 232.2 [M + H]+, 249.2 [M + NH4]+, 254.3 [M + Na]+.
4.1.2. Synthesis of N-Boc-d-Leu-OH
The experimental procedure for the synthesis of N-Boc- d-Leu-OH followed as per described in the synthesis of N-Boc-Leu-OH. In this synthesis, 3.93 g d-Leu-OH were used instead of l-Leu-OH. As a result, 6.74 g white product were obtained with a yield of 97.2%. 1H-NMR (500 MHz, CDCl3), δH 4.87 (m, 1H), 4.31 (m, 1H), 1.75 (m, 2H), 1.54 (m, 1H), 1.45 (s, 9H), 0.95 (d, 6H, J = 6.4 Hz); MS (ESI) m/z: 232.2 [M + H]+, 249.2 [M + NH4]+, 254.3 [M + Na]+.
4.1.3. Synthesis of N-Boc-Phe-OH
The experimental procedure for the synthesis of N-Boc-Phe-OH followed as per described in the synthesis of N-Boc-Leu-OH, and 4.95 g L-Phe-OH were used instead of 3.93 g l-Leu-OH. As a result, 7.72 g white product were obtained with a yield of 97%.
4.1.4. Synthesis of N-Boc-d-Phe-OH
The experimental procedure for the synthesis of N-Boc-d-Phe-OH followed as per described in the synthesis of N-Boc-Leu-OH, and 4.95 g l-Phe-OH (30 mmol) were used instead of 3.93 g l-Leu-OH. As a result, 7.73 g white product were obtained with a yield of 97.2%.
4.1.5. Synthesis of N-Boc-Me-Leu-OH
In a 250-mL round-bottom flask under N2 atmosphere, N-Boc-Leu-OH (4.62 g) and THF (60 mL) were added with stirring within the ice bath. After the solution of N-Boc-Leu-OH in THF become clear, CH3I (6.3 mL, 100 mmol) was added slowly and allowed to react for 30 min. Then, a 60% NaH dispersion in mineral oil (1.6 g, 40 mmol) was added slowly. The temperature of the system was increased slowly, and the solution was stirred overnight. After 24 h, an additional amount of 60% NaH in mineral oil (0.8 g, 20 mmol) was added after being cooled in the ice bath; again, the temperature increased slowly to room temperature, and it was stirred for overnight. After completion of the reaction, it was cooled in an ice bath, and distilled water (15 mL) was added slowly. Further, ethyl acetate (100 mL) was added to the flask under cooling in the ice bath. Then, the organic solvent was removed by vacuum distillation, and the residual was extracted twice using ethyl ether (60 mL). The combined ether phase was washed with saturated NaCl solution (50 mL) and combined with the aqueous phase. Under cooling in the ice bath, the solution was acidified by adding an appropriate amount of 1 M HCl to increase the pH to 2 to 3, and it was extracted three times using ethyl acetate (100 mL). Combining the organic phase together, it was washed by 150 mL 5% Na2S2O3 solution three times and saturated NaCl solution (100 mL). Subsequently, the solution was dried by anhydrous sodium sulfate, filtered, concentrated, dried under vacuum and purified using silica-gel column chromatography (eluant ratio of Vpetroleum ether/Vacetone = 40:1); finally, 4.25 g white needle-like powder were obtained with a yield of 86.3%. m.p.: 62.6~64.3 °C, (c = 0.5, EtOH), 1H-NMR (400 MHz, CDCl3), δH 4.82 (m, 1H), 4.62 (m, 1H), 3.34 (s, 3H), 1.71 (m, 2H), 1.58 (m, 1H), 1.46 (s, 9H), 0.95 (d, 6H, J = 6.4 Hz); MS (ESI) m/z: 246.2 [M + H]+, 263.2 [M + NH4]+, 268.3 [M + Na]+.
4.1.6. Synthesis of N-Boc-Me-d-Leu-OH
The experimental procedure for the synthesis of N-Boc-Me-d-Leu-OH followed as per described in the synthesis of N-Boc-Me-Leu-OH, and N-Boc-d-Leu-OH (4.62 g) was used instead of N-Boc-Leu-OH. As a result, 4.62 g white powder product were obtained with a yield of 87.2%. 1H-NMR (500 MHz, CDCl3), δH 6.04 (m, 1H), 4.45 (m, 1H), 1.78 (m, 2H), 1.50 (m, 1H), 1.40 (s, 9H), 0.91 (d, 6H, J = 6.4 Hz); MS (ESI) m/z: 232.2 [M + H]+, 249.2 [M + NH4]+, 254.3 [M + Na]+.
4.1.7. Synthesis of N-Boc-Me-Phe-OH
The synthesis followed the experimental ways for N-Boc-Me-Leu-OH as above. However, in this synthesis, 5.3 g N-Boc-Phe-OH were used instead of N-Boc-Leu-OH. As a result, 4.7 g colorless oily product were obtained with a yield of 85.0%. 1H-NMR (500 MHz, CDCl3), δH 7.35 (m, 5H), 7.02 (s, 1H), 4.90 (m, 1H), 3.57 (s, 3H), 3.12–3.43 (m, 1H), 1.42 (s, 9H); MS (ESI) m/z: 280.2 [M + H]+, 297.2 [M + NH4]+, 302.3 [M + Na]+.
4.1.8. Synthesis of N-Boc-Me-d-Phe-OH
The experimental procedure for the synthesis of N-Boc-Me-Phe-OH followed as per described in the synthesis of N-Boc-Me-Leu-OH, and 5.3 g N-Boc-D-Phe-OH were used instead of 5.3 g N-Boc-Leu-OH.
4.1.9. Synthesis of Dipeptide N-Boc-Me-Leu-Leu-OBn(D1)
In a 250-mL round-bottom flask under N2 atmosphere, 2.46 g N-Boc-Me-Leu-OH and 10 mL THF were added under stirring and cooled in ice bath condition. Then, DEPBT (4.5 g, 15 mmol) and DIEA (2.6 mL, 15 mmol) was added, and allowed to react for 10 min. After that 4.72 g l-Leu-OBn·TosOH (12 mmol) were added and stirred for complete dissolution. Then, the pH of reaction solution was adjusted to 7 to 8 by adding DIEA slowly. Subsequently, the temperature was increased slowly to room temperature, and the reaction was monitored using thin layer chromatography (TLC). After overnight stirring, the solvent was removed by vacuum distillation, and the residual crude product was purified directly using silica-gel column chromatography (eluant ratio of Vpetroleum ether/Vacetone = 40:1). Finally, 3.76 g product were obtained with a yield of 83.8%. 1H-NMR (500 MHz, CDCl3), δH 7.35 (m, 5H), 7.02 (s, 1H), 4.90 (m, 1H), 3.57 (s, 3H), 3.12–3.43 (m, 1H), 1.42(s, 9H); MS (ESI) m/z: 280.2 [M + H]+, 297.2 [M + NH4]+, 302.3 [M + Na]+.
4.1.10. Synthesis of Dipeptide N-Boc-Me-d-Leu-Leu-OBn(D2)
The experimental procedure for the synthesis of N-Boc-Me-d-Leu-Leu-OBn(D2) followed as per described in the synthesis of N-Boc-Me-Leu-Leu-OBn(D1). In this synthesis, N-Boc-Me-d-Leu-OH was used instead of N-Boc-Me-Leu-OH. As a result, 3.82 g product were obtained with a yield of 85.2%. 1H-NMR (300 MHz, CDCl3), δH 7.45–7.31 (m, 5H), 6.39 (d, J = 78.1 Hz, 1H), 5.24–5.11 (m, 2H), 4.65 (s, 2H), 2.76 (s, 3H), 1.69 (dd, J = 12.4, 6.4 Hz, 2H), 1.65–1.57 (m, 2H), 1.49 (s, 9H), 0.97–0.87 (m, 12H).; 13C-NMR (75 MHz, CDCl3), δC 172.5, 171.1, 167.4, 135.4, 128.6 (2C), 128.4, 128.2 (2C), 99.98, 67.0, 56.1, 50.7 (2C), 36.3, 29.8, 28.4 (3C), 24.8 (2C), 22.9 (2C), 22.1, 21.7; MS (ESI) m/z: 449.3 [M + H]+, 466.5 [M + NH4]+, 471.6 [M + Na]+.
