
Role of Splice Variants of Gtf2i, a Transcription Factor Localizing at Postsynaptic Sites, and Its Relation to Neuropsychiatric Diseases


Abstract
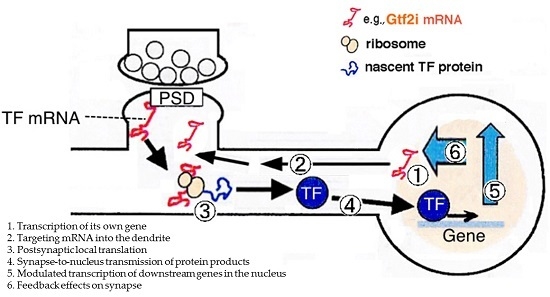
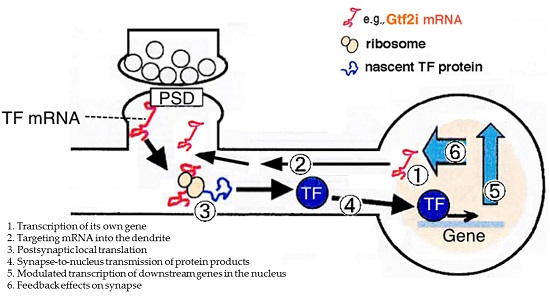
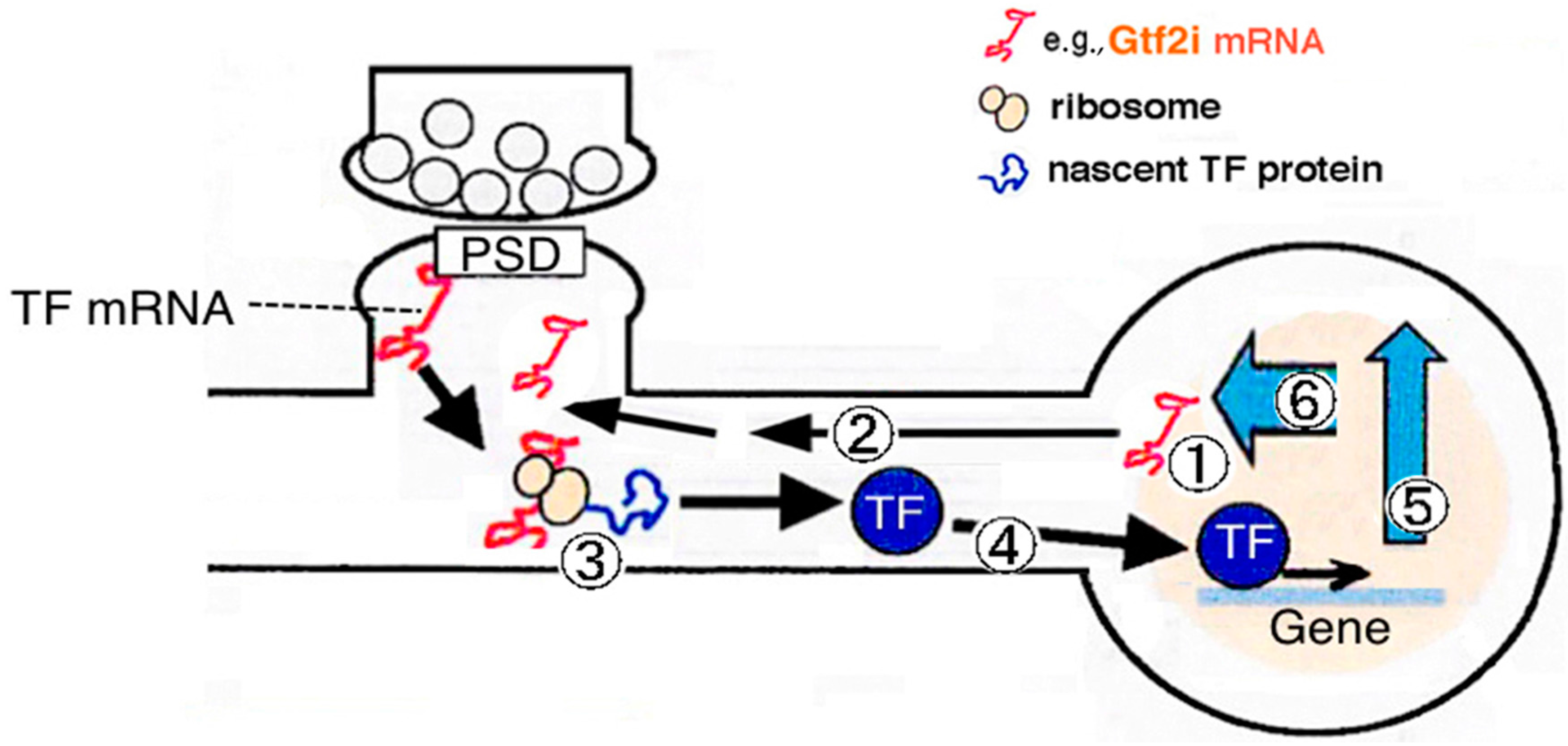
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Role of Splice Variation in 5′ Untranslated Regions of Gtf2i
2.1. Role of 5′ Untranslated Region Variations in Gtf2i mRNA for Regulation of Its Own Transcription
2.2. Role of 5′UTR Variation in Gtf2i mRNA for Subcellular Localization
2.3. Regulation of Translation and Localization of mRNA by RNA-Binding Proteins Interacting with 5′UTRs
2.4. 5′UTR Works as a “Spatial Code” and a “Quantitative Code”
3. Downstream of Gtf2i Local Translation
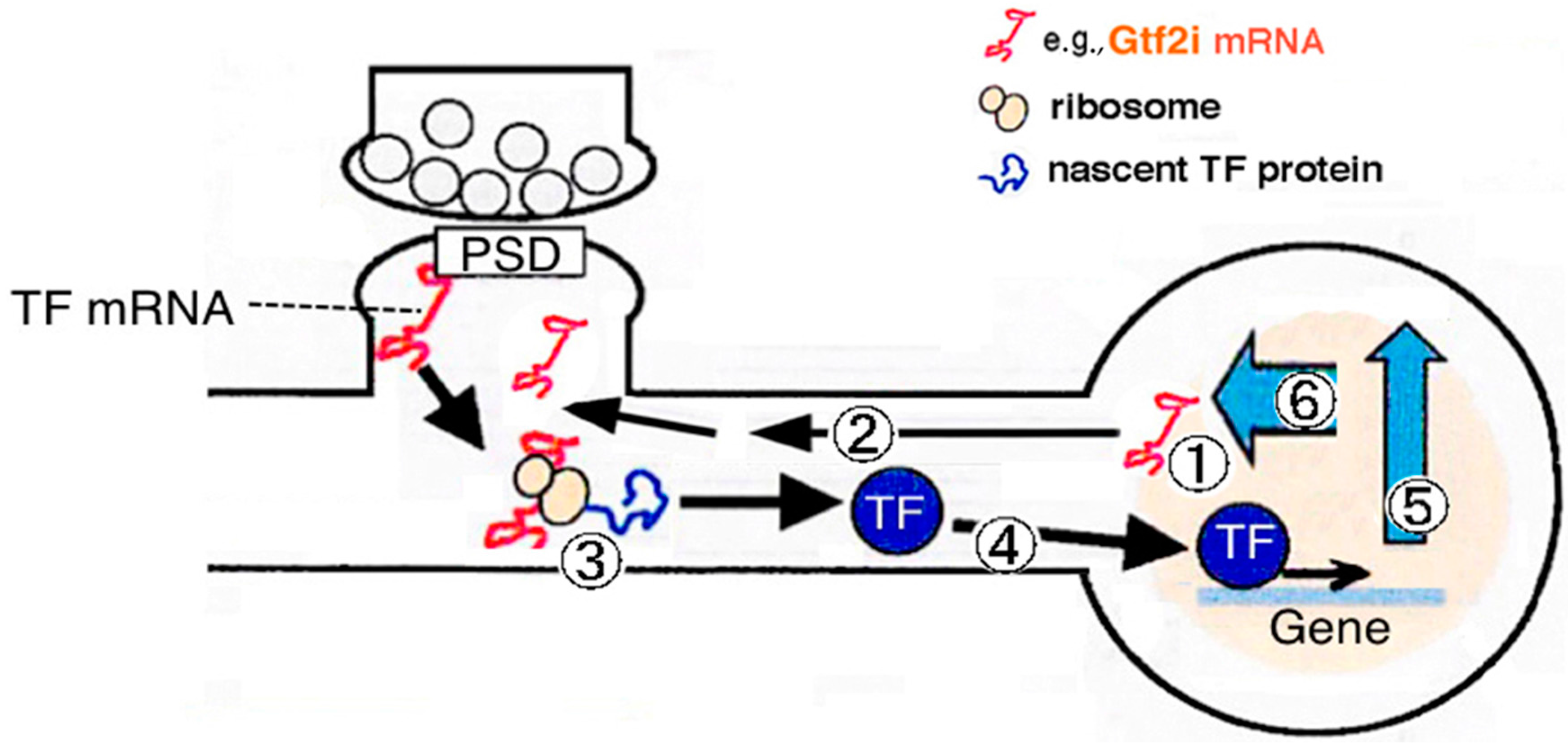
3.1. Synapse-to-Nucleus Signal Transduction via Gtf2i
3.2. Downstream Target Genes and Cellular Functions of Gtf2i
3.3. Coding Variants May Differentially Regulate Downstream Transcription and Cellular Processes
4. Relationship between GTF2I and Neuropsychiatric Abnormality
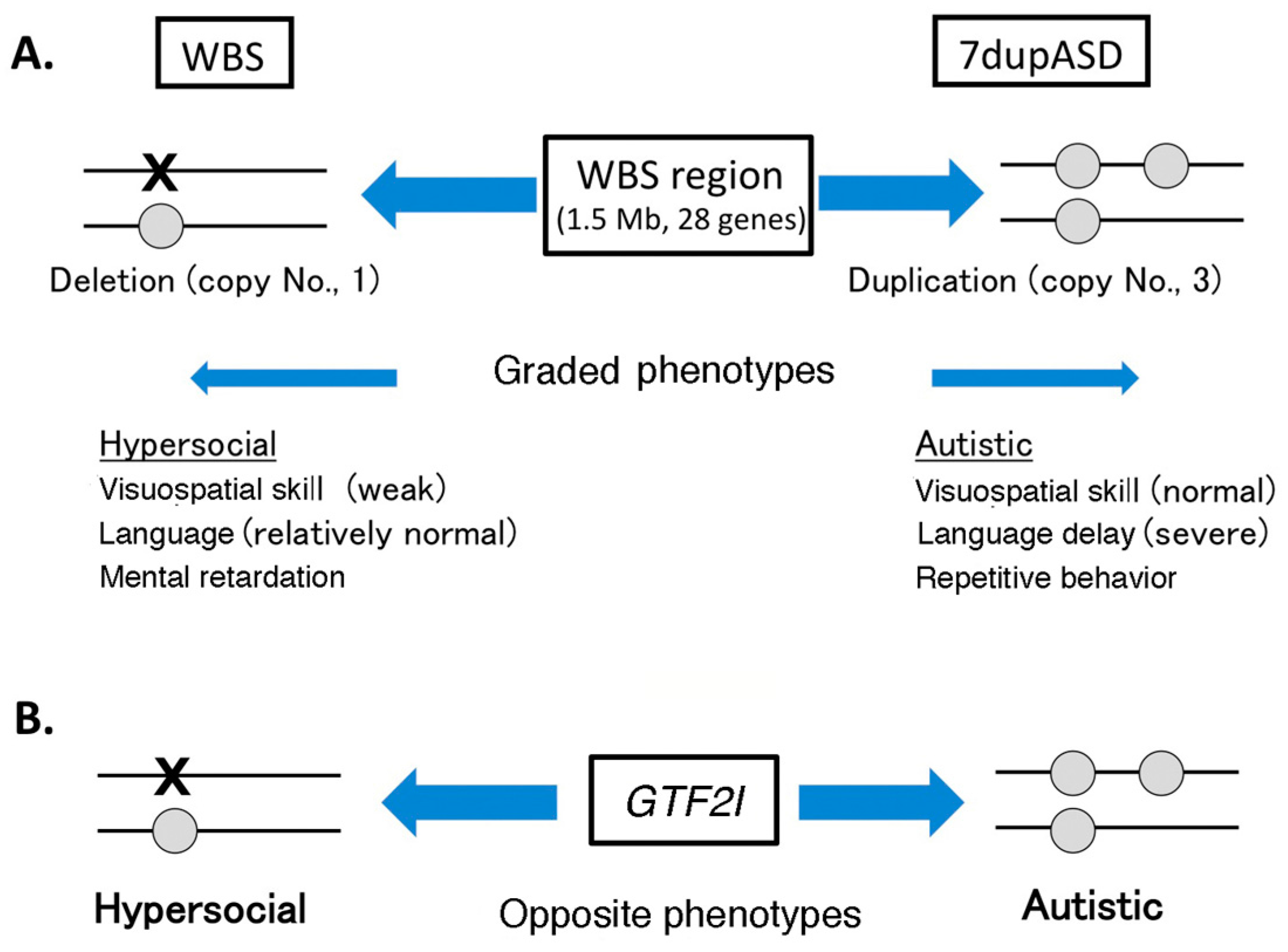
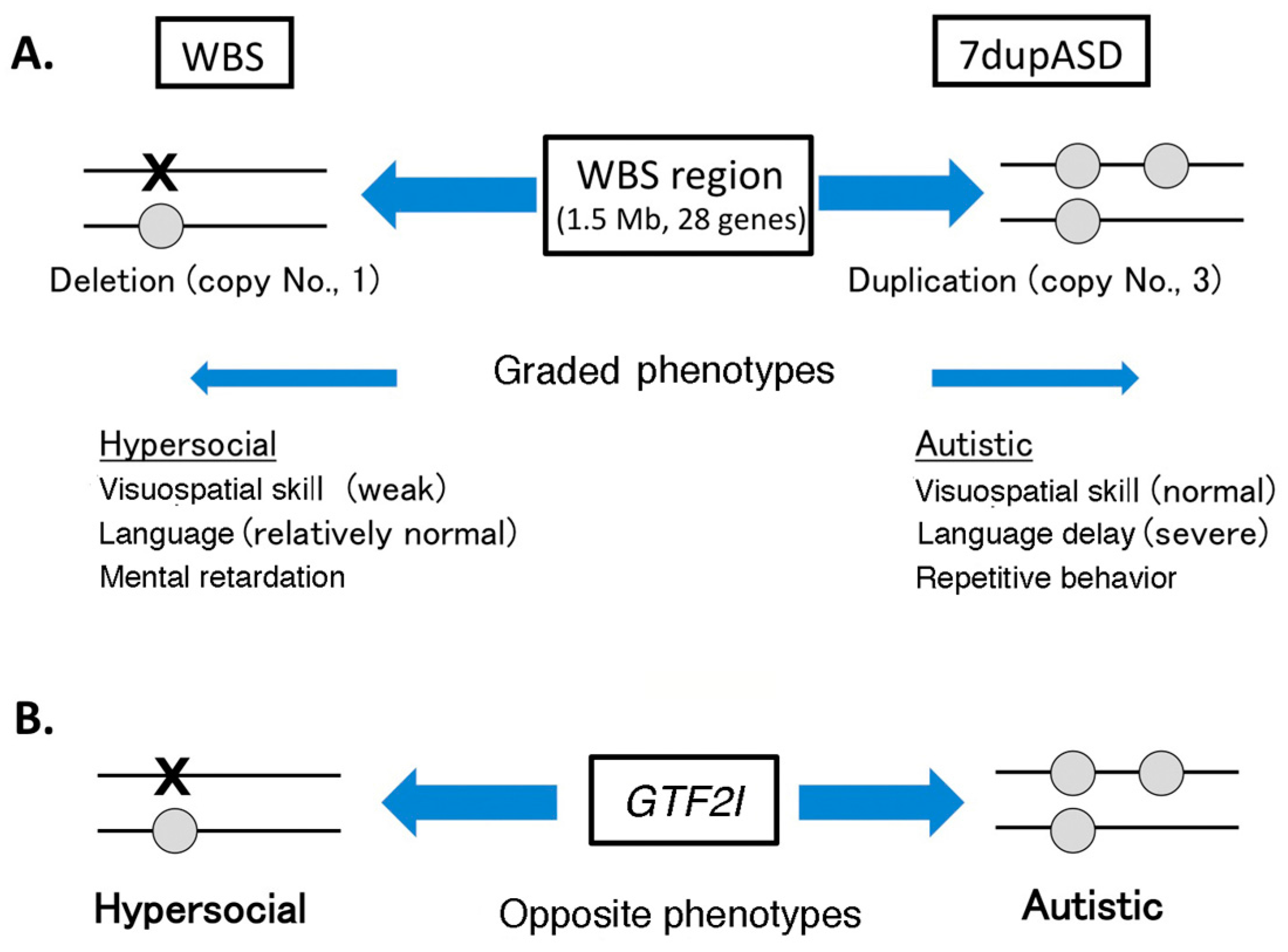
4.1. Copy Number Variation in Williams–Beuren Syndrome Region and Neuropsychiatric Diseases
4.2. GTF2I Abnormality Is Related to Neuropsychiatric Diseases
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 7dupASD | 7q microduplication syndrome |
| ASD | autism spectrum disorder |
| BDNF | brain-derived neurotrophic factor |
| CNV | copy number variation |
| FMRP | fragile X mental retardation protein |
| GTF2I | general transcription factor II-I |
| iPSCs | induced pluripotent stem cells |
| NLS | nuclear localization signal |
| PSD | postsynaptic density |
| RBP | RNA-binding protein |
| SNP | single nucleotide polymorphisms |
| TSS | transcription start site |
| UTR | untranslated region |
| WBS | Williams–Beuren syndrome |
References
- Suzuki, T.; Tian, Q.B.; Kuromitsu, J.; Kawai, T.; Endo, S. Characterization of MRNA species that are associated with postsynaptic density fraction by gene chip microarray analysis. Neurosci. Res. 2007, 57, 61–85. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.B.; Nakayama, K.; Okano, A.; Suzuki, T. Identification of MRNAS localizing in the postsynaptic region. Brain Res. Mol. Brain Res. 1999, 72, 147–157. [Google Scholar] [CrossRef]
