Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance



Abstract
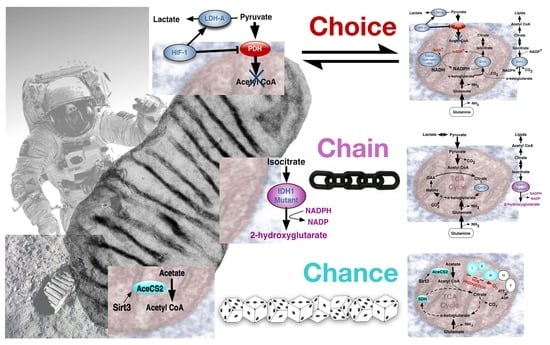
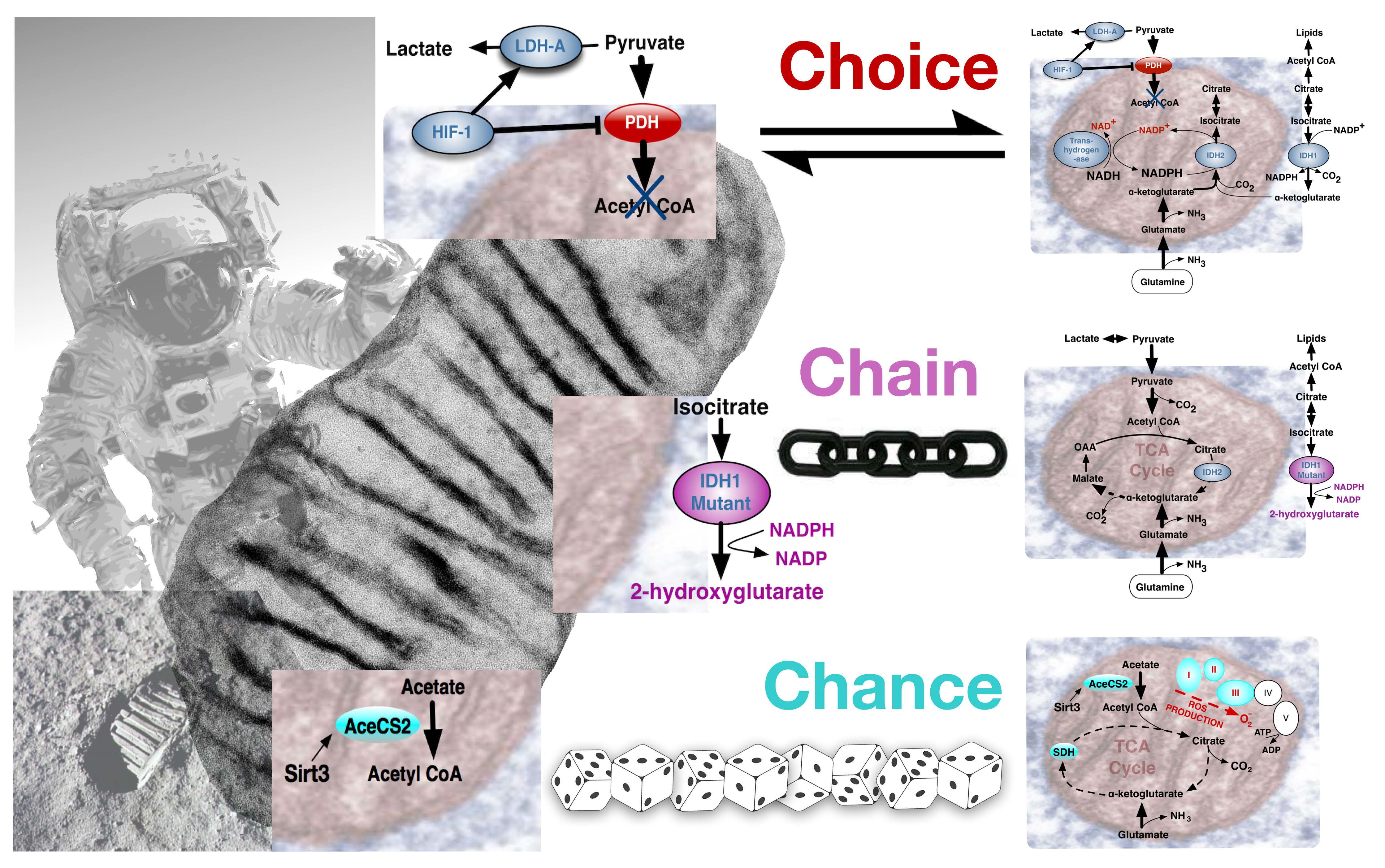{kind=link}
{kind=link}
{kind=link}
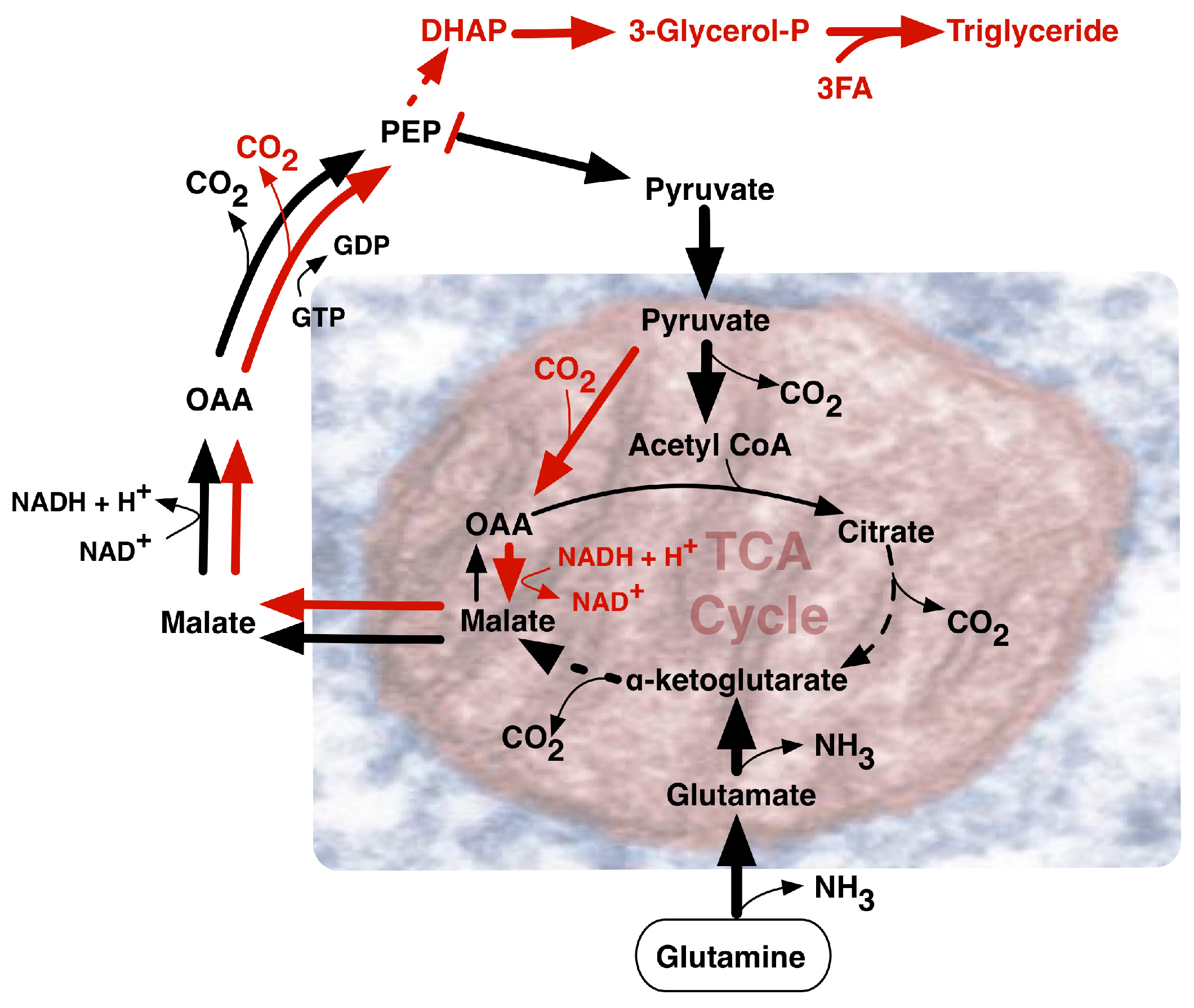{kind=link}
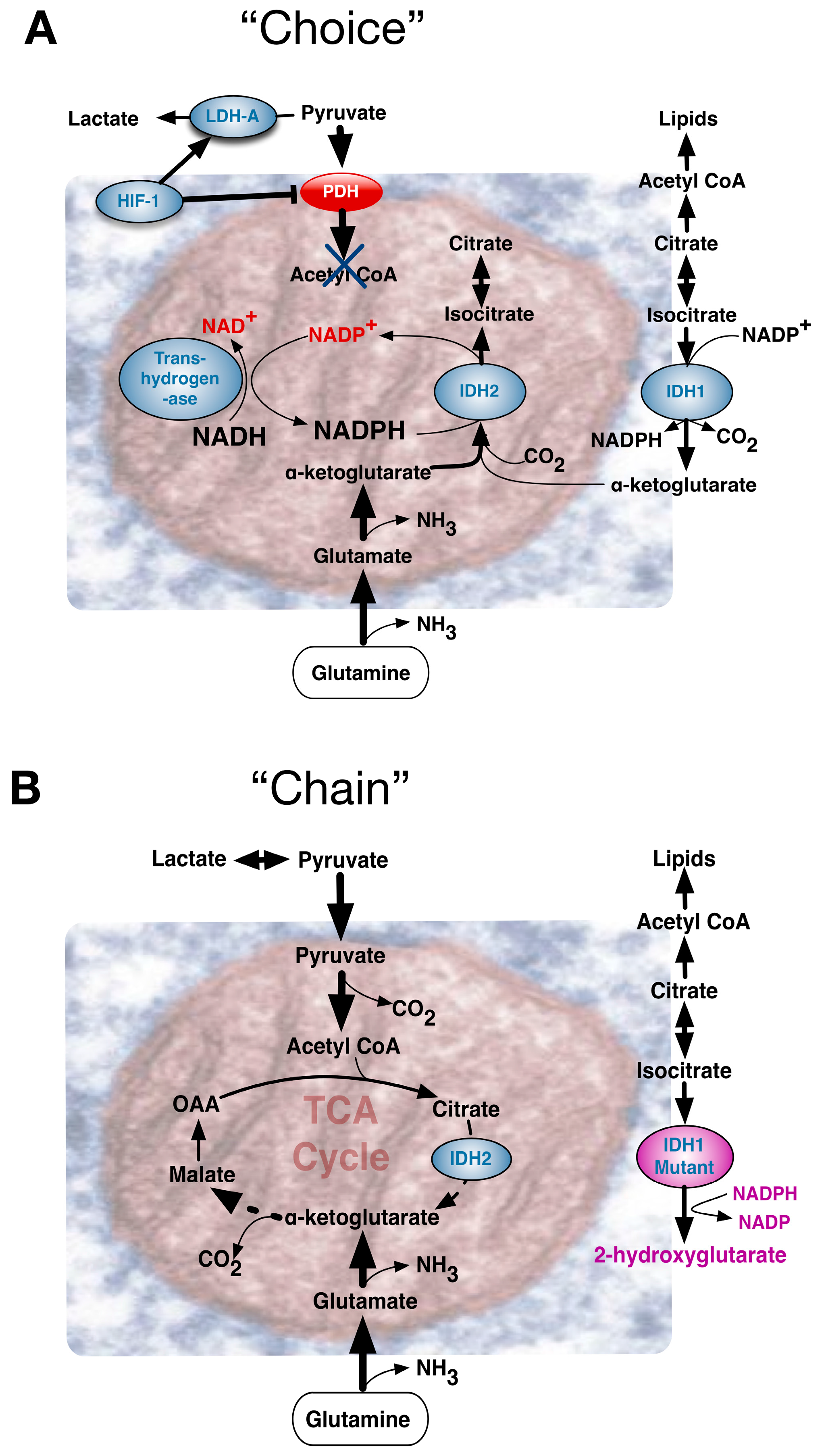{kind=link}
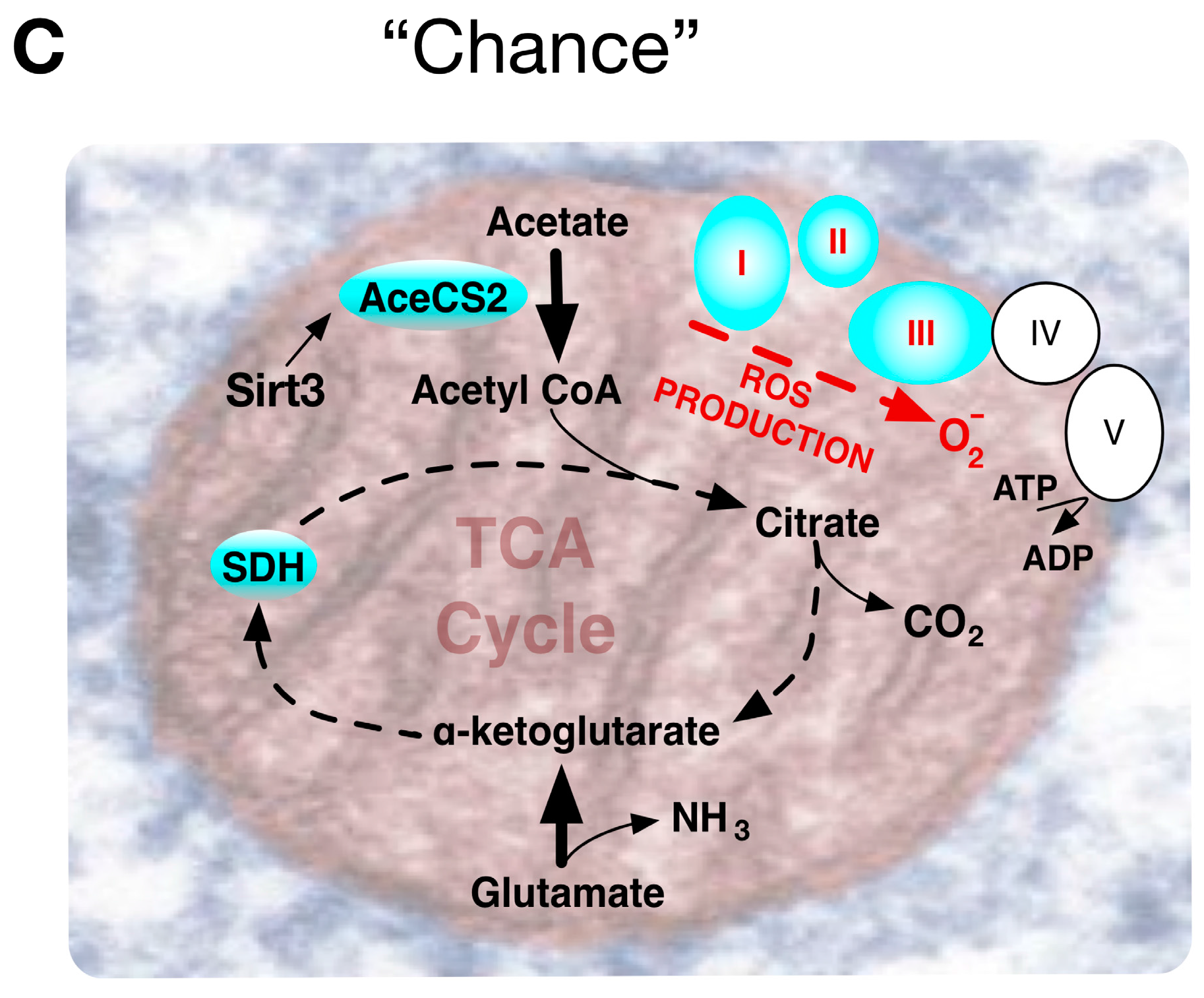{kind=link}
1. Introduction: What Is the Warburg Effect?
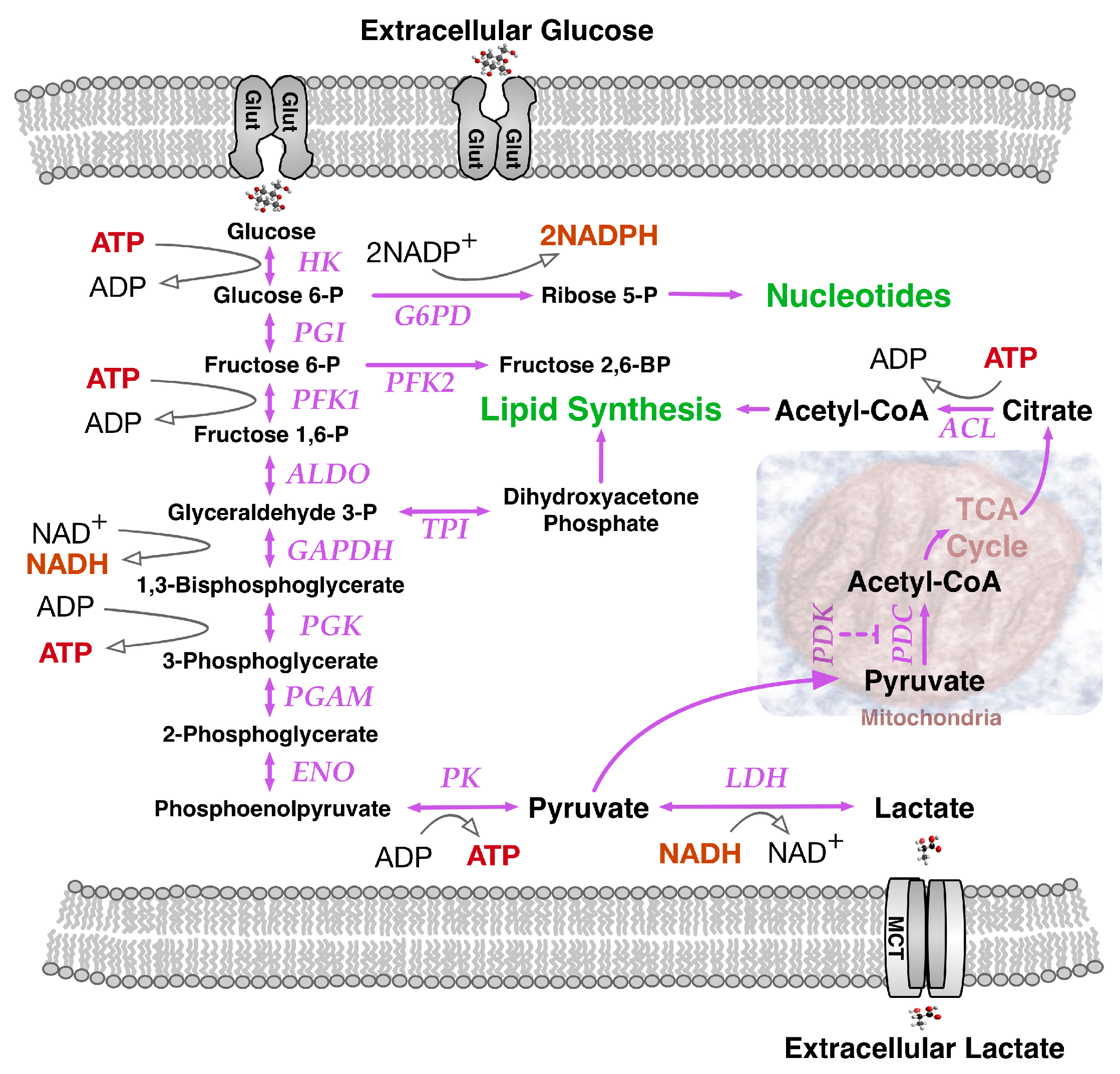
2. Paradoxes of Efficiency within Perpetual Pyruvate Pathways
3. The Need for NADPH and Diversified Carbon Sources
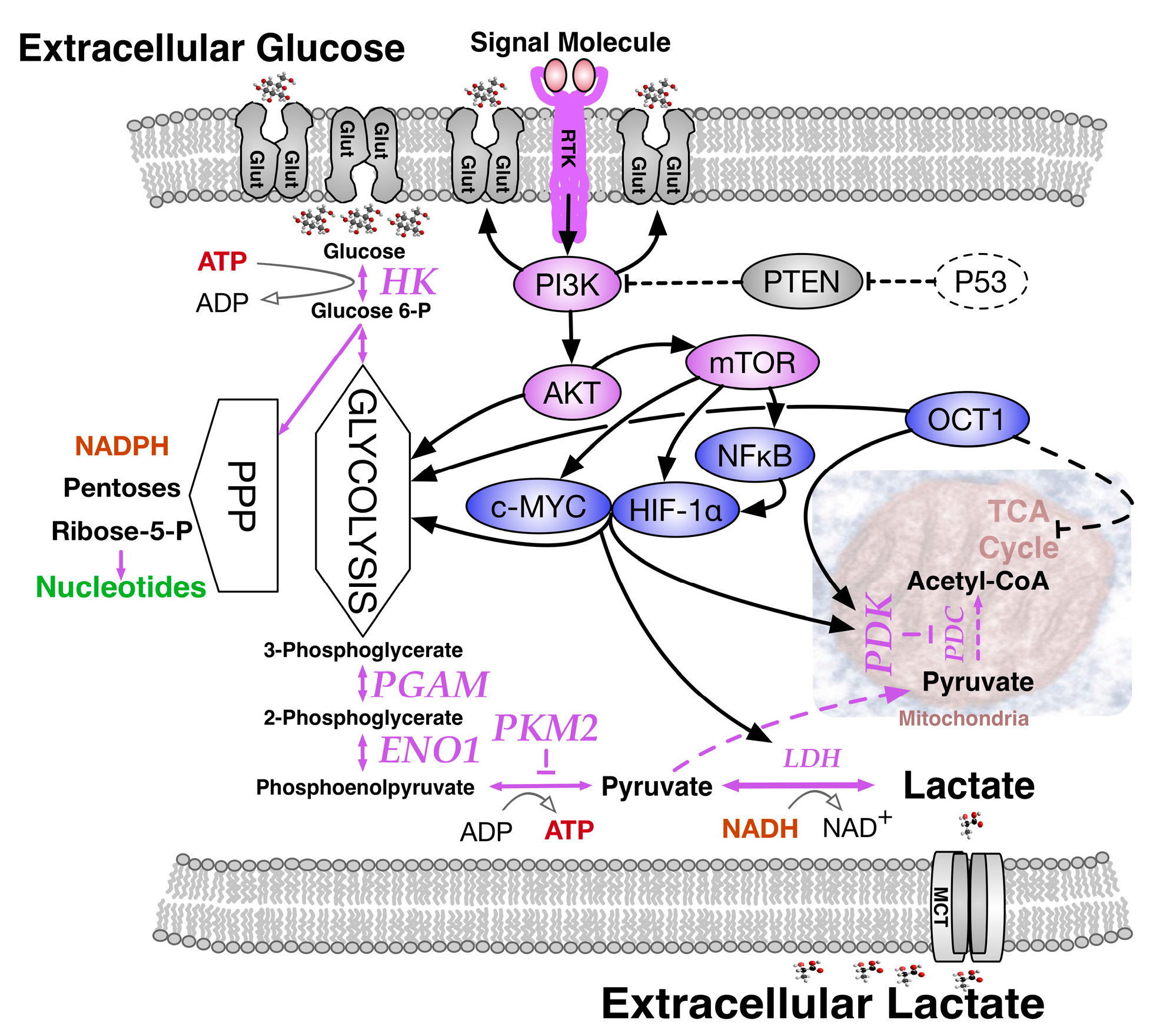
4. The Warburg Effect in Normal and Cancer Cells; Deriving the Choice, Chain or Chance Perspective
4.1. The Warburg Effect by Choice; an Adaptive Response to Oxygen and Nutrient Restrictions
4.2. The Warburg Effect Chained to Oncogenic Events
4.3. The Warburg Effect Evolved by Chance during Ageing
5. The Warburg Effect and Diabetogenesis
5.1. Lessons for T2DM from a Warburg Effect through Normal Cell Metabolic Choice
5.2. Lessons for T2DM from an Oncogenically Chained Warburg Effect
5.3. Lessons for T2DM from a Warburg Effect by Chance Whilst Ageing
6. Not Forgetting the Reverse and Inverse Warburg Hypothesis
7. New Light on Redox Metabolism Pathways in Extreme Conditions
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ehlmann, B.L.; Chowdhury, J.; Marzullo, T.C.; Collins, R.E.; Litzenberger, J.; Ibsen, S.; Krauser, W.R.; DeKock, B.; Hannon, M.; Kinnevan, J.; et al. Humans to Mars: A feasibility and cost–benefit analysis. Acta Astronaut. 2005, 56, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Wells, H.G. The War of the Worlds; William Heinemann: Portsmouth, NH, USA, 2003. [Google Scholar]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B. Introduction to mitochondrial oxidative phosphorylation. Adv. Exp. Med. Biol. 2012, 748, 1–11. [Google Scholar] [PubMed]
- Warburg, O.; Posener, K.; Negelein, E. Über den stoffwechsel der Carcinomzelle. Biochem. Z. 1924, 152, 319–344. [Google Scholar] [CrossRef]
- Wong, J.L. From fertilization to cancer: A lifelong pursuit into how cells use oxygen. Otto Heinrich Warburg (October 8, 1883–August 1, 1970). Mol. Reprod. Dev. 2011, 78. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Yan, L.; Wang, L.; Ma, J.; Jiang, W.; Zhang, Y.; Shi, Y.; Wang, Q. Isoform switch of pyruvate kinase M1 indeed occurs but not to pyruvate kinase M2 in human tumorigenesis. PLoS ONE 2015, 10, e0118663. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Locasale, J.W.; Swanson, K.D.; Sharfi, H.; Heffron, G.J.; Amador-Noguez, D.; Christofk, H.R.; Wagner, G.; Rabinowitz, J.D.; Asara, J.M.; et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010, 329, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.; Lee, H.; Park, J.; Kim, S.H.; Park, J. Targeting cancer metabolism—Revisiting the Warburg effects. Toxicol. Res. 2016, 32, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Redel, B.K.; Brown, A.N.; Spate, L.D.; Whitworth, K.M.; Green, J.A.; Prather, R.S. Glycolysis in preimplantation development is partially controlled by the Warburg Effect. Mol. Reprod. Dev. 2012, 79, 262–271. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; de Figueiredo, L.F.; Schroeter, A.; Kaleta, C. Combining metabolic pathway analysis with Evolutionary Game Theory: Explaining the occurrence of low-yield pathways by an analytic optimization approach. Biosystems 2011, 105, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Golpour, M.; Akhavan Niaki, H.; Khorasani, H.R.; Hajian, A.; Mehrasa, R.; Mostafazadeh, A. Human fibroblast switches to anaerobic metabolic pathway in response to serum starvation: A mimic of warburg effect. Int. J. Mol. Cell. Med. 2014, 3, 74–80. [Google Scholar] [PubMed]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Hwang, C.K.; Song, K.Y.; Law, P.Y.; Wei, L.N.; Loh, H.H. Poly(C)-binding proteins as transcriptional regulators of gene expression. Biochem. Biophys. Res. Commun. 2009, 380, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.; Reick, M.; Wu, L.C.; McKnight, S.L. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 2001, 293, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, V.E.; Howe, L.J. Histone acetylation: Where to go and how to get there. Epigenetics 2009, 4, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Zielke, H.R.; Ozand, P.T.; Tildon, J.T.; Sevdalian, D.A.; Cornblath, M. Reciprocal regulation of glucose and glutamine utilization by cultured human diploid fibroblasts. J. Cell. Physiol. 