4.1.11. Synthesis of Dipeptide N-Boc-Me-Leu-d-Leu-OBn(D3)
The experimental procedure for the synthesis of N-Boc-Me-Leu-d-Leu-OBn(D3) followed as per described in the synthesis of N-Boc-Me-Leu-Leu-OBn(D1). In this synthesis, d-Leu-OBn·TosOH was used instead of l-Leu-OBn·TosOH. As a result, 3.92 g oily product were obtained with a yield of 87.4%. 1H-NMR (300 MHz, CDCl3), δH 7.42–7.29 (m, 5H), 6.64–6.31 (m, 1H), 5.14 (q, J = 12.3 Hz, 2H), 4.84–4.47 (m, 2H), 2.69 (s, 3H), 1.82–1.58 (m, 4H), 1.58–1.50 (m, 2H), 1.48 (s, 9H), 0.94 (t, J = 7.1 Hz, 12H); 13C-NMR (75 MHz, CDCl3), δC 172.6, 171.4, 157.1, 135.5, 128.6 (2C), 128.4, 128.2 (2C), 80.5, 66.9, 55.9, 50.8, 41.1, 36.0, 29.8, 28.3 (3C), 24.9, 24.7, 23.2, 22.8 (2C), 21.8; MS (ESI) m/z: 449.3 [M + H]+, 466.5 [M + NH4]+, 471.6 [M + Na]+.
4.1.12. Synthesis of Dipeptide N-Boc-Me-Phe-Leu-OBn(D4)
The experimental procedure for the synthesis of N-Boc-Me-Phe-Leu-OBn(D4) followed as per described in the synthesis of N-Boc-Me-Leu-Leu-OBn(D1). In this synthesis, N-Boc-Me-Phe-OH was used instead of N-Boc-Me-Leu-OH. As a result, oily 3.98 g product were obtained with a yield of 82.6%. 1H-NMR (300 MHz, CDCl3), δH 7.45–7.30 (m, 5H), 7.25–7.17 (m, 5H), 5.37 (dd, J = 10.1, 5.3 Hz, 2H), 5.15 (q, J = 12.3 Hz, 2H), 4.85 (dd, J = 15.6, 6.7 Hz, 1H), 3.13–2.98 (m, 1H), 2.86 (s, 3H), 2.84–2.77 (m, 1H), 1.87–1.60 (m, 2H), 1.47–1.39 (m, 9H), 1.38–1.24 (m, 2H), 0.92 (dd, J = 8.4, 6.7 Hz, 6H); 13C-NMR (75 MHz, CDCl3), δC 172.7, 171.3, 155.3, 136.3, 135.6, 129.5 (2C), 129.4, 128.6 (2C), 128.4 (2C), 128.3 (2C), 126.8, 79.6, 77.2, 66.9, 54.8, 51.7, 38.7, 37.1, 28.0 (3C), 24.7, 23.3, 21.5; MS (ESI) m/z: 483.7 [M + H]+, 500.6 [M + NH4]+, 505.6 [M + Na]+.
4.1.13. Synthesis of Dipeptide N-Boc-d-Phe-Me-Leu-OBn(D5)
The experimental procedure for the synthesis of N-Boc-d-Phe-Me-Leu-OBn(D5) followed as per described in the synthesis of N-Boc-Me-Leu-Leu-OBn(D1). In this synthesis, N-Boc-Me-d-Phe-OH was used instead of N-Boc-Me-Phe-OH. As a result, oily 4.05 g product were obtained with a yield of 84.1%. 1H-NMR (300 MHz, CDCl3), δH 7.46–7.33 (m, 5H), 7.32–7.16 (m, 5H), 6.48 (dd, J = 95.7, 7.5 Hz, 1H), 5.17 (q, J = 12.3 Hz, 2H), 4.84 (ddd, J = 54.7, 17.6, 8.4 Hz, 2H), 3.54–3.27 (m, 1H), 3.01–2.84 (m, 1H), 2.76 (s, 3H), 1.74–1.60 (m, 1H), 1.54 (dd, J = 14.7, 7.7 Hz, 2H), 1.34 (s, 9H), 0.90 (d, J = 5.4 Hz, 6H); 13C-NMR (75 MHz, CDCl3), δC 172.5, 170.6, 156.7, 137.5, 135.4, 129.1, 129.0, 128.6 (2C), 128.4 (2C), 128.2, 126.4, 80.5, 67.0, 59.4, 50.8, 41.2, 33.9, 28.2 (3C), 25.0, 24.8, 22.8, 21.8; MS (ESI) m/z: 483.7 [M + H]+, 500.6 [M + NH4]+, 505.6 [M + Na]+.
4.1.14. Synthesis of Tripeptide N-Boc-Leu-Me-Leu-Leu-OBn(T1)
D1 (3.14g, 7 mmol) was first dissolved in 32 mL dichloromethane, and then, 1.47 mL anisole were added under N2 protection. The solution was cooled in the ice bath and stirred continuously; TFA (8 mL) was added slowly to the solution and allowed to react for 4 h. The solvent was removed by vacuum distillation, then additional appropriate dichloromethane was added and vacuum distillated again. This step was repeated three times for complete removal of TFA; finally, an oily product named as D2-A was obtained after drying, and it was used for further synthesis.
BOC-Leu-OH (1.85 g, 8 mmol) was dissolved in THF (5 mL) under stirring and cooled in an ice bath in N2 atmosphere. Subsequently, DEPBT (3.00 g, 10 mmol) and DIEA (1.8 mL, 10 mmol) were added into the solution and reacted for 10 min. Thereafter, an appropriate amount of the previously synthesized D2-A THF solution (10 mL) was added, and DIEA was used to adjust the pH of the solution to 7 to 8. The temperature was slowly increased to room temperature, and the reaction was monitored by TLC. After completion of the reaction, the solvent was removed by vacuum distillation, and the residual crude product was purified directly using silica-gel column chromatography (eluant ratio of Vpetroleum ether/Vacetone = 40:1). Finally, 2.95 g colorless oily product were obtained with an overall yield of 80.8%. 1H-NMR (500 MHz, CDCl3), δH 7.32 (m, 5H), 6.32 (d, 1H, J = 4.0 Hz), 5.09 (m, 4H), 4.56 (m, 2H), 2.94 (s, 3H), 1.63 (m, 3H), 1.46 (m, 3H), 1.41 (s, 9H), 1.23 (m, 3H), 0.90 (m, 18H); 13C-NMR (125 MHz, CDCl3), δC 174.1, 172.3, 170.2, 155.7, 135.3, 128.5, 128.4, 128.3, 128.2, 127.9, 79.6, 67.0, 54.3, 50.6, 49.0, 41.9, 40.9, 35.9, 30.1, 28.3 (3C), 24.7, 24.6, 23.4 (2C), 22.9, 22.8, 22.0 21.6,21.5; MS (ESI) m/z: 562.8 [M + H]+, 579.8 [M + NH4]+, 584.9 [M + Na]+.
4.1.15. Synthesis of Tripeptide N-Boc-Leu-Me-Leu-d-Leu-OBn(T2)
The experimental procedure for the synthesis of N-Boc-Leu-Me-Leu-d-Leu-OBn(T2) followed as per described in the synthesis of N-Boc-Leu-Me-Leu-Leu-Obn(T1). In this synthesis, D3 was used instead of D1. 1H-NMR (300 MHz, CDCl3), δH 7.40–7.32 (m, 5H), 5.20–5.19 (m, 1H), 5.10 (dd, J = 12.6, 5.5 Hz, 1H), 4.60–4.56 (m, 1H), 2.90 (s, 3H), 1.70–1.63 (m, 6H), 1.42 (s, 9H), 1.35 (d, J = 10.8 Hz, 3H), 0.97–0.90 (m, 18H); 13C-NMR (75 MHz, CDCl3), δC 174.9, 172.4, 170.6, 155.7, 135.5, 128.5 (2C), 128.5, 128.3 (2C), 128.2, 79.6, 66.9, 54.2, 50.7, 49.2, 41.6, 40.9, 35.6, 30.2, 28.3 (3C), 24.8, 24.6, 23.4, 23.1, 22.8, 22.0, 21.7, 21.6; MS (ESI) m/z: 562.8 [M + H]+, 579.8 [M + NH4]+, 584.9 [M + Na]+.
