Molecular Pathogenesis of Radiation-Induced Cell Toxicity in Stem Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Biologic Effects of Radiation Therapy
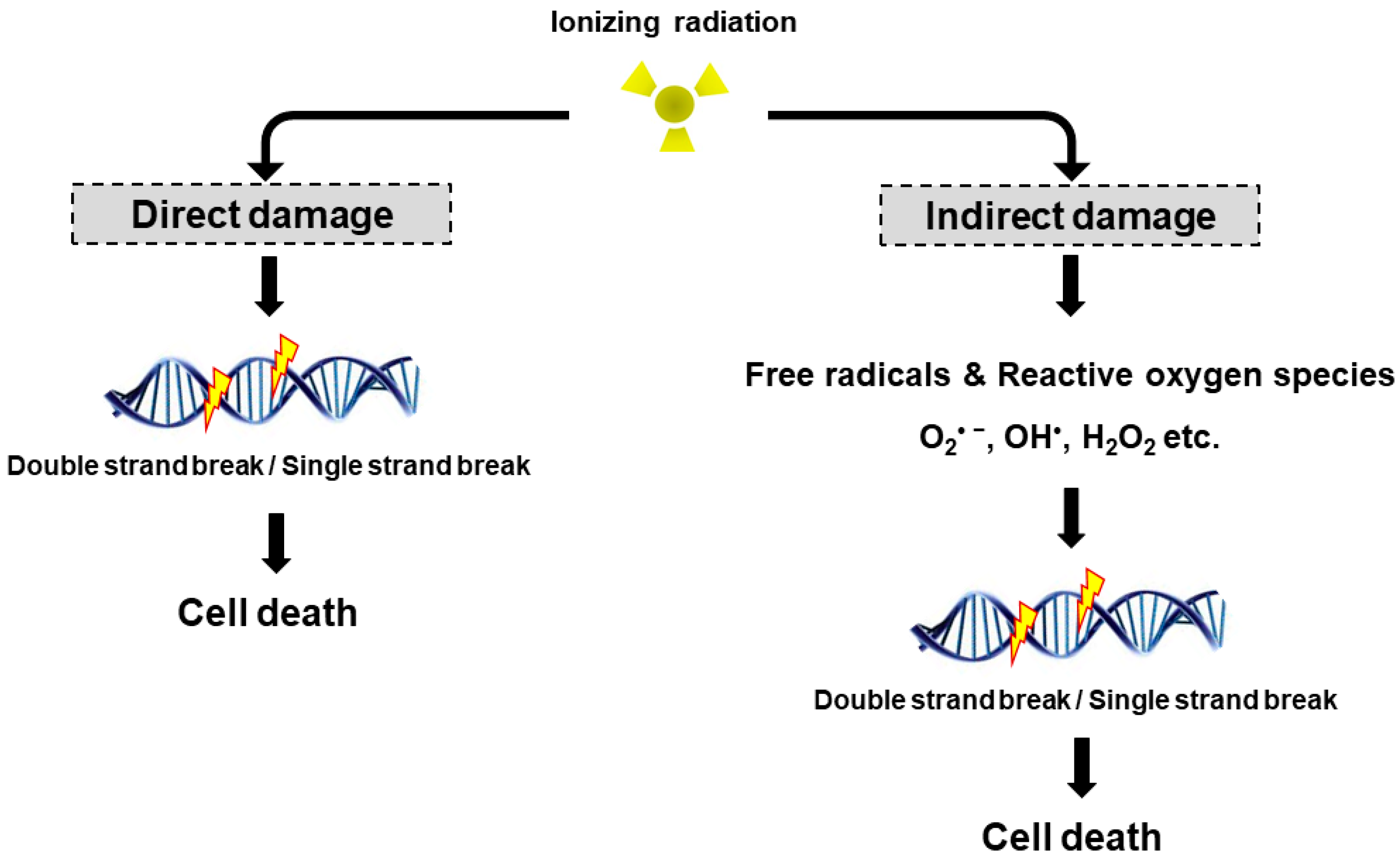
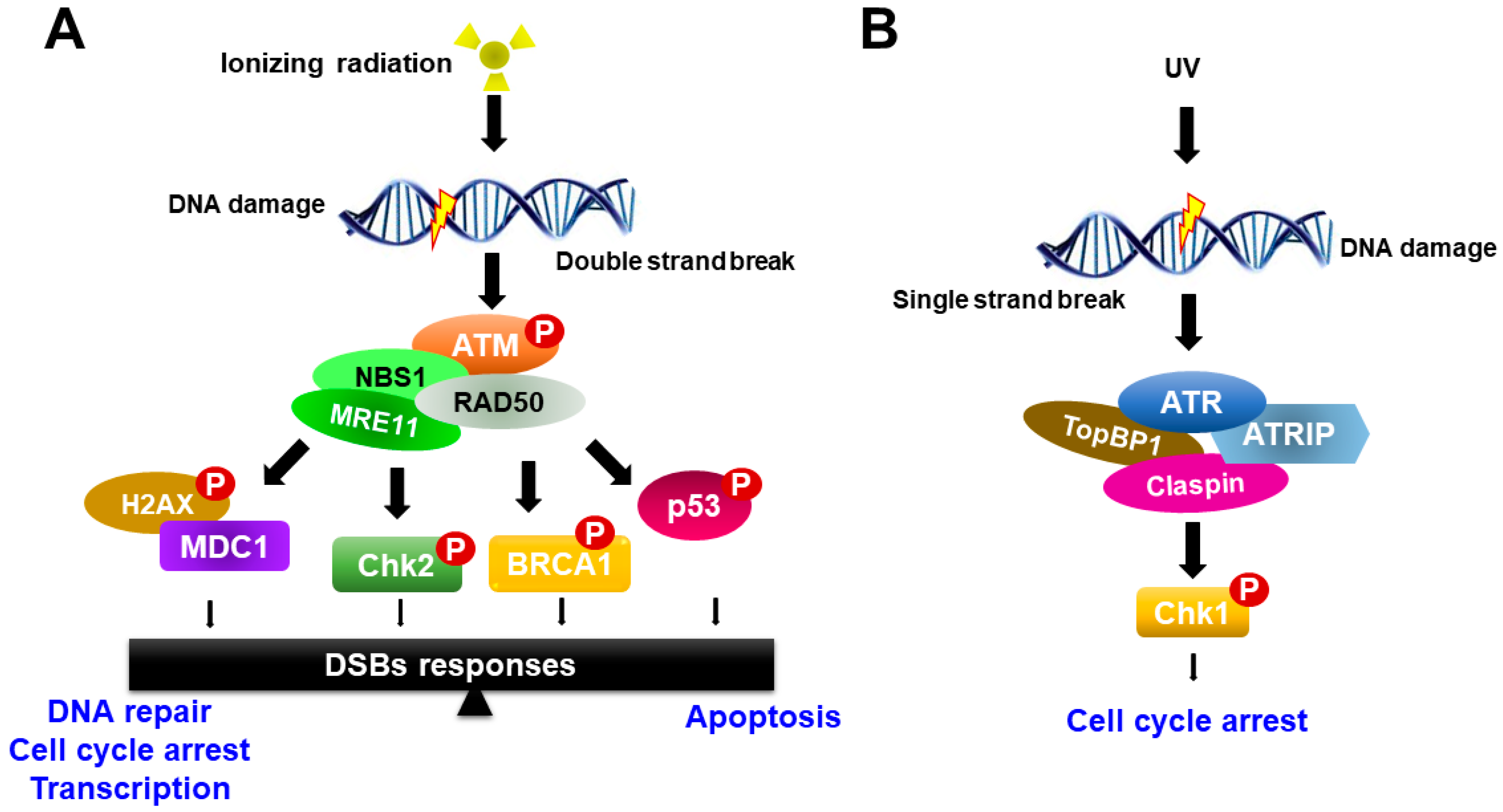
3. Mechanism of Radiation-Induced Cell Toxicity and Radiation Sensitization
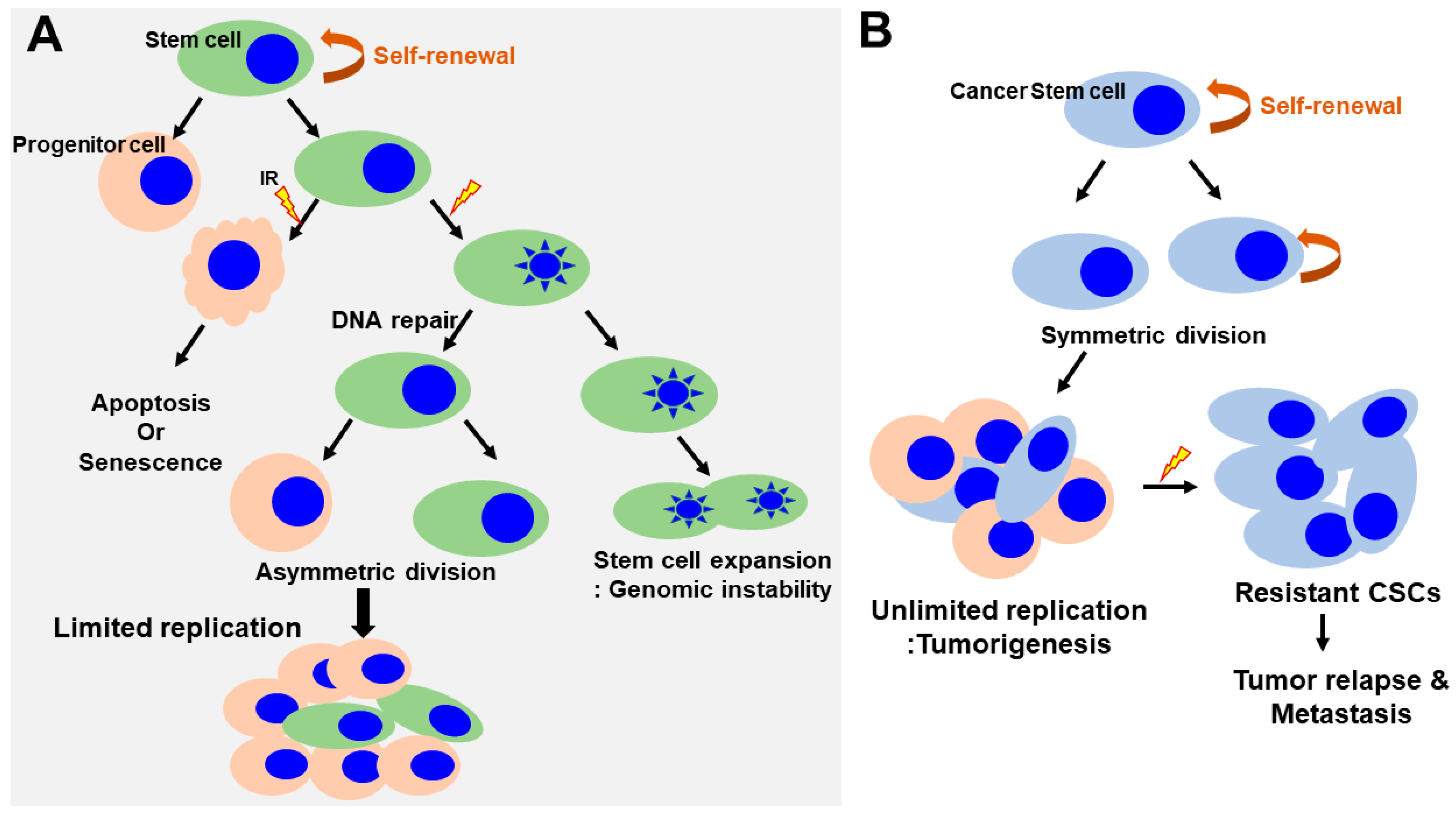
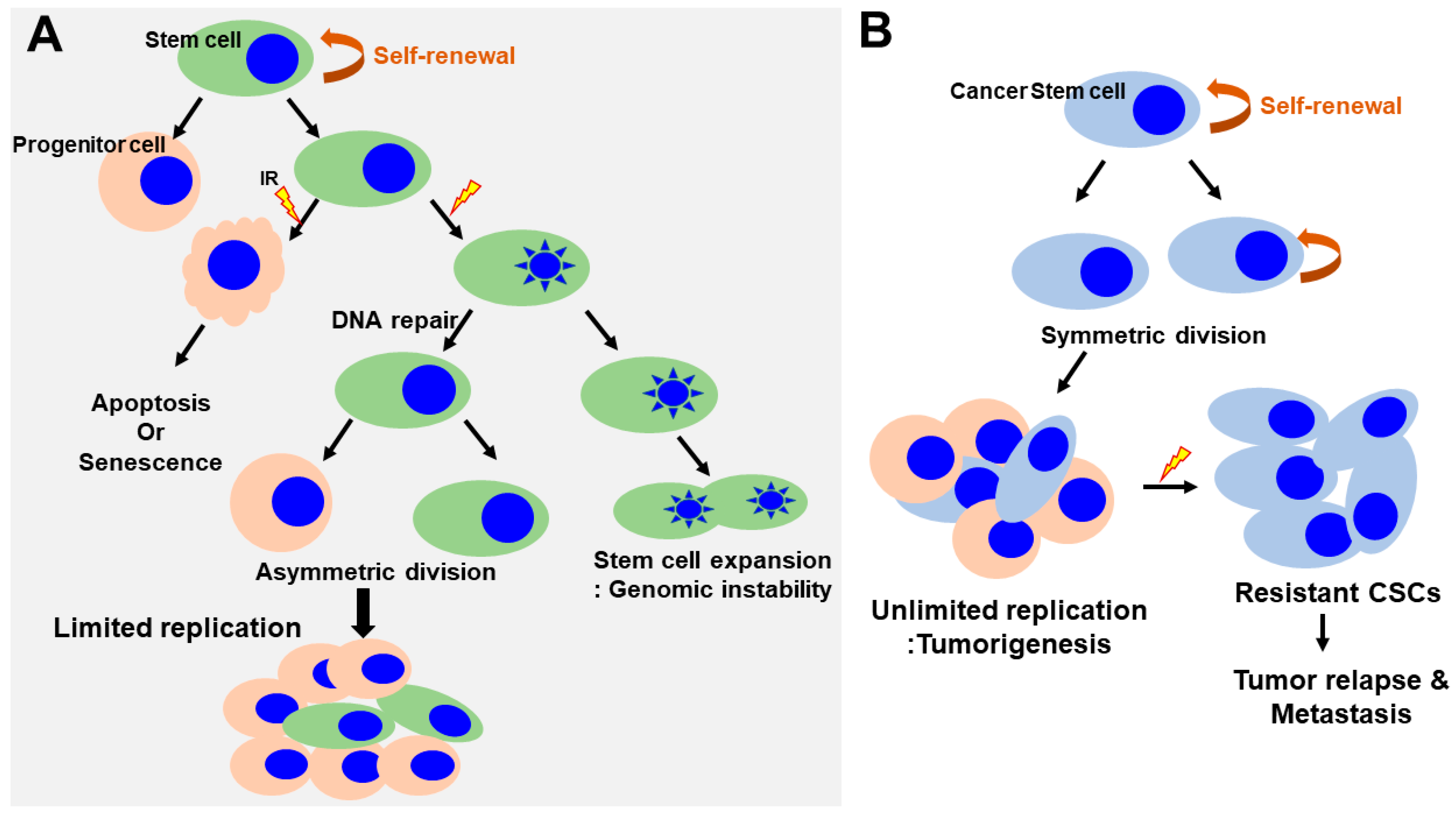
4. Response of Normal Stem Cells to Ionizing Radiation
5. Response of Cancer Stem Cells to Ionizing Radiation
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liauw, S.L.; Connell, P.P.; Weichselbaum, R.R. New paradigms and future challenges in radiation oncology: An update of biological targets and technology. Sci. Transl. Med. 2013, 5, 173sr172. [Google Scholar] [CrossRef] [PubMed]
- Guadagnolo, B.A.; Liao, K.P.; Elting, L.; Giordano, S.; Buchholz, T.A.; Shih, Y.C. Use of radiation therapy in the last 30 days of life among a large population-based cohort of elderly patients in the United States. J. Clin. Oncol. 2013, 31, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.A.; Mutic, S. Advances and future of Radiation Oncology. Rep. Pract. Oncol. Radiother. 2013, 18, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Posner, E. Reception of Rontgen’s discovery in Britain and U.S.A. Br. Med. J. 1970, 4, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Thariat, J.; Hannoun-Levi, J.M.; Sun Myint, A.; Vuong, T.; Gerard, J.P. Past, present, and future of radiotherapy for the benefit of patients. Nat. Rev. Clin. Oncol. 