Abstract
We previously reported that various mRNAs were associated with postsynaptic density (PSD) purified from rat forebrain. Among the thousands of PSD-associated mRNAs, we highlight the biology of the general transcription factor II-I (Gtf2i) mRNA, focusing on the significance of its versatile splicing for targeting its own mRNA into dendrites, regulation of translation, and the effects of Gtf2i expression level as well as its relationship with neuropsychiatric disorders.
1. Introduction
We previously reported that a large number of mRNAs are associated with postsynaptic density (PSD) prepared from rat forebrain [1,2]. Later, deep sequencing of hippocampal neuropil-residing mRNAs supported the abundance of RNAs in dendrites/axons [3]. Among the thousands of PSD-associated mRNAs, more than 100 mRNAs encode nuclear proteins.
De novo synthesis of proteins is necessary for the induction of plastic modification in stimulated synapses, which form the molecular basis of learning and memory [4,5]. Translation of postsynaptically localized mRNAs at the stimulated synapses is one of the mechanisms for spatiotemporally specific modifications in stimulated synapses [6,7]. Short-term plastic modifications in synapses are explained by modification and mobilization into synapse active zones of synaptic proteins, such as receptors, cytoskeletons, and various signaling molecules. However, this may not be enough for the long-term or long-lasting maintenance of the plastic modification in synapses. Long-lasting synaptic modification (maintenance and stabilization) requires novel protein synthesis both at postsynaptic sites and via transcription in the nucleus [8]. For example, transcription of brain-derived neurotrophic factor (BDNF) [9] and Arc [10,11] is induced after neural activation. Synapse-to-nucleus signal transmission should occur ahead of the transcription of these genes.
Dendritic mRNAs encoding nuclear proteins contained mRNAs encoding transcription factors. These transcription factor-encoding mRNAs are supposed to be transferred into the neuronal dendrites and locally translated there upon synaptic stimulation. Their products are translocated back into the nucleus, where they transcribe downstream genes involved in long-term synaptic modification (Figure 1). Maturation of mRNAs may occur in the dendrites, in addition to cell soma [12]. The mRNA encoding general transcription factor II-I (Gtf2i) is one of these PSD-associated mRNAs. We hypothesize that postsynaptic localization of Gtf2i mRNA is related to two events—local translation at the postsynapse and synapse-to-nucleus signal transduction. Locally translated transcription factors at the postsynapse act as signal transduction molecules transmitting synaptic signals into the nucleus. The localization of mRNA encoding a transcription factor at the postsynaptic sites is studied for the first time in Gtf2i. Therefore, until now, there has been no report revealing the fate of this protein at the postsynaptic site and its role in synaptic function. However, our hypothesis is also based on other transcription factors and molecules, such as cAMP responsive element binding protein (CREB), signal transducer and activator of transcription 3 (STAT3), Mothers against dpp (SMAD), huntingtin, and importin, that translocate from postsynaptic sites to the nucleus [7,13,14,15,16,17,18].
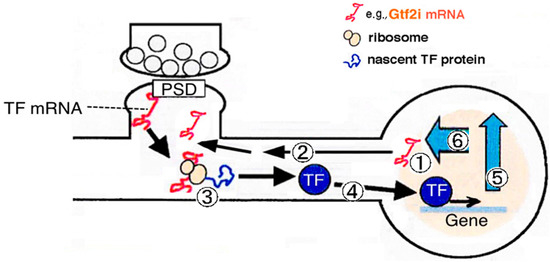
Figure 1.
Transcription, targeting, and the fate of postsynaptically localizing mRNAs that encode transcription factors. The mRNA encoding Gtf2i is one of the postsynaptic density (PSD)-associated mRNAs. It is hypothesized that locally translated transcription factors at the postsynapse act as signal transduction molecules transmitting synaptic signals into the nucleus. 1. Transcription of its own gene; 2. Targeting mRNA into the dendrite; 3. Postsynaptic local translation; 4. Synapse-to-nucleus transmission of protein products; 5. Modulated transcription of downstream genes in the nucleus; 6. Feedback effects on synapse (Multiple processes/pathways are plausible; however, their details are still unknown). TF: transcription factor.
In this review, we would discuss the regulation of the Gtf2i expression in the postsynaptic area, its downstream cellular events, and the association of GTF2I with neuropsychiatric disorders.
2. Role of Splice Variation in 5′ Untranslated Regions of Gtf2i
2.1. Role of 5′ Untranslated Region Variations in Gtf2i mRNA for Regulation of Its Own Transcription
Transcriptional regulation is achieved by promoters localizing upstream of each transcription start site (TSS). A promoter is activated by its specific intrinsic transcription factors in each of the 5′ untranslated regions (5′UTR). Alternative promoters of Gtf2i have been identified in humans and mice. In human GTF2I, three additional exons between exon 1 (encoding the known 5′UTR) and exon 2 (encoding the start codon of GTF2I), three TSSs, and four alternative 5′UTRs have been identified [19]. In mouse Gtf2i, two additional exons between exon 1 and exon 2, three TSSs, and five alternative 5′UTRs have been identified [19]. In rats, we identified seven novel TSSs, six novel exons between the exon 1 and the exon 2, and eight transcripts (including seven novel transcripts) of Gtf2i with different 5′UTRs [20]. Upstream of the TSSs of all of the Gtf2i 5′UTRs identified so far, each Gtf2i 5′UTR has its own intrinsic transcription factor-binding sites [19,20]. Multiple promoters of Gtf2i make transcription responsive to various stimuli in different types of cells. Multiple promoters of Gtf2i also enable production of various splice variants with different 5′UTRs, and multiple 5′UTRs make Gtf2i mRNA translation responsive to various stimuli at different cellular and subcellular locations.