4.1.16. Synthesis of Tripeptide N-Boc-Phe-Me-Leu-d-Leu-OBn(T3)
The experimental procedure for the synthesis of N-Boc-Phe-Me-Leu-d-Leu-OBn(T3) followed as per described in the synthesis of N-Boc-Leu-Me-Leu-Leu-Obn(T1). In this synthesis, Boc-Phe-OH was used instead of Boc-Leu-OH. 1H-NMR (300 MHz, CDCl3), δH 7.37–7.33 (m, 5H), 7.32–7.30 (m, 1H), 7.28–7.19 (m, 5H), 6.20 (d, J = 8.3 Hz, 1H), 5.15–5.03 (m, 2H), 4.81 (dt, J = 9.1, 6.6 Hz, 1H), 4.65–4.52 (m, 2H), 3.09 (dd, J = 13.8, 6.6 Hz, 1H), 2.96–2.85 (m, 01H), 2.81 (s, 3H), 1.95–1.71 (m, 2H), 1.58–1.46 (m, 2H), 1.39 (s, 9H), 1.30 (m, 2H), 0.95 (t, J = 5.4 Hz, 9H), 0.80 (dd, J = 6.6, 1.4 Hz, 3H); MS (ESI) m/z: 595.7 [M + H]+, 612.7 [M + NH4]+, 617.9 [M + Na]+.
4.1.17. Synthesis of Tripeptide N-Boc-Phe-Me-Leu-Leu-OBn(T4)
The experimental procedure for the synthesis of N-Boc-Phe-Me-Leu-Leu-OBn(T4) followed as per described in the synthesis of N-Boc-Leu-Me-Leu-Leu-OBn(T1). In this synthesis, Boc-Phe-OH was used instead of Boc-Leu-OH. 1H-NMR (300 MHz, CDCl3), δH 7.93 (d, J = 7.9 Hz, 1H), 7.42–7.32 (m, 5H), 7.32–7.18 (m, 5H), 6.18 (d, J = 8.0 Hz, 1H), 5.31–5.20 (m, 1H), 5.20–5.13 (m, 2H), 4.85–4.71 (m, 1H), 4.71–4.55 (m, 1H), 3.18–2.99 (m, 1H), 2.90–2.81 (m, 1H), 2.79 (s, 3H), 1.88 (dd, J = 9.5, 3.6 Hz, 1H), 1.76–1.66 (m, 1H), 1.66–1.57 (m, 2H), 1.57–1.44 (m, 2H), 1.40 (d, J = 1.6 Hz, 9H), 0.98–0.76 (m, 12H); 13C-NMR (75 MHz, CDCl3), δC 173.2, 172.3, 170.2, 155.2, 135.4, 129.3, 128.9, 128.6 (2C), 128.5, 128.4, 128.3 (2C), 128.2, 128.0, 127.0, 79.9, 67.1, 55.0, 50.8, 41.1, 39.0, 36.4, 30.6, 28.3 (3C), 24.9, 24.7 (2C) 23.1, 22.8, 22.0, 21.9; MS (ESI) m/z: 595.7 [M + H]+, 612.7 [M + NH4]+, 617.9 [M + Na]+.
4.1.18. Synthesis of Tripeptide N-Boc-Phe-Me-d-Leu-Leu-OBn(T5)
The experimental procedure for the synthesis of N-Boc-Phe-Me-d-Leu-Leu-OBn(T5) followed as per described in the synthesis of N-Boc-Leu-Me-Leu-Leu-OBn(T1). In this synthesis, D2 was used instead of D1. 1H-NMR (300 MHz, CDCl3), δH 7.41–7.33 (m, 5H), 7.33–7.20 (m, 5H), 6.89 (d, J = 7.4 Hz, 1H), 5.25 (d, J = 6.8 Hz, 1H), 5.21–5.13 (m, 2H), 5.08 (d, J = 12.2 Hz, 1H), 4.71 (dd, J = 14.8, 6.8 Hz, 1H), 4.66–4.57 (m, 1H), 3.04–2.94 (m, 1H), 2.60 (s, 3H), 1.79–1.67 (m, 2H), 1.55 (dd, J = 12.9, 5.8 Hz, 2H), 1.43 (s, 9H), 1.39–1.24 (m, 2H), 0.91 (dd, J = 6.0, 2.9 Hz, 6H), 0.86 (d, J = 6.5 Hz, 6H); MS (ESI) m/z: 95.7 [M + H]+, 612.7 [M + NH4]+, 617.9 [M + Na]+.
4.1.19. Synthesis of Linear Pentapeptide N-Boc-Me-Leu-Leu-Phe-Me-d-Leu-Leu-OBn(P1)
In a 50-mL round-bottom flask with N2 protection, D1 (1.68 g) was dissolved in ethyl acetate (20 mL), and then 10% catalyst Pd/C catalyst (0.56 g) was added. The flask was sealed and flushed with H2, the solution was stirred at room temperature and monitored for the progress of the reaction by TLC. After 4–6 h of reaction when the TLC point of the raw materials disappeared, H2 flow was closed, and the reaction was stopped. The solution was first filtered to recycle the catalyst Pd/C, and the rest of the solution was distillated in a vacuum to remove the solvent residues and was dried in the oven. Finally, the colorless product of D1-B was obtained and used for the following step.
The previously-obtained product T5 (1.61 g) was dissolved in CH2Cl2 (20 mL) under N2 atmosphere, then anisole (0.76 mL, 7.2 mmol) was added under stirring and cooled in an ice bath. TFA (8 mL) was added to the solution slowly and stirred for reaction 4 h. Thereafter, the solvent was removed by vacuum distillation, then additional appropriate dichloromethane was added and distillated under vacuum again to remove traces of TFA. This step was repeated three times for complete removal of TFA. Finally, the oily product of T5-A was obtained for the next step.
The above obtained T5-A was dissolved in 5 mL THF under stirring and cooled in an ice bath. Subsequently, DEPBT (2.15 g, 7.2 mmol) and DIEA (1.26 mL, 7.2 mmol) were added into the solution and reacted for 10 min Thereafter, an appropriate amount of the above obtained D1-B THF solution (5 mL) was added, and DIEA was used to adjust the pH of the solution to 7–8. The temperature was increased slowly to room temperature and monitored for the progress of reaction by TLC. The solution was stirred 24 h; the solvent was removed by vacuum distillation; and the residual crude product was purified directly using silica-gel column chromatography (eluant ratio of Vpetroleum ether/Vacetone = 20:1). 1H-NMR (300 MHz, CDCl3), δH 7.31 (m, 7H), 7.23 (m, 3H), 6.76 (d, J = 8.4 Hz, 1H), 6.36 (d, J = 8.4 Hz, 1H), 5.30 (d, J = 8.4 Hz, 1H), 5.13 (s, 2H), 5.01 (m, 1H), 4.83 (m, 2H), 4.70 (m, 1H), 4.56 (m, 1H), 2.96 (s, 3H), 2.73 (m, 2H), 2.67 (s, 3H), 1.64 (m, 2H), 1.59 (m, 6H), 1.49 (m, 4H), 1.41 (s, 9H), 0.91 (m, 24H); 13C-NMR (75 MHz, CDCl3), δC 172.62, 172.5, 171.8, 169.8, 169.5, 137.7, 128.9, 128.8, 128.2 (2C), 128.1, 128.1, 128.0, 128.0, 127.8 (2C), 127.5, 126.6, 76.8, 66.6, 66.2, 54.2, 54.1, 50.2, 47.2, 40.8, 40.7, 39.5, 39.3, 35.5, 30.0, 29.9, 27.9 (3C), 24.4, 24.3, 24.3, 24.2, 22.8, 22.6, 22.5, 22.4, 21.7, 21.6, 21.2, 21.1; MS (ESI) m/z: 836.9 [M + H]+, 853.9 [M + NH4]+, 858.9 [M + Na]+.
4.1.20. Synthesis of Linear Pentapeptide N-Boc-Me-Leu-Leu-Phe-Me-Leu-d-Leu-OBn(P2)
The experimental procedure was followed as per described for the synthesis of P1. In this synthesis, T3 and T3-A were used instead of T5 and T5-A in the corresponding places, respectively. 1H-NMR (300 MHz, CDCl3), δH 7.36–7.27 (m, 5H), 7.27–7.12 (m, 5H), 6.94 (d, J = 8.3 Hz, 1H), 6.55 (d, J = 27.8 Hz, 2H), 5.14 (s, 1H), 5.15–5.09 (m, 2H), 5.04 (dd, J = 19.1, 6.7 Hz, 1H), 4.72–4.54 (m, 2H), 4.45 (dd, J = 18.2, 10.1 Hz, 1H), 3.30–3.05 (m, 1H), 2.98–2.87 (m, 1H), 2.85 (s, 3H), 2.71 (s, 3H), 1.92–1.52 (m, 8H), 1.48 (d, J = 4.5 Hz, 9H), 1.39–1.23 (m, 4H), 0.99–0.74 (m, 24H); MS (ESI) m/z: 836.7 [M + H]+, 853.8 [M + NH4]+, 858.7 [M + Na]+.