1978, 95, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Jauniaux, E.; Gulbis, B.; Burton, G.J. The human first trimester gestational sac limits rather than facilitates oxygen transfer to the foetus—A review. Placenta 2003, 24, S86–S93. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Jauniaux, E.; Murray, A.J. Oxygen and placental development; parallels and differences with tumour biology. Placenta 2017, 56, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Birket, M.J.; Orr, A.L.; Gerencser, A.A.; Madden, D.T.; Vitelli, C.; Swistowski, A.; Brand, M.D.; Zeng, X. A reduction in ATP demand and mitochondrial activity with neural differentiation of human embryonic stem cells. J. Cell Sci. 2011, 124, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Facucho-Oliveira, J.M.; St John, J.C. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev. 2009, 5, 140–158. [Google Scholar] [CrossRef] [PubMed]
- Krisher, R.L.; Prather, R.S. A role for the Warburg effect in preimplantation embryo development: Metabolic modification to support rapid cell proliferation. Mol. Reprod. Dev. 2012, 79, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.C.; Ilott, N.E.; Schödel, J.; Sims, D.; Tumber, A.; Lippl, K.; Mole, D.R.; Pugh, C.W.; Ratcliffe, P.J.; Ponting, C.P.; et al. Tuning the Transcriptional Response to Hypoxia by Inhibiting Hypoxia-inducible Factor (HIF) Prolyl and Asparaginyl Hydroxylases. J. Biol. Chem. 2016, 291, 20661–20673. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 versus HIF-2—Is one more important than the other. Vasc. Pharmacol. 2012, 56, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Doe, M.R.; Ascano, J.M.; Kaur, M.; Cole, M.D. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2012, 72, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.; Gardie, B.; Couvé, S.; Gad, S. Von Hippel-Lindau: How a rare disease illuminates cancer biology. Semin. Cancer Biol. 2013, 23, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.; Okhotin, D.V.; Kim, B.; Okhotina, Y.; Okhotin, D.J.; Miasnikova, G.Y.; Sergueeva, A.I.; Polyakova, L.A.; Maslow, A.; Lee, Y.; et al. Increased size of solid organs in patients with Chuvash polycythemia and in mice with altered expression of HIF-1α and HIF-2α. J. Mol. Med. 2010, 88, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.A.; Chao, Y.; Shiah, S.G.; Lin, W.W. Nutrient deprivation induces the Warburg effect through ROS/AMPK-dependent activation of pyruvate dehydrogenase kinase. Biochim. Biophys. Acta 2013, 1833, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Signaling in control of cell growth and metabolism. Cold Spring Harb. Perspect. Biol. 2012, 4, a006783. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A. Fine-tuning the lipogenic/lipolytic balance to optimize the metabolic requirements of cancer cell growth: Molecular mechanisms and therapeutic perspectives. Biochim. Biophys. Acta 2010, 1801, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Palorini, R.; De Rasmo, D.; Gaviraghi, M.; Sala Danna, L.; Signorile, A.; Cirulli, C.; Chiaradonna, F.; Alberghina, L.; Papa, S. Oncogenic K-ras expression is associated with derangement of the cAMP/PKA pathway and forskolin-reversible alterations of mitochondrial dynamics and respiration. Oncogene 2013, 32, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Lin, T.; Kamarajugadda, S.; Lu, J. Regulation of glycolysis and the Warburg effect by estrogen-related receptors. Oncogene 2013, 32, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, P.; Zhao, P.; Angelis, E.; Ruan, H.; Korge, P.; Olson, A.; Wang, Y.; Jin, E.S.; Jeffrey, F.M.; Portman, M.; et al. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J. Clin. Investig. 2010, 120, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.J.; Ayer, D.E. Myc, mondo, and metabolism. Genes Cancer 2010, 1, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Sans, C.L.; Satterwhite, D.J.; Stoltzman, C.A.; Breen, K.T.; Ayer, D.E. MondoA-Mlx heterodimers are candidate sensors of cellular energy status: Mitochondrial localization and direct regulation of glycolysis. Mol. Cell. Biol. 2006, 26, 4863–4871. [Google Scholar] [CrossRef] [PubMed]