- Cajigas, I.J.; Tushev, G.; Will, T.J.; tom Dieck, S.; Fuerst, N.; Schuman, E.M. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 2012, 74, 453–466. [Google Scholar] [CrossRef]
- Davis, H.P.; Squire, L.R. Protein synthesis and momory: A review. Psychol. Bull. 1984, 96, 518–559. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.A.; Schuman, E.M. Local translational control in dendrites and its role in long-term synaptic plasticity. J. Neurobiol. 2005, 64, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.E.; Schuman, E.M. The central dogma decentralized: New perspectives on RNA function and local translation in neurons. Neuron 2013, 80, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Gkogkas, C.G.; Sonenberg, N.; Holt, C.E. Remote control of gene function by local translation. Cell 2014, 157, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Greer, P.L.; Greenberg, M.E. From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron 2008, 59, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Tongiorgi, E.; Armellin, M.; Giulianini, P.G.; Bregola, G.; Zucchini, S.; Paradiso, B.; Steward, O.; Cattaneo, A.; Simonato, M. Brain-derived neurotrophic factor MRNA and protein are targeted to discrete dendritic laminas by events that trigger epileptogenesis. J. Neurosci. 2004, 24, 6842–6852. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Shepherd, J.D.; Okuno, H.; Lyford, G.; Petralia, R.S.; Plath, N.; Kuhl, D.; Huganir, R.L.; Worley, P.F. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 2006, 52, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Rial Verde, E.M.; Lee-Osbourne, J.; Worley, P.F.; Malinow, R.; Cline, H.T. Increased expression of the immediate-early gene arc/arg3.1 reduces ampa receptor-mediated synaptic transmission. Neuron 2006, 52, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.; Miyashiro, K.Y.; Sul, J.Y.; Barrett, L.; Belt, B.; Haydon, P.; Eberwine, J. RNA splicing capability of live neuronal dendrites. Proc. Natl. Acad. Sci. USA 2005, 102, 16859–16864. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yaakov, K.; Dagan, S.Y.; Segal-Ruder, Y.; Shalem, O.; Vuppalanchi, D.; Willis, D.E.; Yudin, D.; Rishal, I.; Rother, F.; Bader, M.; et al. Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J. 2012, 31, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.J.; Hengst, U.; Gurskaya, N.G.; Lukyanov, K.A.; Jaffrey, S.R. Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat. Cell Biol. 2008, 10, 149–159. [Google Scholar] [CrossRef]
- Ji, S.J.; Jaffrey, S.R. Intra-axonal translation of SMAD1/5/8 mediates retrograde regulation of trigeminal ganglia subtype specification. Neuron 2012, 74, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.A.; Kreutz, M.R. Nucleocytoplasmic protein shuttling: The direct route in synapse-to-nucleus signaling. Trends Neurosci. 2009, 32, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Marcora, E.; Kennedy, M.B. The Huntington’s disease mutation impairs Huntingtin’s role in the transport of nf-kappab from the synapse to the nucleus. Hum. Mol. Genet. 2010, 19, 4373–4384. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.R.; Otis, K.O.; Chen, D.Y.; Zhao, Y.; O’Dell, T.J.; Martin, K.C. Synapse to nucleus signaling during long-term synaptic plasticity; A role for the classical active nuclear import pathway. Neuron 2004, 44, 997–1009. [Google Scholar] [PubMed]
- Makeyev, A.V.; Bayarsaihan, D. Alternative splicing and promoter use in TFII-I genes. Gene 2009, 433, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Watanabe, M.; Sakagami, H.; Suzuki, T. Novel splice variants in the 5′UTR of Gtf2i expressed in the rat brain: Alternative 5′UTRs and differential expression in the neuronal dendrites. J. Neurochem. 2015, 134, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Bramham, C.R.; Wells, D.G. Dendritic MRNA: Transport, translation and function. Nat. Rev. Neurosci. 2007, 8, 776–789. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.; Kiebler, M.A. Mechanisms of dendritic MRNA transport and its role in synaptic tagging. EMBO J. 2011, 30, 3540–3552. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.; Rage, F.; Tabet, R.; Flatter, E.; Mandel, J.L.; Moine, H. G-quadruplex RNA structure as a signal for neurite mrna targeting. EMBO Rep. 2011, 12, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, A.; Balasubramanian, S. 5′-UTR RNA G-quadruplexes: Translation regulation and targeting. Nucleic Acids Res. 2012, 40, 4727–4741. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mrnas linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Jensen, K.B.; Jin, P.; Brown, V.; Warren, S.T.; Darnell, R.B. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 2001, 107, 489–499. [Google Scholar] [CrossRef]
- Laggerbauer, B.; Ostareck, D.; Keide, l.E.M.; Ostareck-Lederer, A.; Fischer, U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 2001, 10, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Ku, L.; Wilkinson, K.D.; Warren, S.T.; Feng, Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001, 29, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, C.; Bardoni, B.; Mandel, J.L.; Ehresmann, B.; Ehresmann, C.; Moine, H. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J. 2001, 20, 4803–4813. [Google Scholar] [CrossRef] [PubMed]
- Comery, T.A.; Harris, J.B.; Willems, P.J.; Oostra, B.A.; Irwin, S.A.; Weiler, I.J.; Greenough, W.T. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 1997, 94, 5401–5404. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.R.; Qian, S.; Bolinger, C.; Fernandez, S.; Schoenberg, D.R.; Boris-Lawrie, K. RNA helicase A is necessary for translation of selected messenger RNAs. Nat. Struct. Mol. Biol. 2006, 13, 509–516. [Google Scholar] [CrossRef]
- Manojlovic, Z.; Stefanovic, B. A novel role of RNA helicase A in regulation of translation of type I collagen mrnas. RNA 2012, 18, 321–334. [Google Scholar] [CrossRef]
- Shirai, Y.; Suzuki, T. Proteins Specifically Bind to the Dendritic 5′UTR of Gtf2i mRNA. Unpublished work. 2017. [Google Scholar]
- Pruunsild, P.; Kazantseva, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Baj, G.; Leone, E.; Chao, M.V.; Tongiorgi, E. Spatial segregation of BDNF transcripts enables BDNF to differentially shape distinct dendritic compartments. Proc. Natl. Acad. Sci. USA 2011, 108, 16813–16818. [Google Scholar] [CrossRef] [PubMed]
