
Inhaled Pollutants: The Molecular Scene behind Respiratory and Systemic Diseases Associated with Ultrafine Particulate Matter


 and
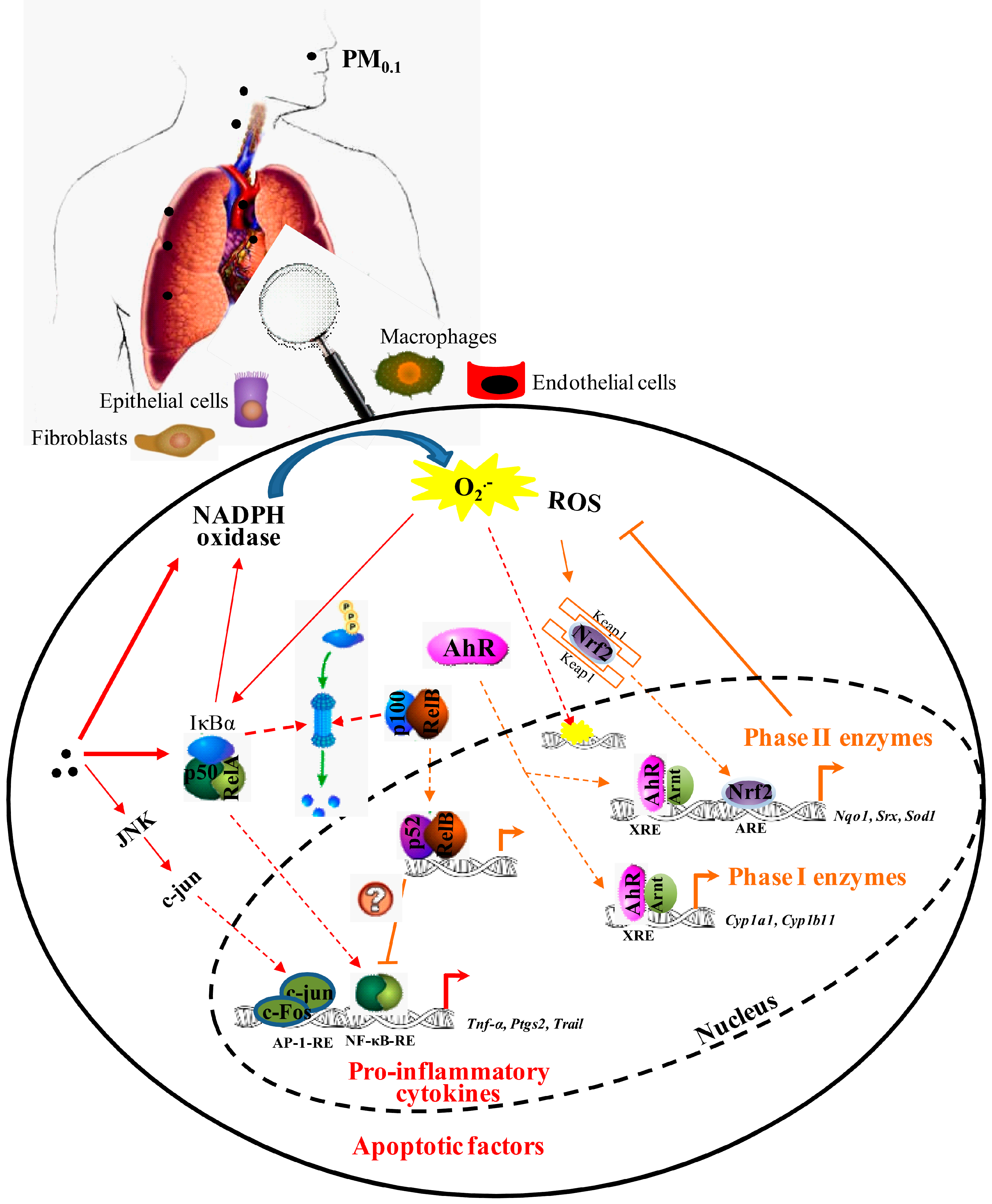
and 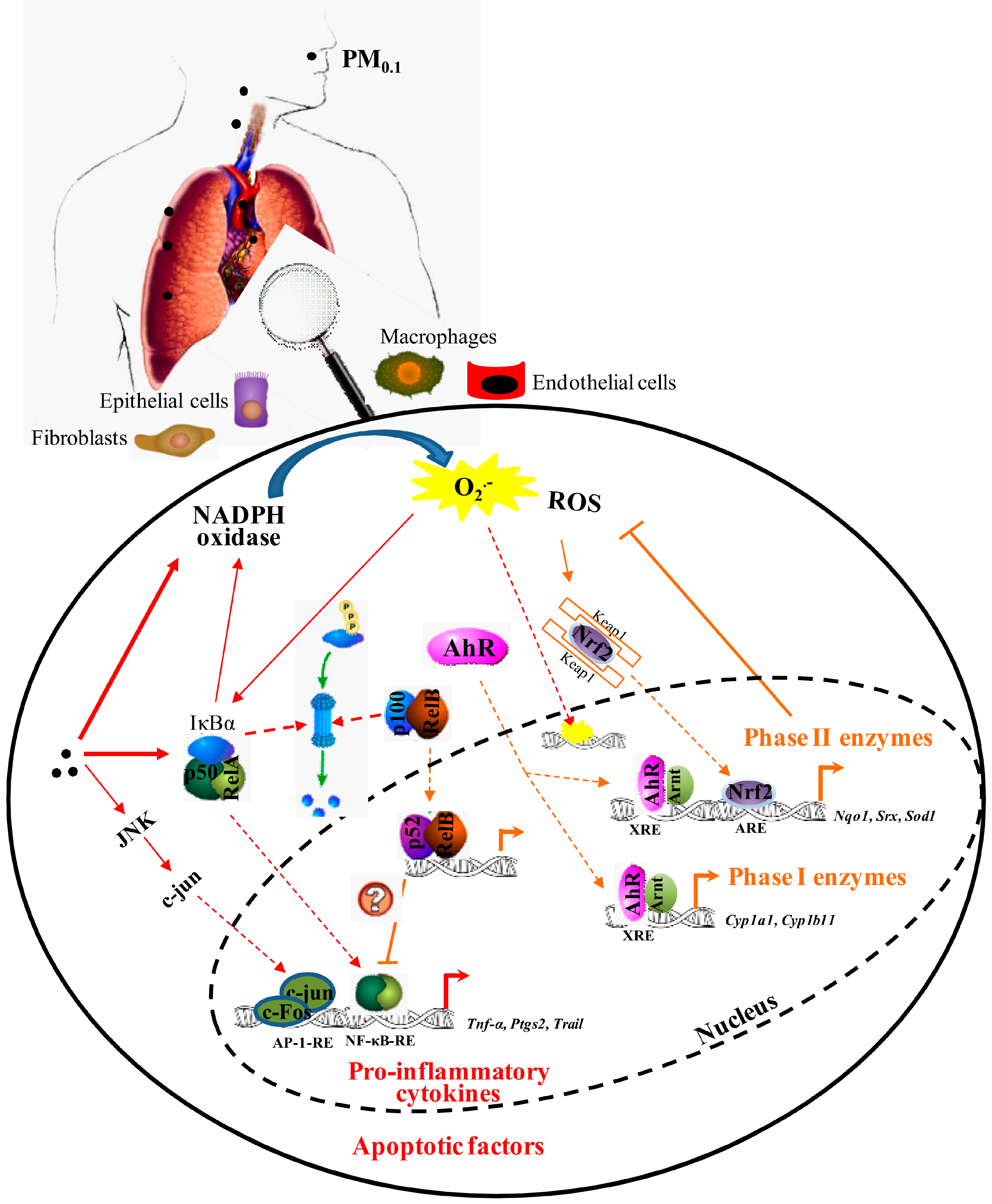{kind=link}
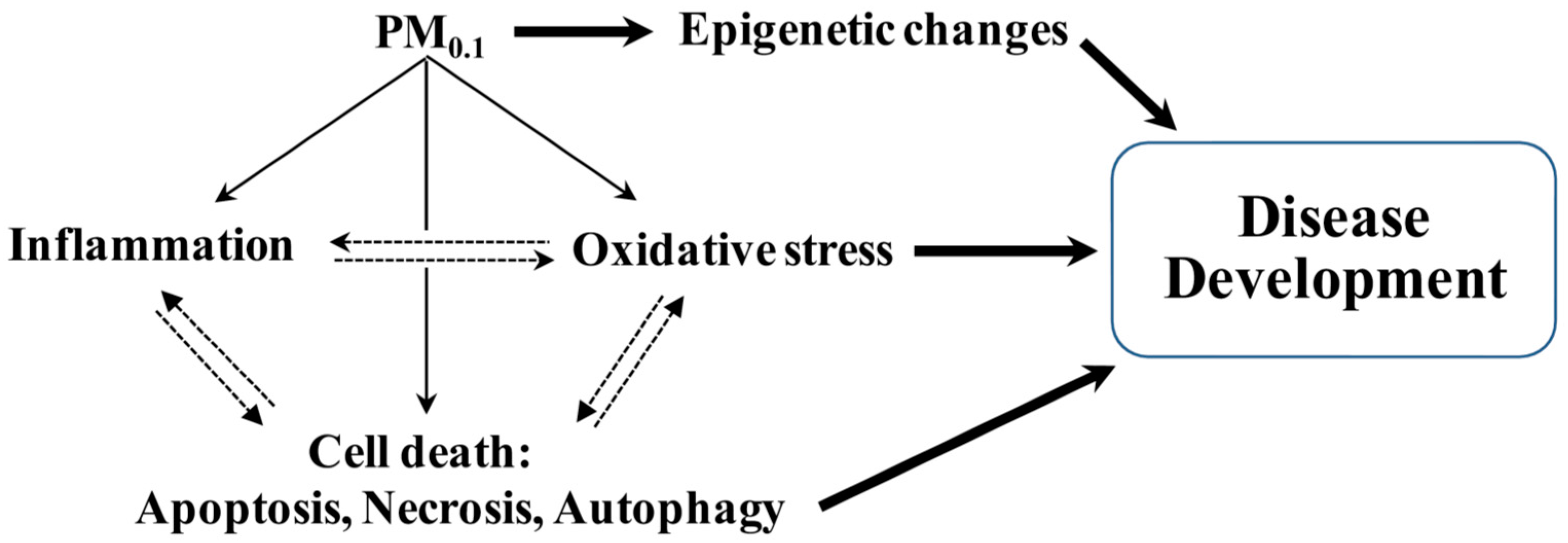
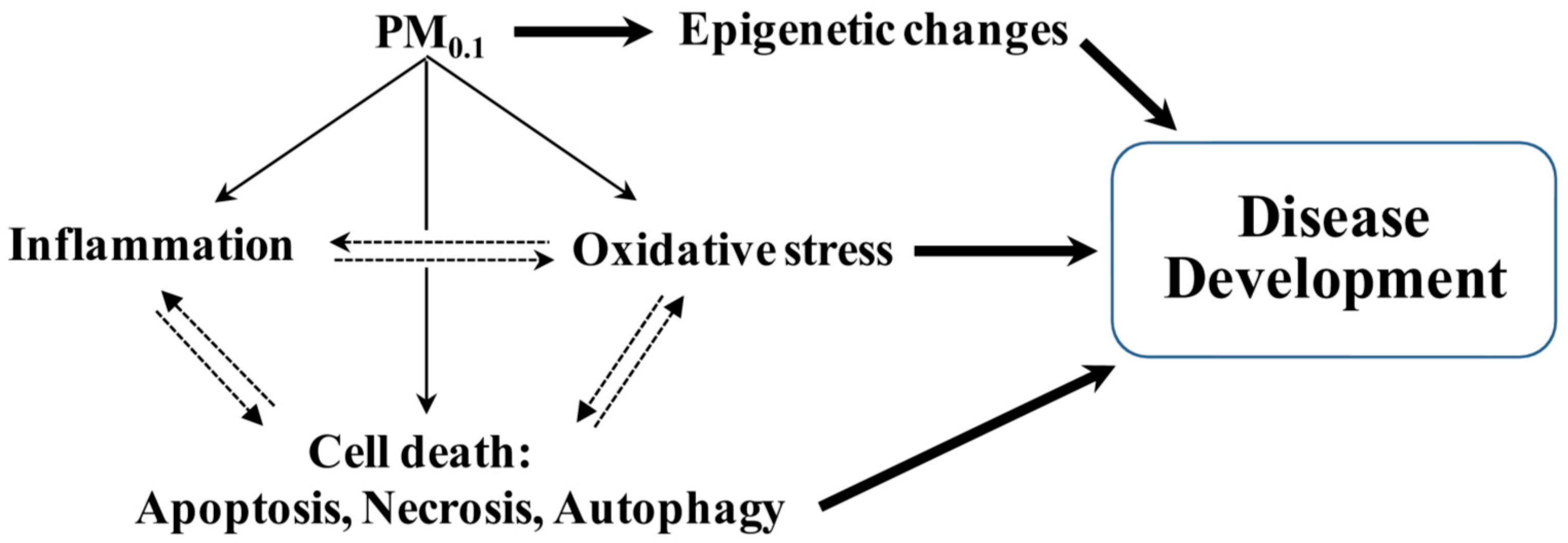{kind=link}
Abstract
:1. Introduction
2. Pathogenic Processes Implicated Following Inhalation of PM0.1
2.1. Inflammation
2.2. Oxidative Stress and Antioxidant Defense
2.3. Cell Death Pathways
2.3.1. Necrosis
2.3.2. Apoptosis
2.4. Autophagy
3. Signal Transduction Pathways Implicated in PM0.1-Induced Inflammation and Cell Death
3.1. Nuclear Factor-κB (NF-κB)
3.2. Aryl Hydrocarbon Receptor (AhR)
3.3. Nuclear Factor (Erythroid-Derived 2)-Like 2 (Nrf2)
3.4. PI3K/Akt/mTOR
3.5. Transcription Factor EB (TFEB)
4. Epigenetic Contribution to Pulmonary and Systemic Disease
4.1. MicroRNA (miRNA)
4.2. Long Non-Coding RNA (lncRNA)
4.3. DNA Methylation
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Paulin, L.; Hansel, N. Particulate air pollution and impaired lung function. F1000 Res. 2016, 5, 201. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Fiotakis, K.; Vlachogianni, T. Airborne particulate matter and human health: Toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2008, 26, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Holme, J.A.; Rosas, I.; Schwarze, P.E.; Alfaro-Moreno, E. Recent advances in particulate matter and nanoparticle toxicology: A review of the in vivo and in vitro studies. BioMed Res. Int. 2013, 2013, 279371. [Google Scholar] [CrossRef] [PubMed]
- Morakinyo, O.M.; Mokgobu, M.I.; Mukhola, M.S.; Hunter, R.P. Health outcomes of exposure to biological and chemical components of inhalable and respirable particulate matter. Int. J. Environ. Res. Public Health 2016, 13, 592. [Google Scholar] [CrossRef] [PubMed]
- Aalapati, S.; Ganapathy, S.; Manapuram, S.; Anumolu, G.; Prakya, B.M. Toxicity and bio-accumulation of inhaled cerium oxide nanoparticles in CD1 mice. Nanotoxicology 2014, 8, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Mills, N.L.; Amin, N.; Robinson, S.D.; Anand, A.; Davies, J.; Patel, D.; de la Fuente, J.M.; Cassee, F.R.; Boon, N.A.; Macnee, W.; et al. Do inhaled carbon nanoparticles translocate directly into the circulation in humans? Am. J. Respir. Crit. Care Med. 2006, 173, 426–431. [Google Scholar] [CrossRef] [PubMed]
- HEI Review Panel on Ultrafine Particles. Understanding the Health Effects of Ambient Ultrafine Particles; Health Effects Institute: Boston, MA, USA, 2013. [Google Scholar]
- Baglole, C.J.; Ray, D.M.; Bernstein, S.H.; Feldon, S.E.; Smith, T.J.; Sime, P.J.; Phipps, R.P. More than structural cells, fibroblasts create and orchestrate the tumor microenvironment. Immunol. Investig. 2006, 35, 297–325. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, A.L.; Sousa, L.P.; Lucas, C.D.; Rossi, A.G.; Pinho, V.; Teixeira, M.M. Resolution of inflammation: Mechanisms and opportunity for drug development. Pharmacol. Ther. 2013, 139, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.R.; Zago, M.; Eidelman, D.H.; Hamid, Q.; Baglole, C.J. Aryl hydrocarbon receptor (AhR) attenuation of subchronic cigarette smoke-induced pulmonary neutrophilia is associated with retention of nuclear RelB and suppression of intercellular adhesion molecule-1 (ICAM-1). Toxicol. Sci. 2014, 140, 204–223. [Google Scholar] [CrossRef] [PubMed]
- Hetland, R.B.; Cassee, F.R.; Refsnes, M.; Schwarze, P.E.; Lag, M.; Boere, A.J.; Dybing, E. Release of inflammatory cytokines, cell toxicity and apoptosis in epithelial lung cells after exposure to ambient air particles of different size fractions. Toxicol. In Vitro 2004, 18, 203–212. [Google Scholar] [CrossRef]
- Val, S.; Martinon, L.; Cachier, H.; Yahyaoui, A.; Marfaing, H.; Baeza-Squiban, A. Role of size and composition of traffic and agricultural aerosols in the molecular responses triggered in airway epithelial cells. Inhal. Toxicol. 2011, 23, 627–640. [Google Scholar] [PubMed]