- Wilde, B.R.; Ayer, D.E. Interactions between Myc and MondoA transcription factors in metabolism and tumourigenesis. Br. J. Cancer 2015, 113, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by MondoA:Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Zhao, F.; Mancuso, A.; Gruber, J.J.; Thompson, C.B. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 21660–21665. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Abu Dawud, R.; Schreiber, K.; Schomburg, D.; Adjaye, J. Human embryonic stem cells and embryonal carcinoma cells have overlapping and distinct metabolic signatures. PLoS ONE 2012, 7, e39896. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Tan, A.S. Effects of mitochondrial gene deletion on tumorigenicity of metastatic melanoma: Reassessing the Warburg effect. Rejuvenation Res. 2010, 13, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Sottnik, J.L.; Lori, J.C.; Rose, B.J.; Thamm, D.H. Glycolysis inhibition by 2-deoxy-d-glucose reverts the metastatic phenotype in vitro and in vivo. Clin. Exp. Metastasis 2011, 28, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; Pergolizzi, B.; Ferro, E.; Trabalzini, L.; Retta, S.F. Molecular Crosstalk between Integrins and Cadherins: Do Reactive Oxygen Species Set the Talk. J. Signal Transduct. 2012, 2012, 807682. [Google Scholar] [CrossRef] [PubMed]
- Smolková, K.; Plecitá-Hlavatá, L.; Bellance, N.; Benard, G.; Rossignol, R.; Ježek, P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 2011, 43, 950–968. [Google Scholar] [CrossRef] [PubMed]
- Ohkouchi, S.; Block, G.J.; Katsha, A.M.; Kanehira, M.; Ebina, M.; Kikuchi, T.; Saijo, Y.; Nukiwa, T.; Prockop, D.J. Mesenchymal stromal cells protect cancer cells from ROS-induced apoptosis and enhance the Warburg effect by secreting STC1. Mol. Ther. 2012, 20, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Roy, A.; Dwarakanath, B.S. Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front. Oncol. 2017, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Ho, Q.S.; Low, J.; Choolani, M.; Wong, K.P. Respiratory competent mitochondria in human ovarian and peritoneal cancer. Mitochondrion 2011, 11, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Tennant, D.A.; Gottlieb, E. IDH1 mutations in gliomas: When an enzyme loses its grip. Cancer Cell 2010, 17, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; Latorre, D.; Cocco, T.; Capuano, F.; Kukat, C.; Seibel, P.; Villani, G. Tight control of mitochondrial membrane potential by cytochrome c oxidase. Mitochondrion 2011, 11, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Groen, A.K.; Wanders, R.J.; Westerhoff, H.V.; van der Meer, R.; Tager, J.M. Quantification of the contribution of various steps to the control of mitochondrial respiration. J. Biol. Chem. 1982, 257, 2754–2757. [Google Scholar] [PubMed]
- Haigis, M.C.; Deng, C.X.; Finley, L.W.; Kim, H.S.; Gius, D. SIRT3 is a mitochondrial tumor suppressor: A scientific tale that connects aberrant cellular ROS, the Warburg effect, and carcinogenesis. Cancer Res. 2012, 72, 2468–2472. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells. Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Ratnaike, T.E.; De Gruyter, H.L.; Jaros, E.; Turnbull, D.M. Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 2210–2214. [Google Scholar] [CrossRef] [PubMed]
- Minet, A.D.; Gaster, M. Cultured senescent myoblasts derived from human vastus lateralis exhibit normal mitochondrial ATP synthesis capacities with correlating concomitant ROS production while whole cell ATP production is decreased. Biogerontology 2012, 13, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, O.; Shimizu, A.; Yokota, M.; Sugiyama, A.; Nakada, K.; Miyoshi, H.; Itami, M.; Ohira, M.; Nagase, H.; Takenaga, K.; et al. Specific mitochondrial DNA mutation in mice regulates diabetes and lymphoma development. Proc. Natl. Acad. Sci. USA 2012, 109, 10528–10533. [Google Scholar] [CrossRef] [PubMed]