Another possible mechanism for control of transcription of the numerous Gtf2i variants themselves is epigenetic control, since the novel 5′UTRs that we identified are embedded within CpG islands [20], where methylated cytosines could be involved in transcriptional repression. Neural activity decreases CpG methylation and activates transcription of target genes via release of repressor proteins [21,22]. This mechanism may enable cell type-specific and/or neural activity-dependent expression of each 5′UTR variant.
2.2. Role of 5′UTR Variation in Gtf2i mRNA for Subcellular Localization
Various mRNAs localize to dendrites in neuronal cells and the 5′UTR and/or 3′UTR of mRNA plays important roles in the localization to as well as local translation in dendrites [6]. The 5′UTR and the 3′UTR of mRNA play important roles in its subcellular localization, translation regulation, and stability [6,7,23,24]. This appears to be the case for Gtf2i. Gtf2i mRNAs with different 5′UTRs showed differential expression patterns in rat brains. Among eight isoforms, one was detected in dendritic processes of neuronal cells, suggesting that dendritic localization of Gtf2i mRNA variants is dependent on 5′UTR variation [20]. This variant may play a synapse-specific role, while other variants may play non-synaptic roles.
2.3. Regulation of Translation and Localization of mRNA by RNA-Binding Proteins Interacting with 5′UTRs
The presence of two cis-acting factors, G-quadruplex [20] and stem-loop structure (our unpublished data), is predicted in the 5′UTR of Gtf2i mRNA variant localizing to neuronal dendrites. The G-quadruplex is a guanine (G)-rich nucleic acid sequence that forms a four-stranded structure. The G-quadruplexes in the 5′UTRs of mRNAs play important roles in the localization of mRNAs to neuronal dendrites [25] and translation regulation of many mRNAs localized postsynaptically [26]. Some RNA-binding proteins (RBPs), as trans-acting factors, regulate translation, subcellular localization, and metabolism of mRNAs via binding to UTRs (both 5′UTRs and 3′UTRs) [27]. A well-known RBP for G-quadruplexes is fragile X mental retardation protein (FMRP), which is localized to dendrites, binds to the 5′UTR and/or 3′UTR of target mRNAs, and negatively regulates translation of the target proteins in dendrites [28,29,30,31]. Thus, FMRP negatively regulates translation of target mRNAs via direct binding to the G-quadruplex structure in 5′UTR[31]. FMRP, via its activity in dendrites, is related to the formation and maintenance of synapse structure and spine shape [32]. Abnormality in FMRP is linked to autistic symptoms of the fragile X syndrome.
The stem-loop structure of mRNA in the 5′UTR is important for translational regulation. One RBP known to interact with this structure is RNA helicase A, which recognizes this structure in the 5′UTR of various mRNAs, unwinds secondary structures, and facilitates ribosome association to enable efficient cap-dependent translation [33,34].
Thus, a mechanism involving specific RBPs is likely to regulate localization and translation of Gtf2i mRNA variants. Identification and functional analysis of RBPs that specifically recognize RNA structures in the dendritic 5′UTR of Gtf2i mRNA is an important next step. Our recent study identified RNA helicase A as one of RBPs that specifically bind to the dendritic 5′UTR of Gtf2i mRNA [35] although the site of interaction has not yet been identified. This study failed to detect FMRP until now. For its detection, standardization of the binding conditions for preservation of the optimum G-quadruplex structure may be required.
2.4. 5′UTR Works as a “Spatial Code” and a “Quantitative Code”
BDNF is an extensively studied example of a gene, similar to Gtf2i, with multiple splice variants with different 5′UTRs. Analogy with BDNF may give us a hint about the functional significance of multiple variations in Gtf2i 5′UTR. Human BDNF has 34 alternative mRNAs, which are combinations of a single coding region, 17 different 5′UTRs, and 2 different 3′UTRs with different polyadenylation sites [36]. Rodent Bdnf has 22 alternatively spliced mRNAs, which are combinations of a single coding region, 11 different 5′UTRs (I, IIA, IIB, IIC, III, IV, V, VI, VII, VIII, and IXa), and 2 different 3′UTRs with different polyadenylation sites [37]. Systematic analysis of subcellular localization of each 5′UTR in cultured rat hippocampal neurons suggests that alternatively spliced 5′UTRs selectively determine the intracellular localization of Bdnf mRNA variants to the soma, proximal dendrites (I, IV), or distal dendrites (IIC, VI) [38]. Furthermore, targeting of Bdnf mRNA variants into distal dendrites is controlled by neural activity [9]. In untreated rat hippocampal tissues, dendritic enrichment of 5′UTR variants (VI, VII in CA1; I, VI, IXa in CA3; V, VI, VII, VIII in DG) was observed. Upon neural stimulation, levels of Bdnf transcripts in dendrites were upregulated or downregulated, depending on variant types and brain region (e.g., in the CA1 region, variant mRNAs II, IV, and VI were upregulated, while variant mRNA III was downregulated) [39].