- Baj, G.; Del Turco, D.; Schlaudraff, J.; Torelli, L.; Deller, T.; Tongiorgi, E. Regulation of the spatial code for BDNF mRNA isoforms in the rat hippocampus following pilocarpine-treatment: A systematic analysis using laser microdissection and quantitative real-time PCR. Hippocampus 2013, 23, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Vaghi, V.; Polacchini, A.; Baj, G.; Pinheiro, V.L.; Vicario, A.; Tongiorgi, E. Pharmacological profile of brain-derived neurotrophic factor (BDNF) splice variant translation using a novel drug screening assay: A “quantitative code”. J. Biol. Chem. 2014, 289, 27702–27713. [Google Scholar] [CrossRef] [PubMed]
- Cheriyath, V.; Desgranges, Z.P.; Roy, A.L. c-Src-dependent transcriptional activation of TFII-I. J. Biol. Chem. 2002, 277, 22798–22805. [Google Scholar] [CrossRef] [PubMed]
- Hakre, S.; Tussie-Luna, M.I.; Ashworth, T.; Novina, C.D.; Settleman, J.; Sharp, P.A.; Roy, A.L. Opposing functions of TFII-I spliced isoforms in growth factor-induced gene expression. Mol. Cell 2006, 24, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.L. Signal-induced functions of the transcription factor TFII-I. Biochim. Biophys. Acta 2007, 1769, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.L. Biochemistry and biology of the inducible multifunctional transcription factor TFII-I: 10 years later. Gene 2012, 492, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Chimge, N.O.; Makeyev, A.V.; Ruddle, F.H.; Bayarsaihan, D. Identification of the TFII-I family target genes in the vertebrate genome. Proc. Natl. Acad. Sci. USA 2008, 105, 9006–9010. [Google Scholar] [CrossRef] [PubMed]
- Enkhmandakh, B.; Makeyev, A.V.; Erdenechimeg, L.; Ruddle, F.H.; Chimge, N.O.; Tussie-Luna, M.I.; Roy, A.L.; Bayarsaihan, D. Essential functions of the williams-beuren syndrome-associated TFII-I genes in embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Poitras, L.; Yu, M.; Lesage-Pelletier, C.; Macdonald, R.B.; Gagne, J.P.; Hatch, G.; Kelly, I.; Hamilton, S.P.; Rubenstein, J.L.; Poirier, G.G.; et al. An SNP in an ultraconserved regulatory element affects DLx5/DLx6 regulation in the forebrain. Development 2010, 137, 3089–3097. [Google Scholar] [CrossRef] [PubMed]
- Zerucha, T.; Stuhmer, T.; Hatch, G.; Park, B.K.; Long, Q.; Yu, G.; Gambarotta, A.; Schultz, J.R.; Rubenstein, J.L.; Ekker, M. A highly conserved enhancer in the DLx5/DLx6 intergenic region is the site of cross-regulatory interactions between DLX genes in the embryonic forebrain. J. Neurosci. 2000, 20, 709–721. [Google Scholar] [PubMed]