2013, 10, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, S.M. Preventing or reducing late side effects of radiation therapy: Radiobiology meets molecular pathology. Nat. Rev. Cancer 2006, 6, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Pellicioli, A.; Lee, S.E.; Lucca, C.; Foiani, M.; Haber, J.E. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol. Cell 2001, 7, 293–300. [Google Scholar] [CrossRef]
- Hall, E.J. Cancer caused by X-rays—A random event? Lancet Oncol. 2007, 8, 369–370. [Google Scholar] [CrossRef]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Spitz, D.R.; Azzam, E.I.; Li, J.J.; Gius, D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: A unifying concept in stress response biology. Cancer Metastasis Rev. 2004, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, H.; Little, J.B. Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Cancer Res. 1992, 52, 6394–6396. [Google Scholar] [PubMed]
- Peng, Y.; Zhang, M.; Zheng, L.; Liang, Q.; Li, H.; Chen, J.T.; Guo, H.; Yoshina, S.; Chen, Y.Z.; Zhao, X.; et al. Cysteine protease cathepsin B mediates radiation-induced bystander effects. Nature 2017, 547, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; de Toledo, S.M.; Little, J.B. Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from alpha -particle irradiated to nonirradiated cells. Proc. Natl. Acad. Sci. USA 2001, 98, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Diegeler, S.; Hellweg, C.E. Intercellular Communication of Tumor Cells and Immune Cells after Exposure to Different Ionizing Radiation Qualities. Front. Immunol. 2017, 8, 664. [Google Scholar] [CrossRef] [PubMed]
- De Toledo, S.M.; Buonanno, M.; Harris, A.L.; Azzam, E.I. Genomic instability induced in distant progeny of bystander cells depends on the connexins expressed in the irradiated cells. Int. J. Radiat. Biol. 2017, 93, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- Le, M.; Fernandez-Palomo, C.; McNeill, F.E.; Seymour, C.B.; Rainbow, A.J.; Mothersill, C.E. Exosomes are released by bystander cells exposed to radiation-induced biophoton signals: Reconciling the mechanisms mediating the bystander effect. PLoS ONE 2017, 12, e0173685. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Tian, W.; Wang, L.; Wang, J.; Zhang, S.; Cao, J.; Yang, H. Radiation quality-dependence of bystander effect in unirradiated fibroblasts is associated with TGF-beta1-Smad2 pathway and miR-21 in irradiated keratinocytes. Sci. Rep. 2015, 5, 11373. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A. DNA damage in cancer therapeutics: A boon or a curse? Cancer Res. 2015, 75, 2133–2138. [Google Scholar] [CrossRef] [PubMed]