Based on these results, the “spatial code hypothesis” suggesting that different 5′UTRs regulate intracellular localization of the corresponding mRNA variants was proposed [38]. This “spatial code hypothesis” may also be applicable to Gtf2i mRNAs and could be generalized, since we found selective localization of certain type of Gtf2i mRNA variants to dendrite [20], although there are no distinctive motifs or sequences conserved between BDNF mRNA and Gtf2i mRNA.
Another functional feature of alternative 5′UTR of Bdnf is translation regulation. Translatability of each 5′UTR was assessed by an in vitro luciferase assay in a neuroblastoma cell culture model. Bdnf mRNAs with different 5′UTRs are translated differently in the basal state, and their translations were modulated differently with synaptic stimulations, such as KCl, BDNF, AMPA, NMDA, dopamine, or 5-HT [40]. Based on this, the “quantitative code hypothesis” was proposed, where translatability of Bdnf mRNA variants, and hence the expression level of BDNF protein, is determined by each 5′UTR in response to specific stimuli [40]. In this hypothesis, the factor that determines the expression level is intrinsic to the 5′UTR. Translatability of the variant mRNAs is an important factor critically affecting the expression level of protein. Therefore, 5′UTR bears a “quantitative code.” This “quantitative code hypothesis” may also be applicable to Gtf2i mRNA and could be generalized, since Gtf2i mRNA also has multiple 5′UTR variants with structures that affect translation efficiency.
3. Downstream of Gtf2i Local Translation
3.1. Synapse-to-Nucleus Signal Transduction via Gtf2i
Gtf2i is a signal-induced transcription factor, responding to signals, such as growth factor stimulation, TGFβ signaling, ER stress signaling, calcium signaling, immune signaling, and c-Src-dependent transcription activation [41,42,43,44]. Thus, this transcription factor is a versatile regulator of numerous cellular processes.
Transcription is induced by neural activity [8]. Several transcription factors, such as CREB, STAT3, and SMAD, are localized away from the soma at distal axons, locally synthesized upon stimulation, and translocated to the nucleus [7,13,14,15]. To our knowledge, there are no reports on locally translated transcription factors at the postsynaptic site. Gtf2i mRNA, at least one type of mRNA splice variant (rDEC4ED sequence-containing mRNA, see [20]), is localized at postsynaptic sites and is likely to be translated in a synaptic stimulation-dependent manner at the synapses. Gtf2i synthesized at postsynaptic sites may translocate into the nucleus, via nuclear localization signals (NLSs), and regulate expression of their downstream target genes. Gtf2i variants play differential roles in signal-induced transcription regulation. For example, the β isoform represses transcription of c-Fos, while the Δ isoform acts as an activator in murine fibroblasts [42].
3.2. Downstream Target Genes and Cellular Functions of Gtf2i
Various downstream genes and cellular functions of Gtf2i have been unveiled. In a promoter binding study, downstream target genes for Gtf2i transcription factor included those involved in axon guidance, neurodevelopmental disorders, calcium signaling, and the cell cycle [45]. Though the knockout in the mouse is embryonically lethal, Gtf2i−/− mouse embryos showed alterations in gene expression related to TgfbrII/Alk1/Smad5 and Vegfr-2 signaling cascades [46], suggesting that Gtf2i is upstream of these signaling pathways during embryogenesis.
Involvement of Gtf2i in the transcription of the Dlx5/Dlx6 homeobox genes has been suggested [47]. Gtf2i binds to the I56i enhancer region of the Dlx5/Dlx6 and possibly modulates affinity of Dlx1 and Dlx2 towards the I56i enhancer region [48] to regulate transcription of Dlx5/Dlx6 [47]. This suggests that Gtf2i is involved in the maturation of inhibitory interneurons since Dlx5 and Dlx6 are involved in the differentiation and migration of GABA-expressing interneurons in the forebrain [47,49]. Thus, Gtf2i is proposed to be a regulator of Dlx5/Dlx6 expression via activation of the I56i enhancer region and may regulate maturation of GABAergic interneurons and finally alter excitatory/inhibitory balance of neural circuits.
Additionally, GTF2I has been found to regulate transcription of DYX1C1 via binding to the promoter (or 5′UTR) region of DYX1C1 [50]. DYX1C1 is implicated in dyslexia [51], neural migration [52], cortical development, and spatial learning [53,54]. Thus, abnormality of GTF2I is related to the pathogenesis of dyslexia possibly via impaired DYX1C1 transcription [50]. These reports suggest that Gtf2i is an upstream regulator of various brain functions, including neuronal development, inhibitory synapse maturation, and neural circuit formation.