- Wang, Y.; Dye, C.A.; Sohal, V.; Long, J.E.; Estrada, R.C.; Roztocil, T.; Lufkin, T.; Deisseroth, K.; Baraban, S.C.; Rubenstein, J.L. Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J. Neurosci. 2010, 30, 5334–5345. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Paez, I.; Tammimies, K.; Massinen, S.; Roy, A.L.; Kere, J. The complex of TFII-I, PARP1, and SFPQ proteins regulates the DYX1C1 gene implicated in neuronal migration and dyslexia. FASEB J. 2008, 22, 3001–3009. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Kaminen, N.; Nopola-Hemmi, J.; Haltia, T.; Myllyluoma, B.; Lyytinen, H.; Muller, K.; Kaaranen, M.; Lindsberg, P.J.; Hannula-Jouppi, K.; et al. A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptide repeat domain protein dynamically regulated in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 11553–11558. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Paramasivam, M.; Thomas, A.; Bai, J.; Kaminen-Ahola, N.; Kere, J.; Voskuil, J.; Rosen, G.D.; Galaburda, A.M.; Loturco, J.J. Dyx1c1 functions in neuronal migration in developing neocortex. Neuroscience 2006, 143, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Rosen, G.D.; Bai, J.; Wang, Y.; Fiondella, C.G.; Threlkeld, S.W.; LoTurco, J.J.; Galaburda, A.M. Disruption of neuronal migration by RNAI of Dyx1c1 results in neocortical and hippocampal malformations. Cereb. Cortex 2007, 17, 2562–2572. [Google Scholar] [CrossRef] [PubMed]
- Threlkeld, S.W.; McClure, M.M.; Bai, J.; Wang, Y.; LoTurco, J.J.; Rosen, G.D.; Fitch, R.H. Developmental disruptions and behavioral impairments in rats following in utero RNAi of Dyx1c1. Brain Res. Bull. 2007, 71, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Cheriyath, V.; Roy, A.L. Structure-function analysis of TFII-I. Roles of the N-terminal end, basic region, and I-repeats. J. Biol. Chem. 2001, 276, 8377–8383. [Google Scholar] [CrossRef] [PubMed]
- Cheriyath, V.; Roy, A.L. Alternatively spliced isoforms of TFII-I. Complex formation, nuclear translocation, and differential gene regulation. J. Biol. Chem. 2000, 275, 26300–26308. [Google Scholar] [CrossRef] [PubMed]