- Hirabara, S.M.; Curi, R.; Maechler, P. Saturated fatty acid-induced insulin resistance is associated with mitochondrial dysfunction in skeletal muscle cells. J. Cell. Physiol. 2010, 222, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Maechler, P.; Wollheim, C.B. Mitochondrial function in normal and diabetic β-cells. Nature 2001, 414, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Strollo, F.; Vassilieva, G.; Ruscica, M.; Masini, M.; Santucci, D.; Borgia, L.; Magni, P.; Celotti, F.; Nikiporuc, I. Changes in stress hormones and metabolism during a 105-day simulated Mars mission. Aviat. Space Environ. Med. 2014, 85, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Tobin, B.W.; Uchakin, P.N.; Leeper-Woodford, S.K. Insulin secretion and sensitivity in space flight: Diabetogenic effects. Nutrition 2002, 18, 842–848. [Google Scholar] [CrossRef]
- Rejeski, W.J.; Ip, E.H.; Bertoni, A.G.; Bray, G.A.; Evans, G.; Gregg, E.W.; Zhang, Q.; Look AHEAD Research Group. Lifestyle change and mobility in obese adults with type 2 diabetes. N. Engl. J. Med. 2012, 366, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H.; Schmidt, L.A.; Brindis, C.D. Public health: The toxic truth about sugar. Nature 2012, 482, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, L.D.; Bruin, J.E.; Greenaway, J.; Campbell, N.E.; Moorehead, R.A.; Holloway, A.C.; Petrik, J. The role of dysregulated glucose metabolism in epithelial ovarian cancer. J. Oncol. 2010, 2010, 514310. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.R.; Um, H.D. Hydrogen peroxide suppresses U937 cell death by two different mechanisms depending on its concentration. Exp. Cell Res. 1999, 248, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, A.D.; Bartlett, T.; Oates, P.J.; Millward, B.A.; Demaine, A.G. The response of antioxidant genes to hyperglycemia is abnormal in patients with type 1 diabetes and diabetic nephropathy. Diabetes 2003, 52, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Brownlee, M. The missing link: A single unifying mechanism for diabetic complications. Kidney Int. Suppl. 2000, 77, S26–S30. [Google Scholar] [CrossRef] [PubMed]
- Ethier-Chiasson, M.; Forest, J.C.; Giguère, Y.; Masse, A.; Marseille-Tremblay, C.; Lévy, E.; Lafond, J. Modulation of placental protein expression of OLR1: Implication in pregnancy-related disorders or pathologies. Reproduction 2008, 136, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Khaidakov, M.; Mitra, S.; Kang, B.Y.; Wang, X.; Kadlubar, S.; Novelli, G.; Raj, V.; Winters, M.; Carter, W.C.; Mehta, J.L. Oxidized LDL receptor 1 (OLR1) as a possible link between obesity, dyslipidemia and cancer. PLoS ONE 2011, 6, e20277. [Google Scholar] [CrossRef] [PubMed]
- Cagnone, G.L.; Dufort, I.; Vigneault, C.; Sirard, M.A. Differential gene expression profile in bovine blastocysts resulting from hyperglycemia exposure during early cleavage stages. Biol. Reprod. 2012, 86, 50. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.K.; Wale, P.L.; Collins, R.; Lane, M. Glucose consumption of single post-compaction human embryos is predictive of embryo sex and live birth outcome. Hum. Reprod. 2011, 26, 1981–1986. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Hitosugi, T.; Chung, T.W.; Xie, J.; Ge, Q.; Gu, T.L.; Polakiewicz, R.D.; Chen, G.Z.; Boggon, T.J.; Lonial, S.; et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD+ redox homeostasis in cancer cells. Mol. Cell. Biol. 2011, 31, 4938–4950. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Malmgren, S.; Nicholls, D.G.; Taneera, J.; Bacos, K.; Koeck, T.; Tamaddon, A.; Wibom, R.; Groop, L.; Ling, C.; Mulder, H.; et al. Tight coupling between glucose and mitochondrial metabolism in clonal β-cells is required for robust insulin secretion. J. Biol. Chem. 2009, 284, 32395–32404. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Fan, J.; Chung, T.W.; Lythgoe, K.; Wang, X.; Xie, J.; Ge, Q.; Gu, T.L.; Polakiewicz, R.D.; Roesel, J.L.; et al. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 2011, 44, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.Y.; Zhou, D.X.; Lu, J.; Zhang, W.J.; Zou, D.J. Down-regulated expression of the protein-tyrosine phosphatase 1B (PTP1B) is associated with aggressive clinicopathologic features and poor prognosis in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2012, 420, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Lessard, L.; Labbé, D.P.; Deblois, G.; Bégin, L.R.; Hardy, S.; Mes-Masson, A.M.; Saad, F.; Trotman, L.C.; Giguère, V.; Tremblay, M.L. PTP1B is an androgen receptor-regulated phosphatase that promotes the progression of prostate cancer. Cancer Res. 2012, 72, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Depetris, R.S.; Barford, D.; Chernoff, J.; Hubbard, S.R. Crystal structure of a complex between protein tyrosine phosphatase 1B and the insulin receptor tyrosine kinase. Structure 2005, 13, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J.; Bittner-Kowalczyk, A.; White, M.F.; Harbeck, M. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the Grb2 adaptor protein. J. Biol. Chem. 2000, 275, 4283–4289. [Google Scholar] [CrossRef] [PubMed]