3.3. Coding Variants May Differentially Regulate Downstream Transcription and Cellular Processes
Combination of four in-frame cassettes of exons 9–12 produces Gtf2i variants in the coding region, and nine such coding variants have been discovered—Gtf2iα (exons 9–10–11), Gtf2iβ (exons 9–11–12), Gtf2iγ (exons 9–10–11–12), Gtf2iΔ (exons 9–11), Gtf2iε (exons 11–12), Gtf2i(9–12), Gtf2i(9), Gtf2i(12), and Gtf2i(−) [19,20]. The Gtf2i protein has an N-terminus leucine zipper, six I-repeat DNA-binding motifs, and two NLSs [44]. The presence and number of these motifs are not changed among these coding variants. Variations in combinations of exons 9–12 may affect complex conformation of Gtf2i proteins [55] and different combinations of Gtf2i variant dimers regulate target genes [42,56]. Each splice variant in the coding region of Gtf2i is possibly involved in the transcription of its own set of target genes [55,56]. Thus, Gtf2i variants in the coding region may be involved in the differential regulation of the expression of target genes via variable Gtf2i dimer organization (see review by Roy [44]).
In addition, variations in the isoforms of the coding region with different combinations of exons 9–12 change the length of the PEST sequence (a protein sequence enriched in proline, glutamate, serine, and threonine), which may lead to changes in the stability of this protein [20]. Furthermore, there are specific combinations of the variants in the 5′UTRs and those in the coding region [20]. Therefore, the properties and functions of Gtf2i are also affected by the corresponding 5′UTR, which regulate localization of Gtf2i mRNA. This suggests that Gtf2i function varies depending on its cellular and subcellular localization.
Phosphorylation of the tyrosine248 residue, which is encoded by exon 9, is involved in translocation to the nucleus and activation of the transcriptional activity of Gtf2i [41,57]. Isoforms lacking exon 9, Gtf2i(12), and Gtf2i(−) in the brain may have functions other than transcriptional regulation in the nucleus. Another function of Gtf2i outside the nucleus is agonist-induced calcium entry, as has been suggested for the Δ isoform [58]. However, this function of Gtf2i may not occur in the brain Gtf2i since the Δ isoform is not expressed in the brain tissue.
4. Relationship between GTF2I and Neuropsychiatric Abnormality
4.1. Copy Number Variation in Williams–Beuren Syndrome Region and Neuropsychiatric Diseases
The human GTF2I is implicated in Williams–Beuren syndrome (WBS) and 7q microduplication syndrome (7dupASD; ASD, autism spectrum disorder). WBS is characterized by altered facial appearance, cardiovascular abnormalities, severe visuospatial cognitive abnormalities, and abnormal social behavior (hypersociability) [59,60]. The linkage of the GTF2I gene to WBS was suggested by the fact that patients with WBS exhibit hemizygous deletion of a 1.5 Mb region in chromosome 7q11.23 (WBS region), containing 28 genes, including GTF2I [59,60]. Thus, GTF2I is regarded as one of genes responsible for human WBS [60,61]. Genotype–phenotype correlation studies of patients with mild WBS phenotype and normal IQ carrying a shorter deletion of this region have shown that GTF2I contributes to the alteration in social behavior of patients with WBS, but not to visual-spatial cognitive impairment, craniofacial features, and other WBS features [59,61]. The linkage between GTF2I and WBS is also supported by a study on haploinsufficiency of Gtf2i mice. Gtf2i heterozygous mice showed increased social interaction, similar to the behavior in patients with WBS, although learning and memory, especially spatial memory, were normal in these mice [62].
The 7dupASD, also known as Somerville–van der Aa syndrome, is caused by duplication of the same WBS region. Symptoms of patients with 7dupASD have cognitive abnormalities, such as language delay and deficits in social interaction [63,64]. Thus, opposite and graded phenotypes have been observed between patients with WBS and patients with 7dupASD (microdeletion and duplication of WBS region, respectively) (Figure 2) [63,64,65], suggesting that dosage of genes in this region is an important determinant of the symptoms in WBS and 7dupASD. Thus, copy number variations (CNVs) of the WBS region are related to some symptoms of WBS and 7dupASD. This suggests that accuracy of the expression level (dosage) of the genes in this region is critical for normal neuronal development related to symptoms in WBS and 7dupASD. This concept is in good agreement with the notion that CNVs (deletions or duplications of small chromosomal regions) are responsible for various neuropsychiatric disorders [65]. Alteration of Gtf2i expression may alter the expression profile of the downstream genes via its role in transcription regulation. This may be one of the molecular mechanisms underlying CNV effects (regulation by GTF2I dosage) in WBS and 7dupASD.
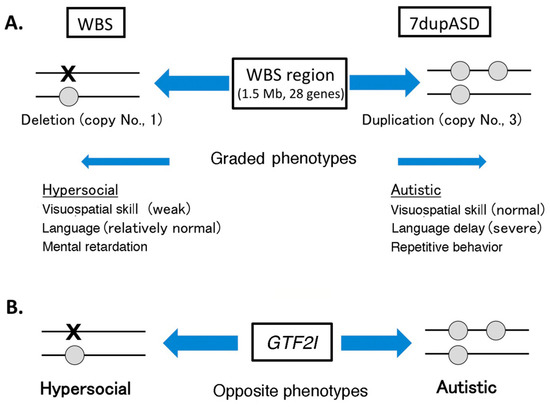
Figure 2.
Psychiatric phenotypes dependent on copy number variations (CNV) of the Williams–Beuren syndrome (WBS) region and GTF2I. (A) Effects of CNV on the WBS region; (B) Effects of CNV on GTF2I. Characteristic features of WBS and 7dupASD are listed. Gray circles indicate presence of a single copy of the GTF2I and X indicates the absence of the gene.