4.1.21. Synthesis of Linear Pentapeptide N-Boc-Me-d-Leu-Leu-Phe-Me-Leu-Leu-OBn(P3)
The experimental procedure was followed as per described for the synthesis of P1. In this synthesis, D2 and D2-B were used instead of D1 and D1-B, and T4 and T4-A were used instead of T5 and T5-A in the corresponding places, respectively. 1H-NMR (300 MHz, CDCl3), δH 7.46–7.33 (m, 5H), 7.20 (dt, J = 12.2, 7.3 Hz, 5H), 7.05 (d, J = 8.1 Hz, 1H), 6.23 (d, J = 4.7 Hz, 1H), 5.70 (dd, J = 11.8, 5.2 Hz, 1H), 5.27–5.16 (m, 2H), 5.16–5.09 (m, 1H), 4.77–4.56 (m, 2H), 4.31 (dd, J = 13.0, 5.3 Hz, 1H), 3.63–3.46 (m, 1H), 2.92 (s, 3H), 2.84 (s, 3H), 2.82–2.71 (m, 1H), 2.10–1.61 (m, 8H), 1.44 (s, 9H), 1.26 (dd, J = 15.0, 5.5 Hz, 1H), 1.02 (d, J = 6.4 Hz, 3H), 1.00–0.92 (m, 12H), 0.91 (d, J = 1.9 Hz, 6H), 0.74–0.58 (m, 6H); 13C-NMR (75 MHz, CDCl3), δC 174.9, 173.3 172.7, 171.6, 170.3, 155.7, 137.3, 135.6, 128.8, 128.6 (2C), 128.5 (2C), 128.4 (2C), 128.3 (2C), 126.5, 79.7, 66.9, 56.9, 54.8, 51.0, 49.2, 48.3, 41.6, 40.4, 40.3, 35.3, 33.6, 30.9, 30.4, 28.3 (3C), 24.8 (2C), 24.6, 24.5, 24.1, 23.4, 23.3, 22.9 (2C), 22.1, 21.7, 21.4; MS (ESI) m/z: 836.7 [M + H]+, 853.8 [M + NH4]+, 858.7 [M + Na]+.
4.1.22. Synthesis of Linear Pentapeptide N-Boc-Me-Leu-d-Leu-Phe-Me-Leu-Leu-OBn(P4)
The experimental procedure was followed as per described for the synthesis of P1. In this synthesis, D3 and D3-A were used instead of D1and D1-A, and T4 and T4-B were used instead of T5 and T5-B in the corresponding places, respectively. 1H-NMR (300 MHz, CDCl3), δH 7.36 (dt, J = 11.0, 3.6 Hz, 5H), 7.30–7.20 (m, 5H), 6.48 (d, J = 8.6 Hz, 1H), 6.38 (d, J = 8.1 Hz, 1H), 5.30 (d, J = 9.1 Hz, 1H), 5.24–5.15 (m, 2H), 5.14–5.03 (m, 2H), 5.00–4.89 (m, 1H), 4.84 (d, J = 9.0 Hz, 1H), 4.63–4.49 (m, 1H), 3.11–2.95 (m, 1H), 2.93 (s, 3H), 2.86 (s, 3H), 2.82–2.67 (m, 1H), 1.81–1.50 (m, 8H), 1.46 (d, J = 7.8 Hz, 4H), 1.40 (s, 9H), 1.08–0.73 (m, 24H); 13C-NMR (75 MHz, CDCl3), δC 173.5, 173.1, 172.4, 170.5, 170.2, 155.2, 135.5, 134.0, 129.3, 128.7 (2C), 128.6 (3C), 128.4, 128.2 (2C), 127.0, 79.8, 66.9, 55.0, 54.3, 51.9, 50.7, 47.7, 41.4, 41.0, 39.0, 35.5, 30.6, 30.2, 28.4, 28.3 (3C), 24.9, 24.8, 24.6, 23.4, 23.1, 23.1, 22.9, 22.8, 22.0, 22.0, 21.7, 21.6; MS (ESI) m/z: 836.7 [M + H]+, 853.8 [M + NH4]+, 858.7 [M + Na]+.
4.1.23. Synthesis of Linear Pentapeptide N-Boc-Leu-Me-Leu-Leu-Me-Phe-Leu-OBn(P5)
The experimental procedure was followed as per described for the synthesis of P1. In this synthesis, T4 and T4-A were used instead of T5 and T5-A in the corresponding places, respectively. 1H-NMR (300 MHz, CDCl3), δH 8.55–8.15 (m, 1H), 7.43–7.33 (m, 5H), 7.32–7.17 (m, 5H), 7.15–7.09 (m, 1H), 5.19 (dd, J = 15.3, 2.9 Hz, 1H), 5.15–5.11 (m, 1H), 4.90–4.70 (m, 1H), 4.69–4.53 (m, 2H), 4.42–4.19 (m, 1H), 3.35–3.20 (m, 1H), 3.07 (dd, J = 18.9, 8.5 Hz, 1H), 2.95 (s, 3H), 2.77 (s, 3H), 1.81–1.65 (m, 8H), 1.44 (s, 9H), 1.32–1.22 (m, 2H), 0.99–0.48 (m, 24H); 13C-NMR (75 MHz, CDCl3), δC 174.1, 172.9, 172.8, 171.4, 169.4, 155.8, 137.9, 135.8, 129.6, 129.2 (2C), 128.9 (2C), 128.6 (2C), 128.2, 128.0, 127.0, 77.2, 67.0, 66.8, 62.7, 53.5, 51.5, 49.0, 47.0, 42.0, 39.9, 35.4, 34.0, 29.9, 29.5, 28.3 (3C), 24.8, 24.6 (2C), 24.3, 23.9, 23.5, 23.4, 23.1, 22.9, 21.7, 21.2, 19.9; MS (ESI) m/z: 836.7 [M + H]+, 853.8 [M + NH4]+, 858.7 [M + Na]+.
4.1.24. Synthesis of Linear Pentapeptide N-Boc-Leu-Me-Leu-Leu-Me-d-Phe-Leu-OBn(P6)
The experimental procedure was followed as per described for the synthesis of P1. In this synthesis, D1 and D1-A were used instead of T1 and T1-A, and T5 and T5-B were used instead of D5 and D5-B in the corresponding places, respectively. 1H-NMR (500 MHz, CDCl3), δH 7.31 (m, 7H), 7.26–7.23 (m, 3H), 6.76 (d, 1H, J = 10.0 Hz), 6.36 (d, 1H, J = 10.0 Hz), 5.30 (d, 1H, J = 10.0 Hz), 5.13 (s, 2H), 5.05–5.01 (m, 1H), 4.90–4.83 (m, 2H), 4.73–4.70 (m, 1H), 4.59–4.56 (m, 1H), 2.96 (s, 3H), 2.77–2.73 (m, 2H), 2.67 (s, 3H), 1.67–1.64(m, 2H), 1.62–1.67 (m, 6H), 1.50–1.46 (m, 4H), 1.41 (s, 9H), 0.98–088 (m, 24H), MS (ESI) m/z: 837.0 [M + H]+, 854.0 [M + NH4]+, 859.0 [M + Na]+.