- Steenhof, M.; Gosens, I.; Strak, M.; Godri, K.J.; Hoek, G.; Cassee, F.R.; Mudway, I.S.; Kelly, F.J.; Harrison, R.M.; Lebret, E.; et al. In vitro toxicity of particulate matter (PM) collected at different sites in the Netherlands is associated with PM composition, size fraction and oxidative potential—The RAPTES project. Part. Fibre Toxicol. 2011, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Osornio-Vargas, A.R.; Bonner, J.C.; Alfaro-Moreno, E.; Martinez, L.; Garcia-Cuellar, C.; Ponce-de-Leon Rosales, S.; Miranda, J.; Rosas, I. Proinflammatory and cytotoxic effects of Mexico City air pollution particulate matter in vitro are dependent on particle size and composition. Environ. Health Perspect. 2003, 111, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, H.M.; Park, M.V.; Gosens, I.; De Jong, W.H.; Cassee, F.R. Physicochemical characteristics of nanomaterials that affect pulmonary inflammation. Part. Fibre Toxicol. 2014, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.A.; Barajas, B.; Kleinman, M.; Wang, X.; Bennett, B.J.; Gong, K.W.; Navab, M.; Harkema, J.; Sioutas, C.; Lusis, A.J.; et al. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ. Res. 2008, 102, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Pedata, P.; Bergamasco, N.; D’Anna, A.; Minutolo, P.; Servillo, L.; Sannolo, N.; Balestrieri, M.L. Apoptotic and proinflammatory effect of combustion-generated organic nanoparticles in endothelial cells. Toxicol. Lett. 2013, 219, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Noel, A.; Xiao, R.; Perveen, Z.; Zaman, H.M.; Rouse, R.L.; Paulsen, D.B.; Penn, A.L. Incomplete lung recovery following sub-acute inhalation of combustion-derived ultrafine particles in mice. Part. Fibre Toxicol. 2016, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Lundborg, M.; Dahlen, S.E.; Johard, U.; Gerde, P.; Jarstrand, C.; Camner, P.; Lastbom, L. Aggregates of ultrafine particles impair phagocytosis of microorganisms by human alveolar macrophages. Environ. Res. 2006, 100, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Shen, Y.; Huang, L.; Lv, G.; Lei, C.; Fan, X.; Lin, F.; Zhang, Y.; Wu, L.; Yang, Y. In vitro cytotoxicity of gold nanorods in A549 cells. Environ. Toxicol. Pharmacol. 2015, 39, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Eom, H.J.; Choi, J. Oxidative stress of CeO2 nanoparticles via p38-Nrf-2 signaling pathway in human bronchial epithelial cell, Beas-2B. Toxicol. Lett. 2009, 187, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Eom, H.J.; Choi, J. Oxidative stress of silica nanoparticles in human bronchial epithelial cell, Beas-2B. Toxicol. In Vitro 2009, 23, 1326–1332. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, M.J.; Lee, S.Y.; Oh, S.M.; Chung, K.H. Genotoxic effects of silver nanoparticles stimulated by oxidative stress in human normal bronchial epithelial (BEAS-2B) cells. Mutat. Res. 2011, 726, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Avalos, A.; Haza, A.I.; Mateo, D.; Morales, P. Effects of silver and gold nanoparticles of different sizes in human pulmonary fibroblasts. Toxicol. Mech. Methods 2015, 25, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, V.S.; Athinarayanan, J.; Al-Hadi, A.M.; Juhaimi, F.A.; Alshatwi, A.A. Effects of titanium dioxide nanoparticles isolated from confectionery products on the metabolic stress pathway in human lung fibroblast cells. Arch. Environ. Contam. Toxicol. 2015, 68, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Davalos, A.; Ventura-Gallegos, J.L.; Alfaro-Moreno, E.; Soria-Castro, E.; Garcia-Latorre, E.; Cabanas-Moreno, J.G.; del Pilar Ramos-Godinez, M.; Lopez-Marure, R. TiO2 nanoparticles induce dysfunction and activation of human endothelial cells. Chem. Res. Toxicol. 