Induced pluripotent stem cells (iPSCs) prepared from both patients with WBS and patients with 7dupASD demonstrated that dosage of the WBS region contributes to transcriptional dysregulation of genes involved in disease-relevant pathways, such as cell adhesion, migration, calcium homeostasis, inner ear morphogenesis, craniofacial phenotypes, and kidney epithelium development. These processes are deeply related to WBS and 7dupASD [66]. For example, an experiment using four different conditions of GTF2I dosage level produced by lentivirus-mediated RNA interference against GTF2I from this patient-derived iPSCs found that expression of the transcription factor BEN domain containing 4 (BEND4), which is immediately downstream of GTF2I, was repressed in a dosage-dependent manner [66]. This further supports the involvement of GTF2I in WBS and 7dupASD.
4.2. GTF2I Abnormality Is Related to Neuropsychiatric Diseases
Some neuropsychiatric disease-related single nucleotide polymorphisms (SNPs) have been found in GTF2I. Two SNPs in GTF2I related to ASD were found in a study on 1142 individuals with ASD, and were observed to be associated with severe deficiency in social skills and repetitive behavior [67]. These two SNPs of GTF2I reside in the intron between the exon 1 (encoding the 5′UTR) and the exon 2 (including the start codon of GTF2I). Because this region contains multiple splice sites and produces various alternative 5′UTRs [19,20], these SNPs may cause abnormal splicing of the GTF2I 5′UTR (in both human and rat). Intriguingly, from a search in the 5′UTR region of the GTF2I genomic sequence using the NCBI SNP database (Available at: https://www.ncbi.nlm.nih.gov/snp/?term=GTF2I), there are more than 10 SNPs in the 5′UTR of GTF2I. These 5′UTR SNPs may be related to altered localization and translation of GTF2I mRNA. These findings shed light on the importance of 5′UTR splice variation of GTF2I for neuronal function.
As previously mentioned, Gtf2i may affect the excitatory/inhibitory balance of brain through Dlx5/Dlx6 regulation. Disruption of this pathway has been suggested to be a possible mechanism that causes autism [68,69]. Indeed, human DLX5/DLX6 are associated with ASD [70]. Furthermore, autism-associated SNPs have been reported in I56i, an enhancer element of the DLX5/DLX6. These mutations may alter the binding properties of this element to GTF2I and DLX2, which would impair the transcriptional activation of I56i [47]. Alteration of the excitation/inhibition balance is a hypothetical basis of ASD [68]. Aberrant development of GABAergic interneurons reportedly results in neurodevelopmental disorders, such as epilepsy, schizophrenia, and ASDs, including tuberous sclerosis, fragile X syndrome, Angelman syndrome, and Rett syndrome [69,71]. Thus, Gtf2i abnormality (e.g., abnormal Gtf2i expression level), via imbalance of the excitatory/inhibitory neuronal balance in the brain, may result in the development of ASD symptoms.
5. Concluding Remarks
Focusing on Gtf2i, here, we reviewed and discussed the roles of 5′UTRs in mRNA targeting into dendrites and translational regulation within dendrites. In many neuropsychiatric diseases, including WBS, the effects of dosage regulation of genes on the pathogenesis of neuropsychiatric disorders have been recently emphasized. Dosage alteration may impair transcription of downstream genes, which may affect neural development, neural structure, and circuit formation related to neuropsychiatric disorders. We suggest that the 5′UTR of Gtf2i mRNA may play a role in the spatiotemporal specific regulation of its protein level via postsynaptic local translation. Direct evidences showing the involvement of 5′UTRs in neuropsychiatric disorders still needs to be established. One powerful method is finding 5′UTR mutation(s) of causal genes related to neuropsychiatric disorders by whole genome sequencing of patient samples. Further studies on dosage regulation of causal genes in neuropsychiatric disorders should be advanced.
Acknowledgments
This work is supported by Distinguished Visiting Professor system of Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, and by a grant-in-aid for JSPS KAKENHI (to Yoshinori Shirai, Grant Number JP23650189, and to Tatsuo Suzuki, Grant Number JP15K06771). We would like to thank Editage (www.editage.jp) for English language editing.