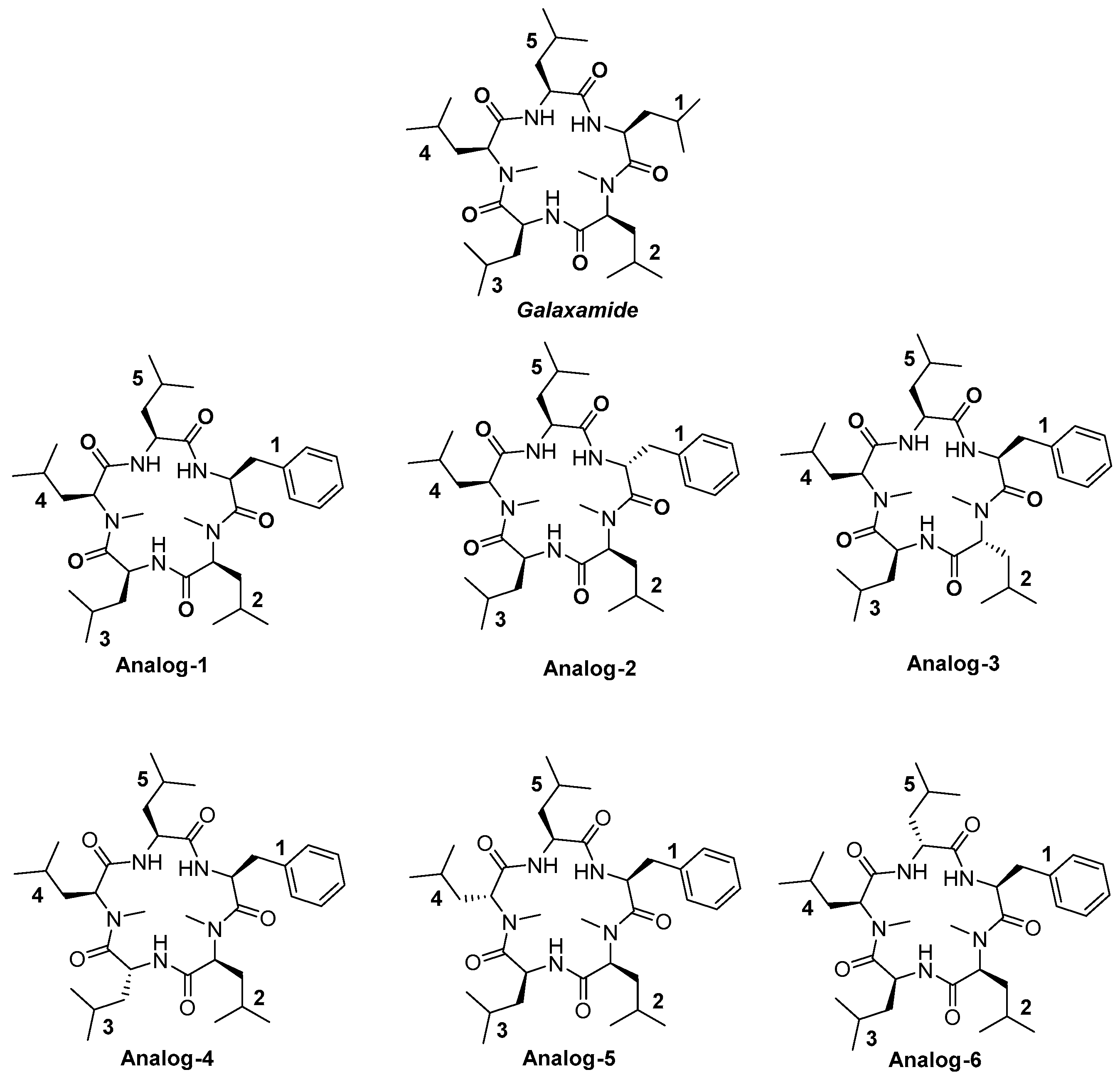
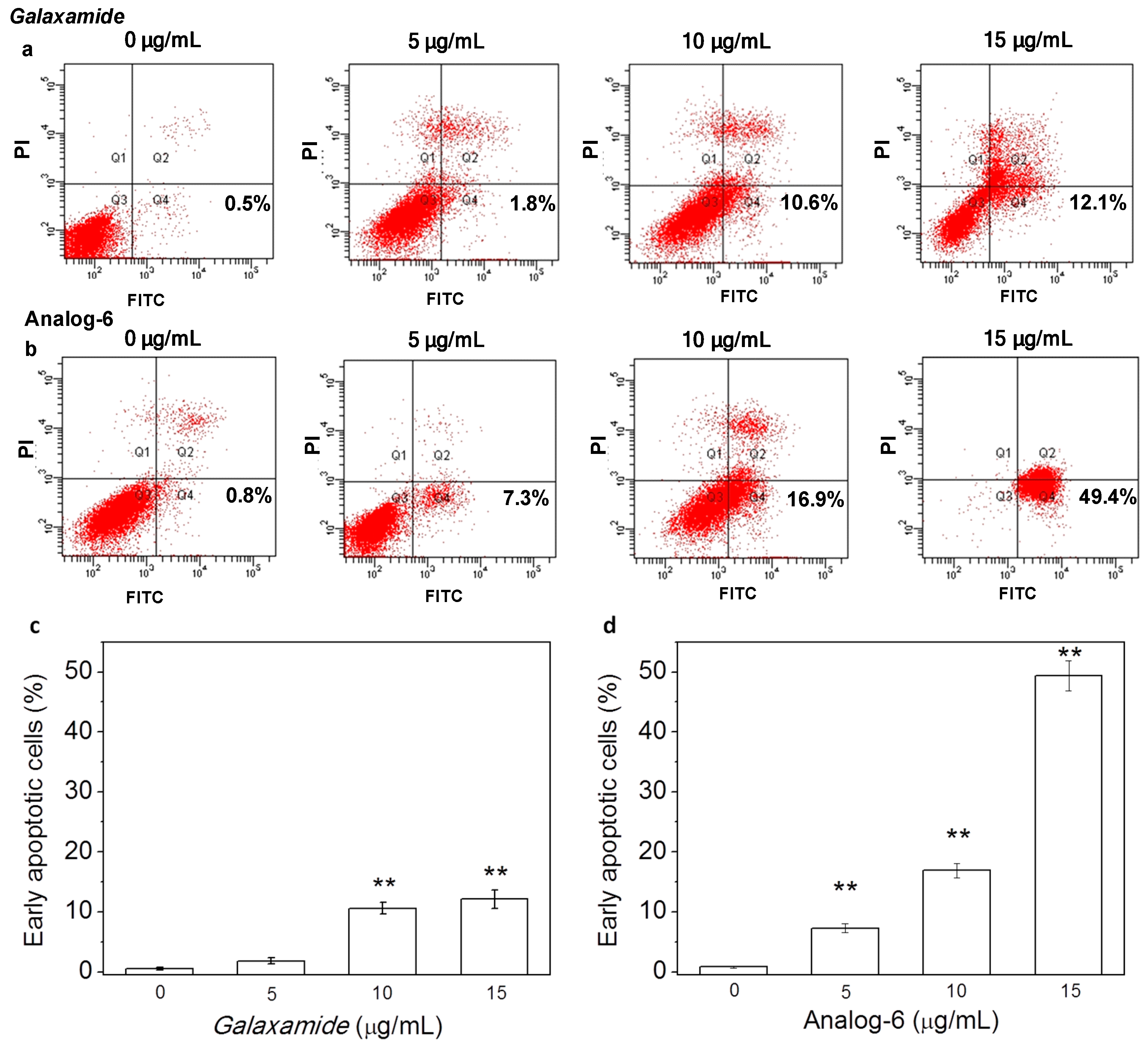
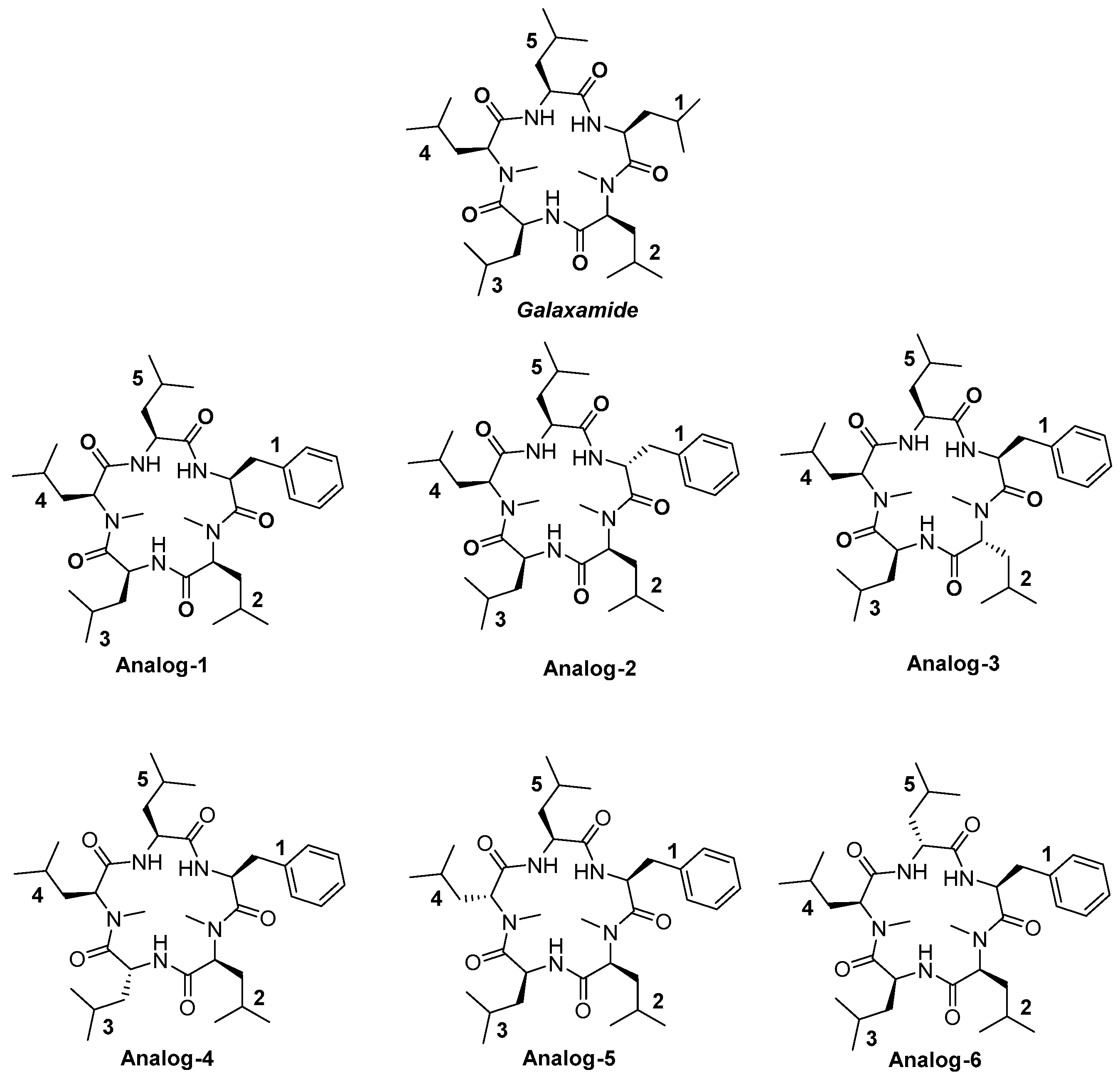
4.1.25. Synthesis of Cyclo(Me-Leu-Leu-Phe-Me-Leu-Leu) (Analog-1)
P5 (0.12 g, 0.15 mmol) was first dissolved in dichloromethane (20 mL) in a 50-mL round-bottom flask, and then, TFA (5 mL) was added under stirring and cooled in an ice bath. After the reaction was stirred for 4 h, the solvent was removed by vacuum distillation, then additional appropriate dichloromethane was added and distillated under vacuum again. This step was repeated three times for complete removal of TFA; finally, an oily product was obtained after drying. Then, the obtained oily product was dissolved in ethyl acetate (20 mL) under N2 atmosphere, and 10% Pd/C (0.2 g) catalyst was added. The flask was sealed and flushed with H2; the solution was stirred under room temperature and monitored for the progress of reaction by TLC. After 4–6 h of reaction when the TLC point of the raw materials disappeared, H2 flow was closed, and the reaction was stopped. The solution was first filtered to recycle the catalyst Pd/C, and the rest of the solution was distillated under vacuum to remove the solvent residues and was dried in the oven. Finally, a colorless product was obtained. The as-obtained product was then dissolved in 50 mL THF/CH2Cl2/CH3CN (2:1:2) mixed solvent under N2 atmosphere; 0.067 g HATU, 0.065 g DEPBT and 0.07 g TBTU were added in sequence and allowed to react for 4 days. The solvent was removed by vacuum distillation, and the solid product was dissolved in CH2Cl2 and washed by saturated NaHCO3 and H2O separately. The resultant organic layer was dried by anhydrous Na2SO4 and concentrated, then purified by RP-HPLC (Agilent Eclipse ZORBAX SB-C18, 5 µm, 9.4 × 250 nm). Finally, 11.2 mg white powder with a yield of 40.2% were obtained. 1H-NMR (300 MHz, CDCl3), δH 8.15 (d, J = 9.7 Hz, 1H), 8.15 (d, J = 9.7 Hz, 1H), 7.53 (d, J = 8.7 Hz, 1H), 7.53 (d, J = 8.7 Hz, 1H), 7.36–7.25 (m, 3H), 7.18–7.11 (m, 2H), 5.80 (d, J = 7.3 Hz, 1H), 4.86–4.72 (m, 2H), 4.27 (ddd, J = 11.1, 7.3, 3.6 Hz, 1H), 3.67 (dd, J = 10.3, 5.8 Hz, 1H), 3.67 (dd, J = 10.3, 5.8 Hz, 1H), 3.45 (d, J = 7.8 Hz, 1H), 3.39 (dd, J = 8.7, 6.4 Hz, 1H), 3.12 (dd, J = 12.9, 5.8 Hz, 1H), 2.95 (s, 3H), 2.81 (s, 3H), 2.32 (ddd, J = 14.2, 8.7, 5.7 Hz, 1H), 2.02–1.90 (m, 1H), 1.90–1.73 (m, 4H), 1.60–1.55 (m, 1H), 1.50–1.32 (m, 4H), 1.26–1.18 (m, 1H), 0.95–0.91 (m, 9H), 0.89 (d, J = 4.7 Hz, 9H), 0.86 (s, 3H), 0.83 (d, J = 6.5 Hz, 3H); 13C-NMR (75 MHz, CDCl3), δC 174.5, 173.0, 172.6, 171.2, 169.7, 136.5 (2C), 129.2 (2C), 129.1, 127.6, 71.1, 68.0, 53.3, 49.3, 48.1, 41.5, 41.1, 40.7, 40.0, 39.6, 38.8, 34.0, 26.1, 25.2, 25.1, 25.0, 23.6, 23.4, 23.2, 23.1, 22.6, 22.1, 21.8, 21.3; MS (ESI) m/z: 628.8 [M + H]+, 645.8 [M + NH4]+, 650.8 [M + Na]+.