- Zinker, B.A.; Rondinone, C.M.; Trevillyan, J.M.; Gum, R.J.; Clampit, J.E.; Waring, J.F.; Xie, N.; Wilcox, D.; Jacobson, P.; Frost, L.; et al. PTP1B antisense oligonucleotide lowers PTP1B protein, normalizes blood glucose, and improves insulin sensitivity in diabetic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11357–11362. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Andersen, J.N.; Salmeen, A.; Barford, D.; Tonks, N.K. Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell 2011, 147, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.F.; Whitaker-Menezes, D.; Lin, Z.; Martinez-Outschoorn, U.E.; Tanowitz, H.B.; Al-Zoubi, M.S.; Howell, A.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Two-compartment tumor metabolism: Autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle 2012, 11, 2545–2556. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.; Zhang, J.; Lam, E.K.; Shin, V.Y.; Cheng, A.S.; Yu, J.; Chan, F.K.; Sung, J.J.; Jin, H.C. Warburg effect revisited: An epigenetic link between glycolysis and gastric carcinogenesis. Oncogene 2010, 29, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jin, H. The epigenetic basis of the Warburg effect. Epigenetics 2010, 5, 566–568. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, A.S.; Kelly, M.; Berg, R.M.; Møller, K.; Pedersen, B.K. Type 2 diabetes is associated with altered NF-κB DNA binding activity, JNK phosphorylation, and AMPK phosphorylation in skeletal muscle after LPS. PLoS ONE 2011, 6, e23999. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Pownall, H.J. The adipocyte IKK/NF-κB pathway: A therapeutic target for insulin resistance. Curr. Opin. Investig. Drugs 2009, 10, 346–352. [Google Scholar] [PubMed]
- Volkmar, M.; Dedeurwaerder, S.; Cunha, D.A.; Ndlovu, M.N.; Defrance, M.; Deplus, R.; Calonne, E.; Volkmar, U.; Igoillo-Esteve, M.; Naamane, N.; et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012, 31, 1405–1426. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.M.; Baddour, A.A.; Kawashita, T.; Allen, B.D.; Syage, A.R.; Nguyen, T.H.; Yoon, N.; Giedzinski, E.; Yu, L.; Parihar, V.K.; et al. Epigenetic determinants of space radiation-induced cognitive dysfunction. Sci. Rep. 2017, 7, 42885. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Lowe, S.W. The microcosmos of cancer. Nature 2012, 482, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Ferland-McCollough, D.; Ozanne, S.E.; Siddle, K.; Willis, A.E.; Bushell, M. The involvement of microRNAs in Type 2 diabetes. Biochem. Soc. Trans. 2010, 38, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Liu, H.; Liu, Y.; Wu, J.; Wang, C.; Hou, X.; Chen, X.; Yang, G.; Zhao, L.; Che, H.; et al. miR-143 inhibits glycolysis and depletes stemness of glioblastoma stem-like cells. Cancer Lett. 2013, 333, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.Y.; Jing, R.; Smalley, K.J.; Taccioli, C.; Fahrmann, J.; Barupal, D.K.; Alder, H.; Farber, J.L.; Fiehn, O.; Croce, C.M. Integration of metabolomics, transcriptomics, and microRNA expression profiling reveals a miR-143-HK2-glucose network underlying zinc-deficiency-associated esophageal neoplasia. Oncotarget 2017, 8, 81910–81925. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.D.; Krüger, M.; Willmes, D.M.; Redemann, N.; Wunderlich, F.T.; Brönneke, H.S.; Merkwirth, C.; Kashkar, H.; Olkkonen, V.M.; Böttger, T.; et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol. 2011, 13, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, J.R. Cu, Fe, Mn, and Zn chelates offer a medicinal chemistry approach to overcoming radiation injury. Curr. Med. Chem. 2002, 9, 639–662. [Google Scholar] [CrossRef] [PubMed]
- Bergouignan, A.; Stein, T.P.; Habold, C.; Coxam, V.; O’ Gorman, D.; Blanc, S. Towards human exploration of space: The THESEUS review series on nutrition and metabolism research priorities. NPJ Microgravity 2016, 2, 16029. [Google Scholar] [CrossRef] [PubMed]