Author Contributions
Yoshinori Shirai and Tatsuo Suzuki conceived and designed the experiments of rat Gtf2i; Yoshinori Shirai performed the experiments; Weidong Li and Tatsuo Suzuki analyzed the data; Yoshinori Shirai wrote the manuscript draft. Weidong Li and Tatsuo Suzuki revised the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 7dupASD | 7q microduplication syndrome |
| ASD | autism spectrum disorder |
| BDNF | brain-derived neurotrophic factor |
| CNV | copy number variation |
| FMRP | fragile X mental retardation protein |
| GTF2I | general transcription factor II-I |
| iPSCs | induced pluripotent stem cells |
| NLS | nuclear localization signal |
| PSD | postsynaptic density |
| RBP | RNA-binding protein |
| SNP | single nucleotide polymorphisms |
| TSS | transcription start site |
| UTR | untranslated region |
| WBS | Williams–Beuren syndrome |
References
- Suzuki, T.; Tian, Q.B.; Kuromitsu, J.; Kawai, T.; Endo, S. Characterization of MRNA species that are associated with postsynaptic density fraction by gene chip microarray analysis. Neurosci. Res. 2007, 57, 61–85. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.B.; Nakayama, K.; Okano, A.; Suzuki, T. Identification of MRNAS localizing in the postsynaptic region. Brain Res. Mol. Brain Res. 1999, 72, 147–157. [Google Scholar] [CrossRef]
- Cajigas, I.J.; Tushev, G.; Will, T.J.; tom Dieck, S.; Fuerst, N.; Schuman, E.M. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 2012, 74, 453–466. [Google Scholar] [CrossRef]
- Davis, H.P.; Squire, L.R. Protein synthesis and momory: A review. Psychol. Bull. 1984, 96, 518–559. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.A.; Schuman, E.M. Local translational control in dendrites and its role in long-term synaptic plasticity. J. Neurobiol. 2005, 64, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.E.; Schuman, E.M. The central dogma decentralized: New perspectives on RNA function and local translation in neurons. Neuron 2013, 80, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Gkogkas, C.G.; Sonenberg, N.; Holt, C.E. Remote control of gene function by local translation. Cell 2014, 157, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Greer, P.L.; Greenberg, M.E. From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron 2008, 59, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Tongiorgi, E.; Armellin, M.; Giulianini, P.G.; Bregola, G.; Zucchini, S.; Paradiso, B.; Steward, O.; Cattaneo, A.; Simonato, M. Brain-derived neurotrophic factor MRNA and protein are targeted to discrete dendritic laminas by events that trigger epileptogenesis. J. Neurosci. 2004, 24, 6842–6852. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Shepherd, J.D.; Okuno, H.; Lyford, G.; Petralia, R.S.; Plath, N.; Kuhl, D.; Huganir, R.L.; Worley, P.F. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 2006, 52, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Rial Verde, E.M.; Lee-Osbourne, J.; Worley, P.F.; Malinow, R.; Cline, H.T. Increased expression of the immediate-early gene arc/arg3.1 reduces ampa receptor-mediated synaptic transmission. Neuron 2006, 52, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.; Miyashiro, K.Y.; Sul, J.Y.; Barrett, L.; Belt, B.; Haydon, P.; Eberwine, J. RNA splicing capability of live neuronal dendrites. Proc. Natl. Acad. Sci. USA 2005, 102, 16859–16864. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yaakov, K.; Dagan, S.Y.; Segal-Ruder, Y.; Shalem, O.; Vuppalanchi, D.; Willis, D.E.; Yudin, D.; Rishal, I.; Rother, F.; Bader, M.; et al. Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J. 2012, 31, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.J.; Hengst, U.; Gurskaya, N.G.; Lukyanov, K.A.; Jaffrey, S.R. Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat. Cell Biol. 2008, 10, 149–159. [Google Scholar] [CrossRef]
- Ji, S.J.; Jaffrey, S.R. Intra-axonal translation of SMAD1/5/8 mediates retrograde regulation of trigeminal ganglia subtype specification. Neuron 2012, 74, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.A.; Kreutz, M.R. Nucleocytoplasmic protein shuttling: The direct route in synapse-to-nucleus signaling. Trends Neurosci. 2009, 32, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Marcora, E.; Kennedy, M.B. The Huntington’s disease mutation impairs Huntingtin’s role in the transport of nf-kappab from the synapse to the nucleus. Hum. Mol. Genet. 2010, 19, 4373–4384. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.R.; Otis, K.O.; Chen, D.Y.; Zhao, Y.; O’Dell, T.J.; Martin, K.C. Synapse to nucleus signaling during long-term synaptic plasticity; A role for the classical active nuclear import pathway. Neuron 2004, 44, 997–1009. [Google Scholar] [PubMed]