2012, 25, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Alinovi, R.; Goldoni, M.; Pinelli, S.; Campanini, M.; Aliatis, I.; Bersani, D.; Lottici, P.P.; Iavicoli, S.; Petyx, M.; Mozzoni, P.; et al. Oxidative and pro-inflammatory effects of cobalt and titanium oxide nanoparticles on aortic and venous endothelial cells. Toxicol. In Vitro 2015, 29, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Petrick, L.; Rosenblat, M.; Paland, N.; Aviram, M. Silicon dioxide nanoparticles increase macrophage atherogenicity: Stimulation of cellular cytotoxicity, oxidative stress, and triglycerides accumulation. Environ. Toxicol. 2014, 31, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Wan, R.; Feng, L.; Chien, S.; Tollerud, D.J.; Zhang, Q. Combination effects of cigarette smoke extract and ambient ultrafine particles on endothelial cells. Toxicol. In Vitro 2012, 26, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Sioutas, C.; Cho, A.; Schmitz, D.; Misra, C.; Sempf, J.; Wang, M.; Oberley, T.; Froines, J.; Nel, A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ. Health Perspect. 2003, 111, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef] [PubMed]
- Brauner, E.V.; Forchhammer, L.; Moller, P.; Simonsen, J.; Glasius, M.; Wahlin, P.; Raaschou-Nielsen, O.; Loft, S. Exposure to ultrafine particles from ambient air and oxidative stress-induced DNA damage. Environ. Health Perspect. 2007, 115, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.Y.; Zhou, Y.M.; Smith, K.R.; Kennedy, I.M.; Chen, C.Y.; Aust, A.E.; Pinkerton, K.E. Oxidative injury in the lungs of neonatal rats following short-term exposure to ultrafine iron and soot particles. J. Toxicol. Environ. Health A 2010, 73, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Staimer, N.; Gillen, D.L.; Tjoa, T.; Schauer, J.J.; Shafer, M.M.; Hasheminassab, S.; Pakbin, P.; Vaziri, N.D.; Sioutas, C.; et al. Associations of oxidative stress and inflammatory biomarkers with chemically-characterized air pollutant exposures in an elderly cohort. Environ. Res. 2016, 150, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.P.; Shertzer, H.G.; Puga, A. Regulation of gene expression by reactive oxygen. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 67–101. [Google Scholar] [CrossRef] [PubMed]
- Sambandam, B.; Devasena, T.; Islam, V.I.; Prakhya, B.M. Characterization of coal fly ash nanoparticles and their induced in vitro cellular toxicity and oxidative DNA damage in different cell lines. Indian J. Exp. Biol. 2015, 53, 585–593. [Google Scholar] [PubMed]
- Chan, J.K.; Kodani, S.D.; Charrier, J.G.; Morin, D.; Edwards, P.C.; Anderson, D.S.; Anastasio, C.; van Winkle, L.S. Age-specific effects on rat lung glutathione and antioxidant enzymes after inhaling ultrafine soot. Am. J. Respir. Cell Mol. Biol. 2013, 48, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Jouan-Lanhouet, S.; Riquet, F.; Duprez, L.; Vanden Berghe, T.; Takahashi, N.; Vandenabeele, P. Necroptosis, in vivo detection in experimental disease models. Semin. Cell Dev. Biol. 2014, 35, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Foldbjerg, R.; Olesen, P.; Hougaard, M.; Dang, D.A.; Hoffmann, H.J.; Autrup, H. PVP-coated silver nanoparticles and silver ions induce reactive oxygen species, apoptosis and necrosis in THP-1 monocytes. Toxicol. Lett. 2009, 190, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Leifert, A.; Ruau, D.; Neuss, S.; Bornemann, J.; Schmid, G.; Brandau, W.; Simon, U.; Jahnen-Dechent, W. Gold nanoparticles of diameter 1.4 nm trigger necrosis by oxidative stress and mitochondrial damage. Small 2009, 5, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, W.; Zhang, R.; Liu, P.; Wang, Q.; Shang, Y.; Wu, M.; Donaldson, K. Comparison of cellular toxicity caused by ambient ultrafine particles and engineered metal oxide nanoparticles. Part. Fibre Toxicol. 2015, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Sydlik, U.; Bierhals, K.; Soufi, M.; Abel, J.; Schins, R.P.; Unfried, K. Ultrafine carbon particles induce apoptosis and proliferation in rat lung epithelial cells via specific signaling pathways both using EGF-R. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L725–L733. [Google Scholar] [CrossRef] [PubMed]