- Novina, C.D.; Kumar, S.; Bajpai, U.; Cheriyath, V.; Zhang, K.; Pillai, S.; Wortis, H.H.; Roy, A.L. Regulation of nuclear localization and transcriptional activity of TFII-I by bruton’s tyrosine kinase. Mol. Cell. Biol. 1999, 19, 5014–5024. [Google Scholar] [CrossRef] [PubMed]
- Caraveo, G.; van Rossum, D.B.; Patterson, R.L.; Snyder, S.H.; Desiderio, S. Action of TFII-I outside the nucleus as an inhibitor of agonist-induced calcium entry. Science 2006, 314, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, G.B.; Howald, C.; Micale, L.; Biamino, E.; Augello, B.; Fusco, C.; Turturo, M.G.; Forzano, S.; Reymond, A.; Merla, G. An atypical 7q11.23 deletion in a normal IQ Williams–Beuren syndrome patient. Eur. J. Hum. Genet. EJHG 2010, 18, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Lindenberg, A.; Mervis, C.B.; Berman, K.F. Neural mechanisms in Williams syndrome: A unique window to genetic influences on cognition and behaviour. Nat. Rev. Neurosci. 2006, 7, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Bellugi, U.; Chen, X.N.; Pulst-Korenberg, A.M.; Jarvinen-Pasley, A.; Tirosh-Wagner, T.; Eis, P.S.; Graham, J.; Mills, D.; Searcy, Y.; et al. Is it Williams syndrome? GTF2IRD1 implicated in visual-spatial construction and GTF2I in sociability revealed by high resolution arrays. Am. J. Med. Genet. Part A 2009, 149A, 302–314. [Google Scholar] [PubMed]
- Sakurai, T.; Dorr, N.P.; Takahashi, N.; McInnes, L.A.; Elder, G.A.; Buxbaum, J.D. Haploinsufficiency of Gtf2i, a gene deleted in williams syndrome, leads to increases in social interactions. Autism Res. 2011, 4, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Merla, G.; Brunetti-Pierri, N.; Micale, L.; Fusco, C. Copy number variants at Williams–Beuren syndrome 7q11.23 region. Hum. Genet. 2010, 128, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Somerville, M.J.; Mervis, C.B.; Young, E.J.; Seo, E.J.; del Campo, M.; Bamforth, S.; Peregrine, E.; Loo, W.; Lilley, M.; Perez-Jurado, L.A.; et al. Severe expressive-language delay related to duplication of the Williams–Beuren locus. N. Engl. J. Med. 2005, 353, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVS, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Atashpaz, S.; Germain, P.L.; Zanella, M.; D’Agostino, G.; Albertin, V.; Chenoweth, J.; Micale, L.; Fusco, C.; Unger, C.; et al. 