- Makeyev, A.V.; Bayarsaihan, D. Alternative splicing and promoter use in TFII-I genes. Gene 2009, 433, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Watanabe, M.; Sakagami, H.; Suzuki, T. Novel splice variants in the 5′UTR of Gtf2i expressed in the rat brain: Alternative 5′UTRs and differential expression in the neuronal dendrites. J. Neurochem. 2015, 134, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Bramham, C.R.; Wells, D.G. Dendritic MRNA: Transport, translation and function. Nat. Rev. Neurosci. 2007, 8, 776–789. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.; Kiebler, M.A. Mechanisms of dendritic MRNA transport and its role in synaptic tagging. EMBO J. 2011, 30, 3540–3552. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.; Rage, F.; Tabet, R.; Flatter, E.; Mandel, J.L.; Moine, H. G-quadruplex RNA structure as a signal for neurite mrna targeting. EMBO Rep. 2011, 12, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, A.; Balasubramanian, S. 5′-UTR RNA G-quadruplexes: Translation regulation and targeting. Nucleic Acids Res. 2012, 40, 4727–4741. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mrnas linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Jensen, K.B.; Jin, P.; Brown, V.; Warren, S.T.; Darnell, R.B. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 2001, 107, 489–499. [Google Scholar] [CrossRef]
- Laggerbauer, B.; Ostareck, D.; Keide, l.E.M.; Ostareck-Lederer, A.; Fischer, U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 2001, 10, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Ku, L.; Wilkinson, K.D.; Warren, S.T.; Feng, Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001, 29, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, C.; Bardoni, B.; Mandel, J.L.; Ehresmann, B.; Ehresmann, C.; Moine, H. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J. 2001, 20, 4803–4813. [Google Scholar] [CrossRef] [PubMed]
- Comery, T.A.; Harris, J.B.; Willems, P.J.; Oostra, B.A.; Irwin, S.A.; Weiler, I.J.; Greenough, W.T. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 1997, 94, 5401–5404. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.R.; Qian, S.; Bolinger, C.; Fernandez, S.; Schoenberg, D.R.; Boris-Lawrie, K. RNA helicase A is necessary for translation of selected messenger RNAs. Nat. Struct. Mol. Biol. 2006, 13, 509–516. [Google Scholar] [CrossRef]
- Manojlovic, Z.; Stefanovic, B. A novel role of RNA helicase A in regulation of translation of type I collagen mrnas. RNA 2012, 18, 321–334. [Google Scholar] [CrossRef]
- Shirai, Y.; Suzuki, T. Proteins Specifically Bind to the Dendritic 5′UTR of Gtf2i mRNA. Unpublished work. 2017. [Google Scholar]
- Pruunsild, P.; Kazantseva, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Baj, G.; Leone, E.; Chao, M.V.; Tongiorgi, E. Spatial segregation of BDNF transcripts enables BDNF to differentially shape distinct dendritic compartments. Proc. Natl. Acad. Sci. USA 2011, 108, 16813–16818. [Google Scholar] [CrossRef] [PubMed]
- Baj, G.; Del Turco, D.; Schlaudraff, J.; Torelli, L.; Deller, T.; Tongiorgi, E. Regulation of the spatial code for BDNF mRNA isoforms in the rat hippocampus following pilocarpine-treatment: A systematic analysis using laser microdissection and quantitative real-time PCR. Hippocampus 2013, 23, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Vaghi, V.; Polacchini, A.; Baj, G.; Pinheiro, V.L.; Vicario, A.; Tongiorgi, E. Pharmacological profile of brain-derived neurotrophic factor (BDNF) splice variant translation using a novel drug screening assay: A “quantitative code”. J. Biol. Chem. 2014, 289, 27702–27713. [Google Scholar] [CrossRef] [PubMed]
- Cheriyath, V.; Desgranges, Z.P.; Roy, A.L. c-Src-dependent transcriptional activation of TFII-I. J. Biol. Chem. 2002, 277, 22798–22805. [Google Scholar] [CrossRef] [PubMed]
- Hakre, S.; Tussie-Luna, M.I.; Ashworth, T.; Novina, C.D.; Settleman, J.; Sharp, P.A.; Roy, A.L. Opposing functions of TFII-I spliced isoforms in growth factor-induced gene expression. Mol. Cell 2006, 24, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.L. Signal-induced functions of the transcription factor TFII-I. Biochim. Biophys. Acta 2007, 1769, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.L. Biochemistry and biology of the inducible multifunctional transcription factor TFII-I: 10 years later. Gene 2012, 492, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Chimge, N.O.; Makeyev, A.V.; Ruddle, F.H.; Bayarsaihan, D. Identification of the TFII-I family target genes in the vertebrate genome. Proc. Natl. Acad. Sci. USA 2008, 105, 9006–9010. [Google Scholar] [CrossRef] [PubMed]
- Enkhmandakh, B.; Makeyev, A.V.; Erdenechimeg, L.; Ruddle, F.H.; Chimge, N.O.; Tussie-Luna, M.I.; Roy, A.L.; Bayarsaihan, D. Essential functions of the williams-beuren syndrome-associated TFII-I genes in embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Poitras, L.; Yu, M.; Lesage-Pelletier, C.; Macdonald, R.B.; Gagne, J.P.; Hatch, G.; Kelly, I.; Hamilton, S.P.; Rubenstein, J.L.; Poirier, G.G.; et al. An SNP in an ultraconserved regulatory element affects DLx5/DLx6 regulation in the forebrain. Development 2010, 137, 3089–3097. [Google Scholar] [CrossRef] [PubMed]
- Zerucha, T.; Stuhmer, T.; Hatch, G.; Park, B.K.; Long, Q.; Yu, G.; Gambarotta, A.; Schultz, J.R.; Rubenstein, J.L.; Ekker, M. A highly conserved enhancer in the DLx5/DLx6 intergenic region is the site of cross-regulatory interactions between DLX genes in the embryonic forebrain. J. Neurosci. 2000, 20, 709–721. [Google Scholar] [PubMed]