- Gaster, M.; Nehlin, J.O.; Minet, A.D. Impaired TCA cycle flux in mitochondria in skeletal muscle from type 2 diabetic subjects: Marker or maker of the diabetic phenotype. Arch. Physiol. Biochem. 2012, 118, 156–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Michikawa, Y.; Mallidis, C.; Bai, Y.; Woodhouse, L.; Yarasheski, K.E.; Miller, C.A.; Askanas, V.; Engel, W.K.; Bhasin, S.; et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc. Natl. Acad. Sci. USA 2001, 98, 4022–4027. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sirtuins, Aging, and Medicine. N. Engl. J. Med. 2011, 364, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.; Han, M.J.; Cha, H.J.; Zoldan, J.; Burkart, A.; Jung, J.H.; Jang, Y.; Kim, C.H.; Jeong, H.C.; Kim, B.G.; et al. Metabolic control of primed human pluripotent stem cell fate and function by the miR-200c-SIRT2 axis. Nat. Cell Biol. 2017, 19, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Seok, S.; Yau, P.; Li, X.; Kemper, B.; Kemper, J.K. Obesity and aging diminish sirtuin 1 (SIRT1)-mediated deacetylation of SIRT3, leading to hyperacetylation and decreased activity and stability of SIRT3. J. Biol. Chem. 2017, 292, 17312–17323. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, Á.; Más-Gutierrez, J.A.; Mirasierra, M.; Fernandez-Pérez, A.; Lee, Y.J.; Ko, H.J.; Kim, J.K.; Romanos, E.; Carrascosa, J.M.; Ros, M.; et al. Essential role of protein tyrosine phosphatase 1B in obesity-induced inflammation and peripheral insulin resistance during aging: Role of PTP1B in aging-related insulin resistance. Aging Cell 2012, 11, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Tran, P.O.T.; Harmon, J.; Robertson, R.P. A role for glutathione peroxidase in protecting pancreatic cells against oxidative stress in a model of glucose toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 12363–12368. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; DePinho, R.A. Axis of ageing: Telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 2012, 13, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Tavi, P.; Korhonen, T.; Hänninen, S.L.; Bruton, J.D.; Lööf, S.; Simon, A.; Westerblad, H. Myogenic skeletal muscle satellite cells communicate by tunnelling nanotubes. J. Cell. Physiol. 2010, 223, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Tarnopolsky, M.A. Mitochondrial DNA shifting in older adults following resistance exercise training. Appl. Physiol. Nutr. Metab. 2009, 34, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Jash, S.; Adhya, S. Induction of muscle regeneration by RNA-mediated mitochondrial restoration. FASEB J. 2012, 26, 4187–4197. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Davidson, M.M.; Hei, T.K. Mitochondria regulate DNA damage and genomic instability induced by high LET radiation. Life Sci. Space Res. 2014, 1, 80–88. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Curry, J.M.; Ko, Y.H.; Lin, Z.; Tuluc, M.; Cognetti, D.; Birbe, R.C.; Pribitkin, E.; Bombonati, A.; Pestell, R.G.; et al. Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: RAS and NFκB target stromal MCT4. Cell Cycle 2013, 12, 2580–2597. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N.; Skinner, A.M.; Pommier, R.F.; Schillace, R.V.; O’Neill, S.; Peckham, J.L.; Muller, P.; Condron, M.E.; Donovan, C.; Naik, A.; et al. Gene expression signatures of breast cancer stem and progenitor cells do not exhibit features of Warburg metabolism. Stem Cell Res. Ther. 2015, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Shao, S.X.; Jiang, H.P.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Wang, Y.L.; Wang, X.S.; Niu, H.T. Warburg effect or reverse Warburg effect? A review of cancer metabolism. Oncol. Res. Treat. 2015, 38, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Manda, G.; Isvoranu, G.; Comanescu, M.V.; Manea, A.; Debelec Butuner, B.; Korkmaz, K.S. The redox biology network in cancer pathophysiology and therapeutics. Redox Biol. 2015, 5, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Knatko, E.V.; Higgins, M.; Fahey, J.W.; Dinkova-Kostova, A.T. Loss of Nrf2 abrogates the protective effect of Keap1 downregulation in a preclinical model of cutaneous squamous cell carcinoma. Sci. Rep. 2016, 6, 25804. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.K.; Warfel, N.A. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants 2017, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat. Res. 2015, 779, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Wakabayashi, N.; Kensler, T.W. Keap1/Nrf2 pathway in the frontiers of cancer and non-cancer cell metabolism. Biochem. Soc. Trans. 2015, 43, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Indo, H.P.; Majima, H.J.; Terada, M.; Suenaga, S.; Tomita, K.; Yamada, S.; Higashibata, A.; Ishioka, N.; Kanekura, T.; Nonaka, I.; et al. Changes in mitochondrial homeostasis and redox status in astronauts following long stays in space. Sci. Rep. 2016, 6, 39015. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, M.; Li, L.; Chen, L. Involvement of the Warburg effect in non-tumor diseases processes. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Tsirigos, A.; Vera, I.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; Martinez-Outschoorn, U.E.; Howell, A.; et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: Similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging 2010, 2, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Friedland, K.; Eckert, A. Mitochondrial dysfunction: The missing link between aging and sporadic Alzheimer’s disease. Biogerontology 2016, 17, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Gavrilova, O.; Brown, A.L.; Soto, J.E.; Bremner, S.; Kim, J.; Xu, X.; Yang, S.; Um, J.H.; Koch, L.G.; et al. DNA-PK promotes the mitochondrial, metabolic, and physical decline that occurs during aging. Cell Metab. 2017, 25, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.; Costalat, R.; Pellerin, L. Determinants of brain cell metabolic phenotypes and energy substrate utilization unraveled with a modeling approach. PLoS Comput. Biol. 2012, 8, e1002686. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; de Bari, L.; Bobba, A.; Amadoro, G. A disease with a sweet tooth: Exploring the Warburg effect in Alzheimer’s disease. Biogerontology 2017, 18, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; de Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure. J. Neurochem. 2016, 139, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Gunderman, R.B.; Gonda, A.S. Radium girls. Radiology 2015, 274, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Setlow, R.B. The hazards of space travel. EMBO Rep. 2003, 4, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Grosse, J.; Wehland, M.; Mann, V.; Reseland, J.E.; Sundaresan, A.; Corydon, T.J. The impact of microgravity on bone in humans. Bone 2016, 87, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Saha, R.; Palanisamy, A.; Ghosh, M.; Biswas, A.; Roy, S.; Pal, A.; Sarkar, K.; Bagh, S. A systems biology pipeline identifies new immune and disease related molecular signatures and networks in human cells during microgravity exposure. Sci. Rep. 2016, 6, 25975. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Liu, B.; Frost, J.L.; Lemere, C.A.; Williams, J.P.; Olschowka, J.A.; O’Banion, M.K. Galactic cosmic radiation leads to cognitive impairment and increased aβ plaque accumulation in a mouse model of Alzheimer’s disease. PLoS ONE 2012, 7, e53275. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Verslegers, M.; Quintens, R.; Samari, N.; de Saint-Georges, L.; van Oostveldt, P.; Baatout, S.; Benotmane, M.A. Combined exposure to simulated microgravity and acute or chronic radiation reduces neuronal network integrity and survival. PLoS ONE 2016, 11, e0155260. [Google Scholar] [CrossRef] [PubMed]
- Pecaut, M.J.; Mao, X.W.; Bellinger, D.L.; Jonscher, K.R.; Stodieck, L.S.; Ferguson, V.L.; Bateman, T.A.; Mohney, R.P.; Gridley, D.S. Is spaceflight-induced immune dysfunction linked to systemic changes in metabolism. PLoS ONE 2017, 12, e0174174. [Google Scholar] [CrossRef] [PubMed]
- Gohil, V.M.; Sheth, S.A.; Nilsson, R.; Wojtovich, A.P.; Lee, J.H.; Perocchi, F.; Chen, W.; Clish, C.B.; Ayata, C.; Brookes, P.S.; et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat. Biotechnol. 2010, 28, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C.; et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Capello, M.; Fredolini, C.; Racanicchi, L.; Piemonti, L.; Liotta, L.A.; Novelli, F.; Petricoin, E.F. Proteomic analysis reveals Warburg effect and anomalous metabolism of glutamine in pancreatic cancer cells. J. Proteome Res. 2012, 11, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012, CD004426. [Google Scholar] [CrossRef]