For the detailed characterization of mass spectra, mass-mass spectra,
1H-NMR and HPLC regarding Analog-1, refer to
Figures S2A, S3A, S4A and S5A.
4.1.26. Synthesis of Cyclo(Me-Leu-Leu-d-Phe-Me-Leu-Leu) (Analog-2)
The experimental procedure was followed as described in the synthesis of Analog-1. In this synthesis, P6 was used instead of P5. Finally, 31.8 mg white powder with a yield of 42.3% were obtained. 1H-NMR (500 MHz, CDCl3), δH 8.13 (s, 1H), 7.74–7.71 (m, 1H), 7.29–7.19 (m, 5H), 6.91 (d, J = 10.0 Hz, 1H), 5.18 (t, J = 10.0Hz, 1H), 4.91–4.87 (m, 1H), 4.79–4.74 (m, 3H), 3.09–3.05 (m, 2H), 2.94–2.91 (m, 1H), 2.84 (s, 1H), 2.80 (s, 2H), 2.62 (s, 3H), 1.93–1.88 (m, 1H), 1.82–1.77 (m, 1H), 1.61–1.50 (m, 6H), 1.43–1.37 (m, 1H), 1.12–1.06 (m, 1H), 0.97–0.89 (m, 18H), 0.73–0.66 (m, 6H); 13C-NMR (125 MHz, CDCl3), δC 175.9, 171.7, 171.5, 170.1, 170.0, 136.2, 129.1 (2C), 128.8 (2C), 127.2, 57.8, 53.7 (2C), 48.7, 47.8, 41.5, 41.1, 37.7, 37.5, 34.5, 31.4, 29.5, 24.9 (2C), 24.8, 24.5, 23.2, 23.1, 22.9 (2C), 22.2, 21.9, 21.8, 21.6; MS (ESI) m/z: 628.8 [M + H]+, 645.9 [M + NH4]+, 650.8 [M + Na]+.
4.1.27. Synthesis of Cyclo(Me-Leu-Leu-Phe-Me-d-Leu-Leu) (Analog-3)
The experimental procedure was followed as described in the synthesis of Analog-1. In this synthesis, P1 was used instead of P5. Finally, 17.0 mg white powder with a yield of 38.5% were obtained. 1H-NMR (300 MHz, CDCl3), δH 7.97 (d, J = 9.1 Hz, 1H), 7.73 (dd, J = 5.7, 3.3 Hz, 1H), 7.55 (dd, J = 5.7, 3.3 Hz, 1H), 7.25 (dd, J = 7.6, 3.4 Hz, 4H), 6.20 (d, J = 9.4 Hz, 1H), 4.24 (dd, J = 5.9, 3.7 Hz, 1H), 3.59 (dt, J = 9.6, 6.7 Hz, 1H), 3.26 (dd, J = 8.1, 5.2 Hz, 1H), 3.10 (s, 2H), 2.96 (s, 2H), 1.59–1.40 (m, 8H), 1.32 (dd, J = 19.1, 8.3 Hz, 6H), 0.94 (dd, J = 8.7, 4.4 Hz, 20H), 0.83 (dd, J = 8.1, 6.7 Hz, 6H); 13C-NMR (75 MHz, CDCl3), δC 172.9, 172.8, 171.3, 171.1, 168.8, 136.1, 130.9, 129.5, 128.8 (2C), 128.6 (2C), 127.0, 68.2, 64.0, 59.0, 55.1, 49.7, 47.3, 41.7, 39.1, 38.7, 37.1, 30.6, 30.4, 28.9, 25.3, 25.0, 24.8, 23.8, 23.3, 23.0, 22.9, 22.7, 22.6, 22.5, 21.8; MS (ESI) m/z: 628.8 [M + H]+, 645.8 [M + NH4]+, 650.8 [M + Na]+.