- Kaewamatawong, T.; Shimada, A.; Okajima, M.; Inoue, H.; Morita, T.; Inoue, K.; Takano, H. Acute and subacute pulmonary toxicity of low dose of ultrafine colloidal silica particles in mice after intratracheal instillation. Toxicol. Pathol. 2006, 34, 958–965. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Soo Lee, S.; Savini, M.; Popp, L.; Colvin, V.L.; Segatori, L. Ceria nanoparticles stabilized by organic surface coatings activate the lysosome-autophagy system and enhance autophagic clearance. ACS Nano 2014, 8, 10328–10342. [Google Scholar] [CrossRef] [PubMed]
- Neibert, K.D.; Maysinger, D. Mechanisms of cellular adaptation to quantum dots--the role of glutathione and transcription factor EB. Nanotoxicology 2012, 6, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Hartono, D.; Ong, C.N.; Bay, B.H.; Yung, L.Y. Autophagy and oxidative stress associated with gold nanoparticles. Biomaterials 2010, 31, 5996–6003. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Wu, Y.F.; Wang, P.L.; Wu, Y.P.; Li, Z.Y.; Zhao, Y.; Zhou, J.S.; Zhu, C.; Cao, C.; Mao, Y.Y.; et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy 2016, 12, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, M.; Nakamura, Y.; Grumont, R.; Gerondakis, S. New insights into the roles of ReL/NF-κB transcription factors in immune function, hemopoiesis and human disease. Int. J. Biochem. Cell Biol. 1999, 31, 1209–1219. [Google Scholar] [CrossRef]
- Martey, C.A.; Pollock, S.J.; Turner, C.K.; O’Reilly, K.M.; Baglole, C.J.; Phipps, R.P.; Sime, P.J. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: Implications for lung inflammation and cancer. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L981–L991. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, J. Endothelial cells dysfunction induced by silica nanoparticles through oxidative stress via JNK/P53 and NF-κB pathways. Biomaterials 2010, 31, 8198–8209. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Ghosh, M.; Ghosh, J.; Das, J.; Sil, P.C. Contribution of nano-copper particles to in vivo liver dysfunction and cellular damage: Role of IκBα/NF-κB, MAPKs and mitochondrial signal. Nanotoxicology 2012, 6, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Nishanth, R.P.; Jyotsna, R.G.; Schlager, J.J.; Hussain, S.M.; Reddanna, P. Inflammatory responses of RAW 264.7 macrophages upon exposure to nanoparticles: Role of ROS-NFκB signaling pathway. Nanotoxicology 2011, 5, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Samet, J.M.; Peden, D.B.; Bromberg, P.A. Phosphorylation of p65 is required for zinc oxide nanoparticle-induced interleukin 8 expression in human bronchial epithelial cells. Environ. Health Perspect. 2010, 118, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Lu, S.L.; Hu, C.W.; Yeh, C.S.; Lee, G.B.; Lei, H.Y. Size-dependent attenuation of TLR9 signaling by gold nanoparticles in macrophages. J. Immunol. 2012, 188, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Bren, G.D.; Solan, N.J.; Miyoshi, H.; Pennington, K.N.; Pobst, L.J.; Paya, C.V. Transcription of the RelB gene is regulated by NF-κB. Oncogene 2001, 20, 7722–7733. [Google Scholar] [CrossRef] [PubMed]
- Beinke, S.; Ley, S.C. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem. J. 2004, 382 Pt 2, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E. The alternative NF-κB pathway from biochemistry to biology: Pitfalls and promises for future drug development. Biochem. Pharmacol. 2006, 72, 1161–1179. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Morgan, M.; Kim, D.G.; Lee, J.Y.; Bai, L.; Lin, Y.; Liu, Z.G.; Kim, Y.S. TNFα induced noncanonical NF-κB activation is attenuated by RIP1 through stabilization of TRAF2. J. Cell Sci. 2011, 124 Pt 4, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Yoza, B.K.; Hu, J.Y.; Cousart, S.L.; Forrest, L.M.; McCall, C.E. Induction of RelB participates in endotoxin tolerance. J. Immunol. 2006, 177, 4080–4085. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.M.; Lenihan, C.R.; Bruning, U.; Cheong, A.; Laffey, J.G.; McLoughlin, P.; Taylor, C.T.; Cummins, E.P. Hypercapnia induces cleavage and nuclear localization of RelB protein, giving insight into CO2 sensing and signaling. J. Biol. Chem. 2012, 287, 14004–14011. [Google Scholar] [CrossRef] [PubMed]