- Wang, Y.; Dye, C.A.; Sohal, V.; Long, J.E.; Estrada, R.C.; Roztocil, T.; Lufkin, T.; Deisseroth, K.; Baraban, S.C.; Rubenstein, J.L. Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J. Neurosci. 2010, 30, 5334–5345. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Paez, I.; Tammimies, K.; Massinen, S.; Roy, A.L.; Kere, J. The complex of TFII-I, PARP1, and SFPQ proteins regulates the DYX1C1 gene implicated in neuronal migration and dyslexia. FASEB J. 2008, 22, 3001–3009. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Kaminen, N.; Nopola-Hemmi, J.; Haltia, T.; Myllyluoma, B.; Lyytinen, H.; Muller, K.; Kaaranen, M.; Lindsberg, P.J.; Hannula-Jouppi, K.; et al. A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptide repeat domain protein dynamically regulated in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 11553–11558. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Paramasivam, M.; Thomas, A.; Bai, J.; Kaminen-Ahola, N.; Kere, J.; Voskuil, J.; Rosen, G.D.; Galaburda, A.M.; Loturco, J.J. Dyx1c1 functions in neuronal migration in developing neocortex. Neuroscience 2006, 143, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Rosen, G.D.; Bai, J.; Wang, Y.; Fiondella, C.G.; Threlkeld, S.W.; LoTurco, J.J.; Galaburda, A.M. Disruption of neuronal migration by RNAI of Dyx1c1 results in neocortical and hippocampal malformations. Cereb. Cortex 2007, 17, 2562–2572. [Google Scholar] [CrossRef] [PubMed]
- Threlkeld, S.W.; McClure, M.M.; Bai, J.; Wang, Y.; LoTurco, J.J.; Rosen, G.D.; Fitch, R.H. Developmental disruptions and behavioral impairments in rats following in utero RNAi of Dyx1c1. Brain Res. Bull. 2007, 71, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Cheriyath, V.; Roy, A.L. Structure-function analysis of TFII-I. Roles of the N-terminal end, basic region, and I-repeats. J. Biol. Chem. 2001, 276, 8377–8383. [Google Scholar] [CrossRef] [PubMed]
- Cheriyath, V.; Roy, A.L. Alternatively spliced isoforms of TFII-I. Complex formation, nuclear translocation, and differential gene regulation. J. Biol. Chem. 2000, 275, 26300–26308. [Google Scholar] [CrossRef] [PubMed]
- Novina, C.D.; Kumar, S.; Bajpai, U.; Cheriyath, V.; Zhang, K.; Pillai, S.; Wortis, H.H.; Roy, A.L. Regulation of nuclear localization and transcriptional activity of TFII-I by bruton’s tyrosine kinase. Mol. Cell. Biol. 1999, 19, 5014–5024. [Google Scholar] [CrossRef] [PubMed]
- Caraveo, G.; van Rossum, D.B.; Patterson, R.L.; Snyder, S.H.; Desiderio, S. Action of TFII-I outside the nucleus as an inhibitor of agonist-induced calcium entry. Science 2006, 314, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, G.B.; Howald, C.; Micale, L.; Biamino, E.; Augello, B.; Fusco, C.; Turturo, M.G.; Forzano, S.; Reymond, A.; Merla, G. An atypical 7q11.23 deletion in a normal IQ Williams–Beuren syndrome patient. Eur. J. Hum. Genet. EJHG 2010, 18, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Lindenberg, A.; Mervis, C.B.; Berman, K.F. Neural mechanisms in Williams syndrome: A unique window to genetic influences on cognition and behaviour. Nat. Rev. Neurosci. 2006, 7, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Bellugi, U.; Chen, X.N.; Pulst-Korenberg, A.M.; Jarvinen-Pasley, A.; Tirosh-Wagner, T.; Eis, P.S.; Graham, J.; Mills, D.; Searcy, Y.; et al. Is it Williams syndrome? GTF2IRD1 implicated in visual-spatial construction and GTF2I in sociability revealed by high resolution arrays. Am. J. Med. Genet. Part A 2009, 149A, 302–314. [Google Scholar] [PubMed]
- Sakurai, T.; Dorr, N.P.; Takahashi, N.; McInnes, L.A.; Elder, G.A.; Buxbaum, J.D. Haploinsufficiency of Gtf2i, a gene deleted in williams syndrome, leads to increases in social interactions. Autism Res. 2011, 4, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Merla, G.; Brunetti-Pierri, N.; Micale, L.; Fusco, C. Copy number variants at Williams–Beuren syndrome 7q11.23 region. Hum. Genet. 2010, 128, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Somerville, M.J.; Mervis, C.B.; Young, E.J.; Seo, E.J.; del Campo, M.; Bamforth, S.; Peregrine, E.; Loo, W.; Lilley, M.; Perez-Jurado, L.A.; et al. Severe expressive-language delay related to duplication of the Williams–Beuren locus. N. Engl. J. Med. 2005, 353, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVS, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Atashpaz, S.; Germain, P.L.; Zanella, M.; D’Agostino, G.; Albertin, V.; Chenoweth, J.; Micale, L.; Fusco, C.; Unger, C.; et al. 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat. Genet. 2015, 47, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Malenfant, P.; Liu, X.; Hudson, M.L.; Qiao, Y.; Hrynchak, M.; Riendeau, N.; Hildebrand, M.J.; Cohen, I.L.; Chudley, A.E.; Forster-Gibson, C.; et al. Association of Gtf2i in the Williams–Beuren syndrome critical region with autism spectrum disorders. J. Autism Dev. Disord. 2012, 42, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.B.; Valakh, V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.P.; Woo, J.M.; Carlson, E.J.; Ghanem, N.; Ekker, M.; Rubenstein, J.L. Analysis of four DLX homeobox genes in autistic probands. BMC Genet. 2005, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Dani, V.S.; Chang, Q.; Maffei, A.; Turrigiano, G.G.; Jaenisch, R.; Nelson, S.B. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of rett syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12560–12565. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).