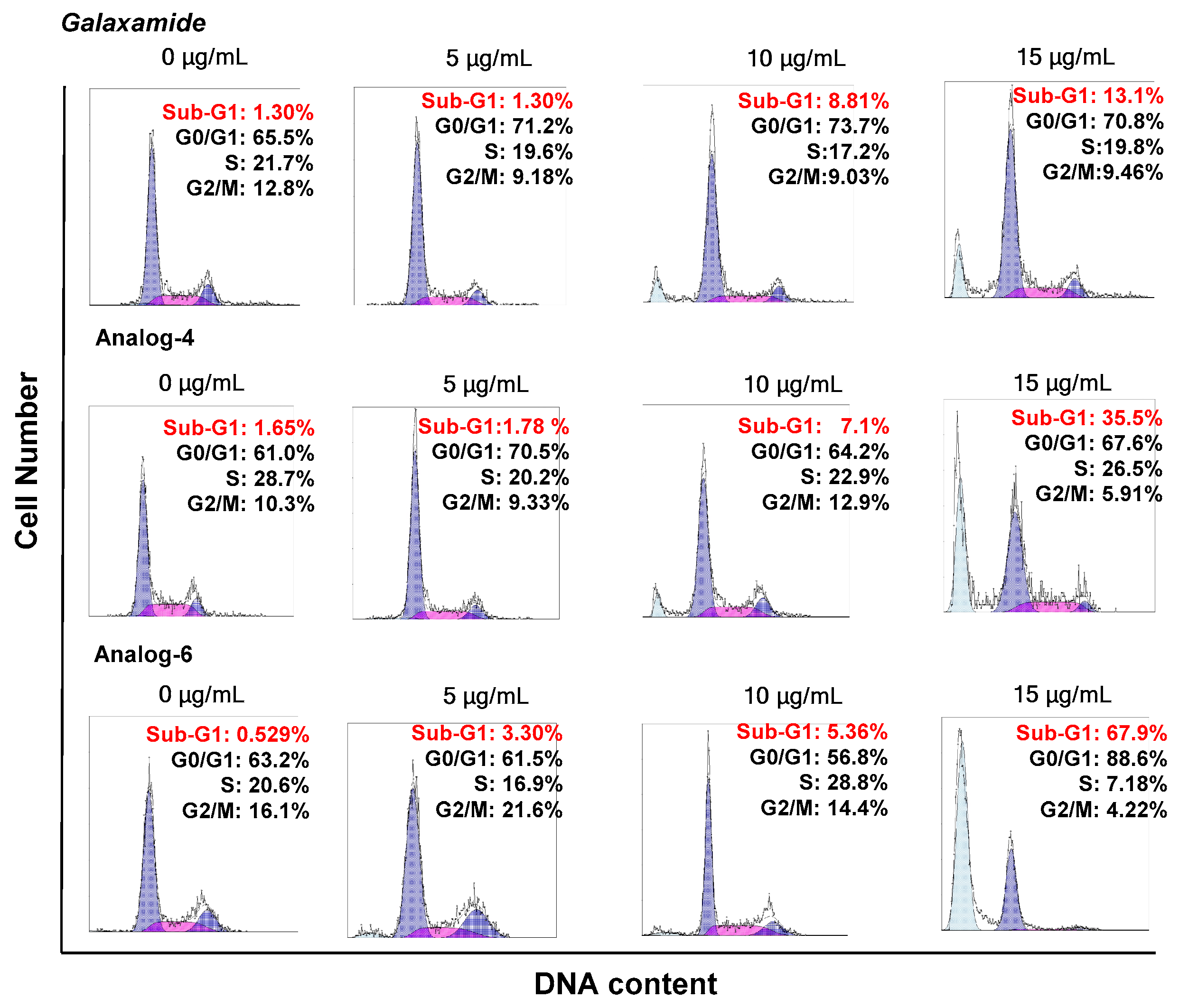
4.1.28. Synthesis of Cyclo(Me-Leu-Leu-Phe-Me-Leu-d-Leu) (Analog-4)
The experimental procedure was followed as described in the synthesis of Analog-1. In this synthesis, P2 was used instead of P5. Finally, 17.6 mg white powder with a yield of 34.2% were obtained. 1H-NMR (300 MHz, CDCl3), δH 7.28–7.17 (m, 5H), 6.95 (d, J = 9.8 Hz, 1H), 6.69 (d, J = 9.9 Hz, 1H), 5.28 (dd, J = 17.0, 8.1 Hz, 1H), 5.12 (t, J = 7.7 Hz, 1H), 4.90 (dd, J = 16.4, 8.6 Hz, 1H), 4.76 (dd, J = 15.4, 7.1 Hz, 1H), 4.59 (dd, J = 8.7, 6.1 Hz, 1H), 4.37 (dd, J = 17.4, 7.7 Hz, 1H), 3.22 (dd, J = 13.1, 8.3 Hz, 1H), 3.05–2.96 (m, 1H), 2.81 (s, 3H), 2.61 (s, 3H), 1.66–1.41 (m, 8H), 1.39–1.21 (m, 4H), 1.00–0.79 (m, 24H); 13C-NMR (75 MHz, CDCl3), δC 173.4, 172.1, 170.6, 169.6, 167.8, 136.2, 129.2 (2C), 128.8, 128.7, 127.1, 58.5, 53.8, 51.7, 49.2, 48.4, 40.9, 40.6, 38.7, 38.0, 34.2, 29.1 (2C), 25.0, 24.9, 24.8, 24.7, 24.6, 23.0, 22.9, 22.7, 22.6, 22.5, 22.2, 22.1; MS (ESI) m/z: 628.8 [M + H]+, 645.8 [M + NH4]+, 650.8 [M + Na]+.
4.1.29. Synthesis of Cyclo(Me-d-Leu-Leu-Phe-Me-Leu-Leu (Analog-5)
The experimental procedure was followed as described in the synthesis of Analog-1. In this synthesis, P3 was used instead of P5. Finally, 39 mg white powder with a yield of 36.5% were obtained. 1H-NMR (300 MHz, CDCl3), δH 7.66 (d, J = 8.7 Hz, 1H), 7.28–7.17 (m, 5H), 6.74 (d, J = 9.8 Hz, 1H), 5.26–5.15 (m, 1H), 5.11 (dd, J = 9.4, 6.2 Hz, 1H), 5.00 (td, J = 8.4, 6.4 Hz, 1H), 4.85–4.79 (m, 1H), 4.63–4.50 (m, 1H), 4.46 (dd, J = 10.4, 6.6 Hz, 1H), 3.30–3.17 (m, 1H), 2.96 (t, J = 5.7 Hz, 1H), 2.84 (s, 3H), 2.54 (s, 3H), 2.12–1.76 (m, 4H), 1.62–1.48 (m, 8H), 1.01–0.75 (m, 24H); 13C-NMR (75 MHz, CDCl3), δC 173.5, 171.8, 171.5, 169.6, 167.9, 137.3, 129.6, 129.4, 128.3 (2C), 126.5, 58.3, 53.8, 51.7, 51.1, 45.8, 41.0, 38.6, 37.8, 37.2, 34.0, 29.7, 29.1, 25.4 24.9, 24.8, 24.7, 24.4, 23.3, 23.0, 22.9, 22.6, 22.4, 22.2, 21.9; MS (ESI) m/z: 628.8 [M + H]+, 645.8 [M + NH4]+, 650.8 [M + Na]+.