- Waterworth, W.M.; Footitt, S.; Bray, C.M.; Finch-Savage, W.E.; West, C.E. DNA damage checkpoint kinase ATM regulates germination and maintains genome stability in seeds. Proc. Natl. Acad. Sci. USA 2016, 113, 9647–9652. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Burrell, K.; Buczkowicz, P.; Remke, M.; Golbourn, B.; Chornenkyy, Y.; Gajadhar, A.; Fernandez, N.A.; Clarke, I.D.; Barszczyk, M.S.; et al. ATM regulates 3-methylpurine-DNA glycosylase and promotes therapeutic resistance to alkylating agents. Cancer Discov. 2014, 4, 1198–1213. [Google Scholar] [CrossRef] [PubMed]
- Stagni, V.; Santini, S.; Barila, D. ITCH E3 ligase in ATM network. Oncoscience 2014, 1, 394–395. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Shah, N.M.; Anderson, D.J. Regulatory mechanisms in stem cell biology. Cell 1997, 88, 287–298. [Google Scholar] [CrossRef]
- Ito, K.; Ito, K. Metabolism and the Control of Cell Fate Decisions and Stem Cell Renewal. Annu. Rev. Cell Dev. Biol. 2016, 32, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer 2003, 3, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegue, E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I. Accumulation of DNA damage in the aged hematopoietic stem cell compartment. Semin. Hematol. 2017, 54, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, J.S.; Epperly, M. Bone marrow-derived stem cells and radiation response. Semin. Radiat. Oncol. 