- Ran, F.; An, L.; Fan, Y.; Hang, H.; Wang, S. Simulated microgravity potentiates generation of reactive oxygen species in cells. Biophys. Rep. 2016, 2, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Dang, B.; Yang, Y.; Zhang, E.; Li, W.; Mi, X.; Meng, Y.; Yan, S.; Wang, Z.; Wei, W.; Shao, C.; et al. Simulated microgravity increases heavy ion radiation-induced apoptosis in human B lymphoblasts. Life Sci. 2014, 97, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Held, K.D.; Blakely, E.A.; Story, M.D.; Lowenstein, D.I. Use of the NASA space radiation laboratory at Brookhaven national laboratory to conduct charged particle radiobiology studies relevant to ion therapy. Radiat. Res. 2016, 185, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Souda, H.; Puspitasari, A.; Held, K.D.; Hidema, J.; Nikawa, T.; Yoshida, Y.; Kanai, T.; Takahashi, A. Development and performance evaluation of a three-dimensional clinostat synchronized heavy-ion irradiation system. Life Sci. Space Res. 2017, 12, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Hidding, B.; Karger, O.; Königstein, T.; Pretzler, G.; Manahan, G.G.; McKenna, P.; Gray, R.; Wilson, R.; Wiggins, S.M.; Welsh, G.H.; et al. Laser-plasma-based space radiation reproduction in the laboratory. Sci. Rep. 2017, 7, 42354. [Google Scholar] [CrossRef] [PubMed]
- Popovici, M.A.; Mitu, I.O.; Căta-Danil, G.; Negoit Ă, F.; Ivan, C. Shielding assessment of high field (QED) experiments at the ELI-NP 10 PW laser system. J. Radiol. Prot. 2017, 37, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P.; Floridi, L.; Boraschi, D.; Cuadrado, A.; Manda, G.; Levic, S.; D’Acquisto, F.; Hamilton, A.; Athersuch, T.J.; Selley, L. Oxidative stress and inflammation induced by environmental and psychological stressors: A biomarker perspective. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Neveu, M.A.; De Preter, G.; Marchand, V.; Bol, A.; Brender, J.R.; Saito, K.; Kishimoto, S.; Porporato, P.E.; Sonveaux, P.; Grégoire, V.; et al. Multimodality imaging identifies distinct metabolic profiles in vitro and in vivo. Neoplasia 2016, 18, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.W. Mutation not universally linked with diabetes. Nature 2002, 416, 677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Moreno-Villanueva, M.; Krieger, S.; Ramesh, G.T.; Neelam, S.; Wu, H. Transcriptomics, NF-κB pathway, and their potential spaceflight-related health consequences. Int. J. Mol. Sci. 2017, 18, 1166. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Zois, C.E.; Kalamida, D.; Pouliliou, S.; Karagounis, I.V.; Yeh, T.; Abboud, M.I.; Claridge, T.D.W.; Schofield, C.J.; et al. Normal tissue radioprotection by amifostine via Warburg-type effects. Sci. Rep. 2016, 6, 30986. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, C.E.; Spitta, L.F.; Henschenmacher, B.; Diegeler, S.; Baumstark-Khan, C. Transcription Factors in the Cellular Response to Charged Particle Exposure. Front. Oncol. 2016, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Liu, W.; Wang, Z.; Ren, D.; Peng, W. Characterizing Alzheimer’s disease through metabolomics and investigating anti-Alzheimer’s disease effects of natural products. Ann. N. Y. Acad. Sci. 2017, 1398, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.A.; Goodwin, T.J.; Pelligra, R. Incorporation of omics analyses into artificial gravity research for space exploration countermeasure development. Metabolomics 2016, 12, 36. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burns, J.S.; Manda, G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int. J. Mol. Sci. 2017, 18, 2755. https://doi.org/10.3390/ijms18122755
Burns JS, Manda G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. International Journal of Molecular Sciences. 2017; 18(12):2755. https://doi.org/10.3390/ijms18122755
Chicago/Turabian StyleBurns, Jorge S., and Gina Manda. 2017. "Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance" International Journal of Molecular Sciences 18, no. 12: 2755. https://doi.org/10.3390/ijms18122755
APA StyleBurns, J. S., & Manda, G. (2017). Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. International Journal of Molecular Sciences, 18(12), 2755. https://doi.org/10.3390/ijms18122755