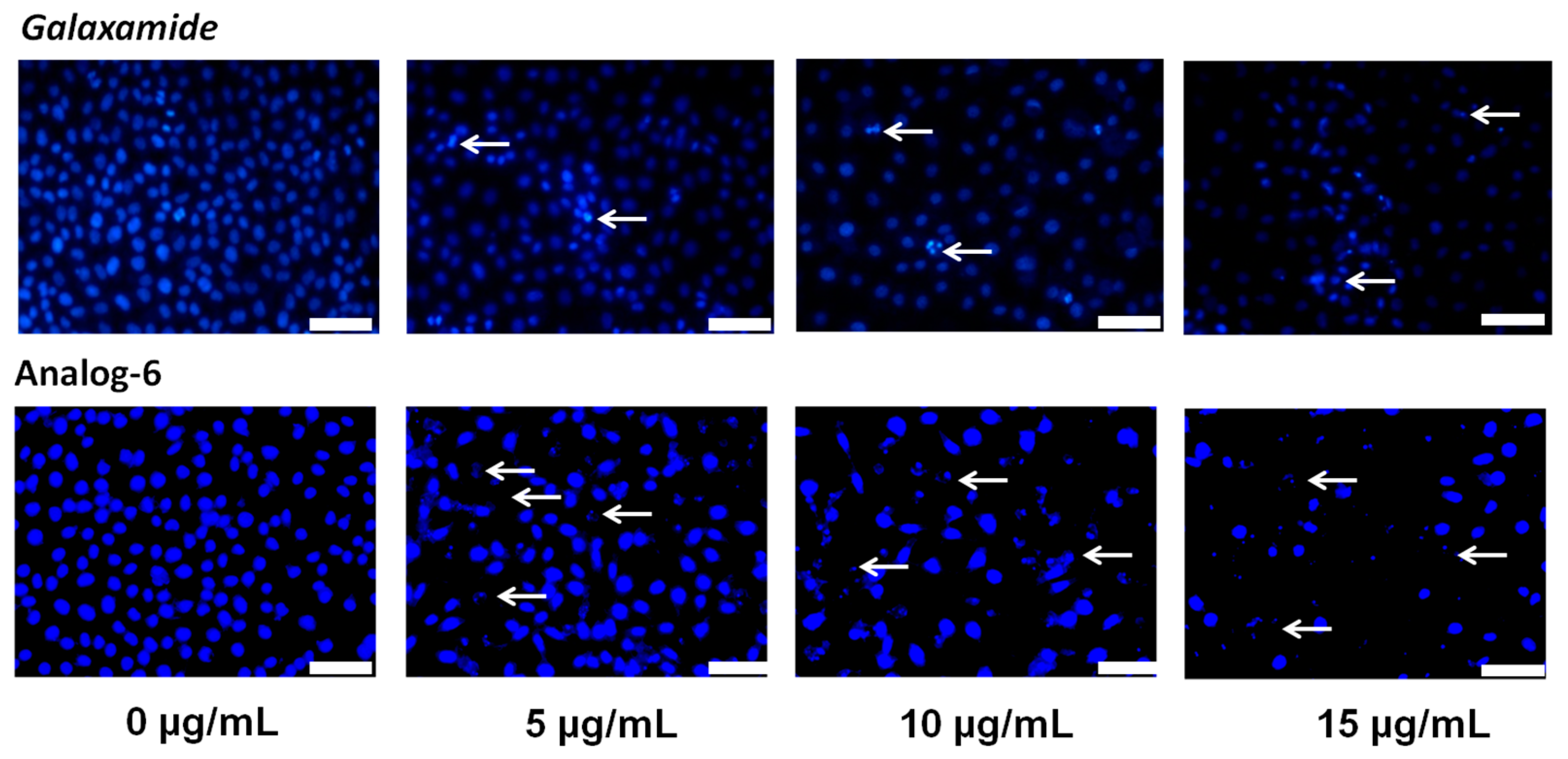
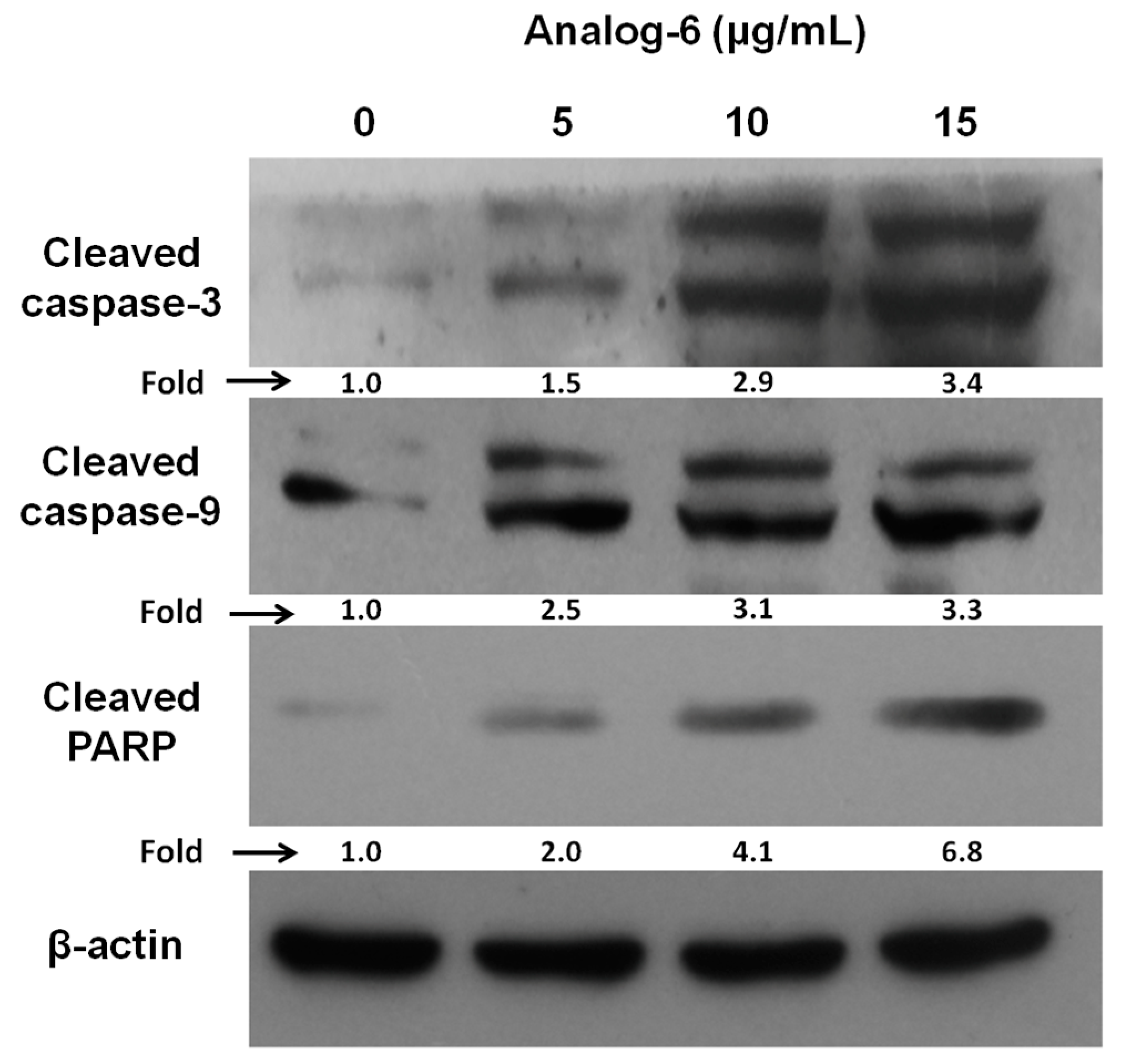
4.1.30. Synthesis of Cyclo(Me-Leu-d-Leu-Phe-Me-Leu-Leu) (Analog-6)
The experimental procedure was followed as described in the synthesis of Analog-1. In this synthesis,P4 was used instead of P5. Yield: 42.5%. 1H-NMR (300 MHz, CDCl3), δH 7.28–7.17 (m, 5H), 7.03–6.87 (m, 1H), 6.71 (d, J = 9.6 Hz, 1H), 5.25 (dd, J = 16.3, 8.2 Hz, 1H), 5.11 (t, J = 7.6 Hz, 1H), 4.91 (t, J = 7.6 Hz, 1H), 4.76 (dd, J = 15.5, 7.0 Hz, 1H), 4.57 (dd, J = 8.8, 5.9 Hz, 1H), 4.34 (dd, J = 17.1, 7.6 Hz, 1H), 3.21 (dd, J = 13.0, 8.6 Hz, 2H), 2.79 (s, 3H), 2.60 (s, 3H), 1.53 (ddd, J = 17.6, 12.8, 7.0 Hz, 8H), 1.42–1.22 (m, 4H), 1.00–0.79 (m, 24H); 13C-NMR (75 MHz, CDCl3), δC 173.3, 172.0, 170.9, 169.9, 167.7, 136.1, 129.2 (2C), 128.8 (2C), 127.2, 58.5, 53.8, 51.8, 50.2, 49.3, 48.4, 40.9, 40.4, 37.9, 34.2, 29.1, 25.1, 24.9, 24.8, 24.6, 23.0, 22.9, 22.7, 22.6, 22.4, 22.4, 22.2, 22.1, 21.7; MS (ESI) m/z: 628.8 [M + H]+, 645.8 [M + NH4]+, 650.8 [M + Na]+.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}