- Brustle, A.; Brenner, D.; Knobbe, C.B.; Lang, P.A.; Virtanen, C.; Hershenfield, B.M.; Reardon, C.; Lacher, S.M.; Ruland, J.; Ohashi, P.S.; et al. The NF-κB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J. Clin. Investig. 2012, 122, 4698–4709. [Google Scholar] [CrossRef] [PubMed]
- Lo, D.; Quill, H.; Burkly, L.; Scott, B.; Palmiter, R.D.; Brinster, R.L. A recessive defect in lymphocyte or granulocyte function caused by an integrated transgene. Am. J. Pathol. 1992, 141, 1237–1246. [Google Scholar] [PubMed]
- Weih, F.; Carrasco, D.; Durham, S.K.; Barton, D.S.; Rizzo, C.A.; Ryseck, R.P.; Lira, S.A.; Bravo, R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/Rel family. Cell 1995, 80, 331–340. [Google Scholar] [CrossRef]
- McMillan, D.H.; Baglole, C.J.; Thatcher, T.H.; Maggirwar, S.; Sime, P.J.; Phipps, R.P. Lung-targeted overexpression of the NF-κB member RelB inhibits cigarette smoke-induced inflammation. Am. J. Pathol. 2011, 179, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999, 13, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, T.; Harada, N.; Mimura, J.; Motohashi, H.; Takahashi, S.; Nakajima, O.; Morita, M.; Kawauchi, S.; Yamamoto, M.; Fujii-Kuriyama, Y. Inducibility of cytochrome P450 1A1 and chemical carcinogenesis by benzo[a]pyrene in AhR repressor-deficient mice. Biochem. Biophys. Res. Commun. 2008, 365, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Baglole, C.J.; Maggirwar, S.B.; Gasiewicz, T.A.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-κB family member RelB. J. Biol. Chem. 2008, 283, 28944–28957. [Google Scholar] [CrossRef] [PubMed]
- Hecht, E.; Zago, M.; Sarill, M.; Rico de Souza, A.; Gomez, A.; Matthews, J.; Hamid, Q.; Eidelman, D.H.; Baglole, C.J. Aryl hydrocarbon receptor-dependent regulation of miR-196a expression controls lung fibroblast apoptosis but not proliferation. Toxicol. Appl. Pharmacol. 2014, 280, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Sarill, M.; Zago, M.; Sheridan, J.A.; Nair, P.; Matthews, J.; Gomez, A.; Roussel, L.; Rousseau, S.; Hamid, Q.; Eidelman, D.H.; et al. The aryl Hydrocarbon receptor suppresses cigarette-smoke-induced oxidative stress in association with dioxin response element (DRE)-independent regulation of sulfiredoxin 1. Free Radic. Biol. Med. 2015, 89, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, W.D.; Gopal, S.; Scheepers, P.T. Nanoparticles in cigarette smoke; real-time undiluted measurements by a scanning mobility particle sizer. Anal. Bioanal. Chem. 2011, 399, 3573–3578. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Vogel, C.F.; Baek, J.; Kodani, S.D.; Uppal, R.S.; Bein, K.J.; Anderson, D.S.; Van Winkle, L.S. Combustion derived ultrafine particles induce cytochrome P-450 expression in specific lung compartments in the developing neonatal and adult rat. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L665–L677. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Tan, D.; Zhou, Q.; Liu, X.; Cheng, Z.; Liu, G.; Zhu, M.; Sang, X.; Gui, S.; Cheng, J.; et al. Oxidative damage of lung and its protective mechanism in mice caused by long-term exposure to titanium dioxide nanoparticles. J. Biomed. Mater. Res. A 2012, 100, 2554–2562. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, M.; Barajas, B.; Sioutas, C.; Williams, M.A.; Nel, A.E. Nrf2 deficiency in dendritic cells enhances the adjuvant effect of ambient ultrafine particles on allergic sensitization. J. Innate Immun. 2013, 5, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, H.; Davies, K.J.; Sioutas, C.; Finch, C.E.; Morgan, T.E.; Forman, H.J. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic. Biol. Med. 2012, 52, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Singh, S.K.; Chauhan, L.K.; Das, M.; Tripathi, A.; Dwivedi, P.D. Zinc oxide nanoparticles induce apoptosis by enhancement of autophagy via PI3K/Akt/mTOR inhibition. Toxicol. Lett. 2014, 227, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Yu, Y.; Li, Y.; Wang, J.; Geng, W.; Jiang, L.; Li, Q.; Zhou, X.; Sun, Z. Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/mTOR signaling pathway. Int. J. Nanomed. 2014, 9, 5131–5141. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Zhang, Y.L.; Yang, N.; Zhang, Y.X.; Liu, X.Q.; Li, C.G.; Zhao, Y.; Wang, Y.G.; Zhang, G.G.; Yang, P.; et al. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt-TSC2-mTOR signaling. Cell Death Dis. 2011, 2, e159. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Wang, F.; Savini, M.; Ake, A.; di Ronza, A.; Sardiello, M.; Segatori, L. TFEB regulates lysosomal proteostasis. Hum. Mol. Genet. 2013, 22, 1994–2009. [Google Scholar] [CrossRef] [PubMed]
- Reamon-Buettner, S.M.; Mutschler, V.; Borlak, J. The next innovation cycle in toxicogenomics: Environmental epigenetics. Mutat. Res. 2008, 659, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Karlsson, H.L.; Coppede, F.; Migliore, L. Epigenetic effects of nano-sized materials. Toxicology 2013, 313, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Martello, G.; Piccolo, S. MicroRNA control of signal transduction. Nat. Rev. Mol. Cell Biol. 2010, 11, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.A.; Zago, M.; Nair, P.; Li, P.Z.; Bourbeau, J.; Tan, W.C.; Hamid, Q.; Eidelman, D.H.; Benedetti, A.L.; Baglole, C.J. Decreased expression of the NF-κB family member RelB in lung fibroblasts from Smokers with and without COPD potentiates cigarette smoke-induced COX-2 expression. Respir. Res. 2015, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, E.; Navarro, A.; Vinolas, N.; Marrades, R.M.; Diaz, T.; Gel, B.; Quera, A.; Bandres, E.; Garcia-Foncillas, J.; Ramirez, J.; et al. miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis 2009, 30, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101, 2087–2092. [Google Scholar] [CrossRef] [PubMed]
- Leidinger, P.; Keller, A.; Borries, A.; Huwer, H.; Rohling, M.; Huebers, J.; Lenhof, H.P.; Meese, E. Specific peripheral miRNA profiles for distinguishing lung cancer from COPD. Lung Cancer 2011, 74, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Zago, M.; Rico de Souza, A.; Hecht, E.; Rousseau, S.; Hamid, Q.; Eidelman, D.H.; Baglole, C.J. The NF-κB family member RelB regulates microRNA miR-146a to suppress cigarette smoke-induced COX-2 protein expression in lung fibroblasts. Toxicol. Lett. 2014, 226, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Calin, G.A.; Arrigo, P.; Steele, V.E.; Croce, C.M.; de Flora, S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009, 23, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Pottelberge, G.R.; Mestdagh, P.; Bracke, K.R.; Thas, O.; Durme, Y.M.; Joos, G.F.; Vandesompele, J.; Brusselle, G.G. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Jardim, M.J.; Fry, R.C.; Jaspers, I.; Dailey, L.; Diaz-Sanchez, D. Disruption of microRNA expression in human airway cells by diesel exhaust particles is linked to tumorigenesis-associated pathways. Environ. Health Perspect. 2009, 117, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Ronkko, T.; Pirjola, L.; Ntziachristos, L.; Heikkila, J.; Karjalainen, P.; Hillamo, R.; Keskinen, J. Vehicle engines produce exhaust nanoparticles even when not fueled. Environ. Sci. Technol. 2014, 48, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.S.; Poh, K.W.; Siddiqi, N.J.; Alhomida, A.S.; Yu, L.E.; Ong, W.Y. Short- and long-term changes in blood miRNA levels after nanogold injection in rats—Potential biomarkers of nanoparticle exposure. Biomarkers 2012, 17, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Echavarria, R.; Mayaki, D.; Neel, J.C.; Harel, S.; Sanchez, V.; Hussain, S.N. Angiopoietin-1 inhibits toll-like receptor 4 signalling in cultured endothelial cells: Role of miR-146b-5p. Cardiovasc. Res. 2015, 106, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Halappanavar, S.; Jackson, P.; Williams, A.; Jensen, K.A.; Hougaard, K.S.; Vogel, U.; Yauk, C.L.; Wallin, H. Pulmonary response to surface-coated nanotitanium dioxide particles includes induction of acute phase response genes, inflammatory cascades, and changes in microRNAs: A toxicogenomic study. Environ. Mol. Mutagen. 