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat. Genet. 2015, 47, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Malenfant, P.; Liu, X.; Hudson, M.L.; Qiao, Y.; Hrynchak, M.; Riendeau, N.; Hildebrand, M.J.; Cohen, I.L.; Chudley, A.E.; Forster-Gibson, C.; et al. Association of Gtf2i in the Williams–Beuren syndrome critical region with autism spectrum disorders. J. Autism Dev. Disord. 2012, 42, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.B.; Valakh, V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.P.; Woo, J.M.; Carlson, E.J.; Ghanem, N.; Ekker, M.; Rubenstein, J.L. Analysis of four DLX homeobox genes in autistic probands. BMC Genet. 2005, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Dani, V.S.; Chang, Q.; Maffei, A.; Turrigiano, G.G.; Jaenisch, R.; Nelson, S.B. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of rett syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12560–12565. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirai, Y.; Li, W.; Suzuki, T. Role of Splice Variants of Gtf2i, a Transcription Factor Localizing at Postsynaptic Sites, and Its Relation to Neuropsychiatric Diseases. Int. J. Mol. Sci. 2017, 18, 411. https://doi.org/10.3390/ijms18020411
Shirai Y, Li W, Suzuki T. Role of Splice Variants of Gtf2i, a Transcription Factor Localizing at Postsynaptic Sites, and Its Relation to Neuropsychiatric Diseases. International Journal of Molecular Sciences. 2017; 18(2):411. https://doi.org/10.3390/ijms18020411
Chicago/Turabian StyleShirai, Yoshinori, Weidong Li, and Tatsuo Suzuki. 2017. "Role of Splice Variants of Gtf2i, a Transcription Factor Localizing at Postsynaptic Sites, and Its Relation to Neuropsychiatric Diseases" International Journal of Molecular Sciences 18, no. 2: 411. https://doi.org/10.3390/ijms18020411
APA StyleShirai, Y., Li, W., & Suzuki, T. (2017). Role of Splice Variants of Gtf2i, a Transcription Factor Localizing at Postsynaptic Sites, and Its Relation to Neuropsychiatric Diseases. International Journal of Molecular Sciences, 18(2), 411. https://doi.org/10.3390/ijms18020411