2011, 52, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.A.; Saber, A.T.; Halappanavar, S.; Jackson, P.A.; Wu, D.; Hougaard, K.S.; Jacobsen, N.R.; Williams, A.; Vogel, U.; Wallin, H.; et al. Carbon black nanoparticle intratracheal installation results in large and sustained changes in the expression of miR-135b in mouse lung. Environ. Mol. Mutagen. 2012, 53, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Bowman, L.; Meighan, T.G.; Shi, X.; Ding, M. Induction of miR-21-PDCD4 signaling by tungsten carbide-cobalt nanoparticles in JB6 cells involves ROS-mediated MAPK pathways. J. Environ. Pathol. Toxicol. Oncol. 2013, 32, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Nabeshi, H.; Yoshikawa, T.; Matsuyama, K.; Nakazato, Y.; Arimori, A.; Isobe, M.; Tochigi, S.; Kondoh, S.; Hirai, T.; Akase, T.; et al. Amorphous nanosilicas induce consumptive coagulopathy after systemic exposure. Nanotechnology 2012, 23, 045101. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Higashisaka, K.; Kunieda, A.; Iwahara, Y.; Tanaka, K.; Nagano, K.; Abe, Y.; Kamada, H.; Tsunoda, S.; Nabeshi, H.; et al. Liver-specific microRNAs as biomarkers of nanomaterial-induced liver damage. Nanotechnology 2013, 24, 405102. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Martianov, I.; Ramadass, A.; Serra Barros, A.; Chow, N.; Akoulitchev, A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 2007, 445, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G., 3rd; Wahlestedt, C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010, 11, R56. [Google Scholar] [CrossRef] [PubMed]
- Granados-Riveron, J.T.; Aquino-Jarquin, G. The complexity of the translation ability of circRNAs. Biochim. Biophys. Acta 2016, 1859, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.F.; Yang, L.; Zhang, Y.; Xiang, J.F.; Wu, Y.W.; Carmichael, G.G.; Chen, L.L. Long noncoding RNAs with snoRNA ends. Mol. Cell 2012, 48, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Dobson, L.; Nyitray, L.; Gaspari, Z. A conserved charged single α-helix with a putative steric role in paraspeckle formation. RNA 2015, 21, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Thai, P.; Statt, S.; Chen, C.H.; Liang, E.; Campbell, C.; Wu, R. Characterization of a novel long noncoding RNA, SCAL1, induced by cigarette smoke and elevated in lung cancer cell lines. Am. J. Respir. Cell Mol. Biol. 2013, 49, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Bollati, V.; Baccarelli, A. Environmental epigenetics. Heredity 2010, 105, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Tarantini, L.; Bonzini, M.; Apostoli, P.; Pegoraro, V.; Bollati, V.; Marinelli, B.; Cantone, L.; Rizzo, G.; Hou, L.; Schwartz, J.; et al. Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environ. Health Perspect. 2009, 117, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Lyn-Cook, L.; Word, B.; George, N.; Lyn-Cook, B.; Hammons, G. Effect of cigarette smoke condensate on gene promoter methylation in human lung cells. Tob. Induc. Dis. 2014, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.; Rivera, I.; Struys, E.A.; Jansen, E.E.; Ravasco, P.; Camilo, M.E.; Blom, H.J.; Jakobs, C.; Tavares de Almeida, I. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin. Chem. 2003, 49, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Zudaire, E.; Cuesta, N.; Murty, V.; Woodson, K.; Adams, L.; Gonzalez, N.; Martinez, A.; Narayan, G.; Kirsch, I.; Franklin, W.; et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J. Clin. Investig. 2008, 118, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Ryan, J.; Pereira, N.; Boughton, B.; Craig, J.M.; Saffery, R. Postnatal stability, tissue, and time specific effects of AHRR methylation change in response to maternal smoking in pregnancy. Epigenetics 2014, 9, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Monick, M.M.; Beach, S.R.; Plume, J.; Sears, R.; Gerrard, M.; Brody, G.H.; Philibert, R.A. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.A.; Beach, S.R.; Brody, G.H. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics 2012, 7, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Zeilinger, S.; Kuhnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Tao, G.; Yang, L.; Liu, J.; Liu, Q.; Zhuang, Z. SiO2 nanoparticles induce global genomic hypomethylation in HaCaT cells. Biochem. Biophys. Res. Commun. 2010, 397, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Tao, G.; Yang, L.; Liu, J.; Liu, Q.; Li, W.; Zhuang, Z. Methylation of PARP-1 promoter involved in the regulation of nano-SiO2-induced decrease of PARP-1 mRNA expression. Toxicol. Lett. 2012, 209, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Rotroff, D.M.; Joubert, B.R.; Marvel, S.W.; Haberg, S.E.; Wu, M.C.; Nilsen, R.M.; Ueland, P.M.; Nystad, W.; London, S.J.; Motsinger-Reif, A. Maternal smoking impacts key biological pathways in newborns through epigenetic modification in Utero. BMC Genom. 2016, 17, 976. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.R.; Haberg, S.E.; Nilsen, R.M.; Wang, X.; Vollset, S.E.; Murphy, S.K.; Huang, Z.; Hoyo, C.; Midttun, O.; Cupul-Uicab, L.A.; et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2012, 120, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.; Shu, C.; Lee, B.K.; Gidaya, N.; Singer, A.; Schieve, L.A.; Schendel, D.E.; Jones, N.; Daniels, J.L.; Windham, G.C.; et al. Presence of an epigenetic signature of prenatal cigarette smoke exposure in childhood. Environ. Res. 2016, 144, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Jaffar, Z.; Pinkerton, K.E.; Porter, V.; Postma, B.; Ferrini, M.; Holian, A.; Roberts, K.; Cho, Y.H. Alterations in DNA methylation and airway hyperreactivity in response to in utero exposure to environmental tobacco smoke. Inhal. Toxicol. 2015, 27, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.G.; Godderis, L.; Pieters, N.; Poels, K.; Kicinski, M.; Cuypers, A.; Fierens, F.; Penders, J.; Plusquin, M.; Gyselaers, W.; et al. Placental DNA hypomethylation in association with particulate air pollution in early life. Part. Fibre Toxicol. 2013, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; You, D.; Saravia, J.; Shen, H.; Cormier, S.A. Maternal exposure to combustion generated PM inhibits pulmonary Th1 maturation and concomitantly enhances postnatal asthma development in offspring. Part. Fibre Toxicol. 2013, 10, 29. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Traboulsi, H.; Guerrina, N.; Iu, M.; Maysinger, D.; Ariya, P.; Baglole, C.J. Inhaled Pollutants: The Molecular Scene behind Respiratory and Systemic Diseases Associated with Ultrafine Particulate Matter. Int. J. Mol. Sci. 2017, 18, 243. https://doi.org/10.3390/ijms18020243
Traboulsi H, Guerrina N, Iu M, Maysinger D, Ariya P, Baglole CJ. Inhaled Pollutants: The Molecular Scene behind Respiratory and Systemic Diseases Associated with Ultrafine Particulate Matter. International Journal of Molecular Sciences. 2017; 18(2):243. https://doi.org/10.3390/ijms18020243
Chicago/Turabian StyleTraboulsi, Hussein, Necola Guerrina, Matthew Iu, Dusica Maysinger, Parisa Ariya, and Carolyn J. Baglole. 2017. "Inhaled Pollutants: The Molecular Scene behind Respiratory and Systemic Diseases Associated with Ultrafine Particulate Matter" International Journal of Molecular Sciences 18, no. 2: 243. https://doi.org/10.3390/ijms18020243
APA StyleTraboulsi, H., Guerrina, N., Iu, M., Maysinger, D., Ariya, P., & Baglole, C. J. (2017). Inhaled Pollutants: The Molecular Scene behind Respiratory and Systemic Diseases Associated with Ultrafine Particulate Matter. International Journal of Molecular Sciences, 18(2), 243. https://doi.org/10.3390/ijms18020243