2009, 19, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Misri, S.; Meyer, B.; Raj, S.; Zobel, C.L.; Sleckman, B.P.; Hallahan, D.E.; Sharma, G.G. Unique epigenetic influence of H2AX phosphorylation and H3K56 acetylation on normal stem cell radioresponses. Mol. Biol. Cell 2016, 27, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Sugrue, T.; Brown, J.A.; Lowndes, N.F.; Ceredig, R. Multiple facets of the DNA damage response contribute to the radioresistance of mouse mesenchymal stromal cell lines. Stem Cells 2013, 31, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Pommier, Y. MDC1 cleavage by caspase-3: A novel mechanism for inactivating the DNA damage response during apoptosis. Cancer Res. 2011, 71, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Deng, J.; Bucci, J.; Malouf, D.; Gillatt, D.; Li, Y. Cancer stem cells and signaling pathways in radioresistance. Oncotarget 2016, 7, 11002–11017. [Google Scholar] [CrossRef] [PubMed]
- Rycaj, K.; Tang, D.G. Cancer stem cells and radioresistance. Int. J. Radiat. Biol. 2014, 90, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Kang, S.K. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. 2007, 16, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Giambra, V.; Jenkins, C.E.; Lam, S.H.; Hoofd, C.; Belmonte, M.; Wang, X.; Gusscott, S.; Gracias, D.; Weng, A.P. Leukemia stem cells in T-ALL require active Hif1alpha and Wnt signaling. Blood 2015, 125, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.; Lam, C.T.; Poon, R.T.; Fan, S.T. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friel, A.M.; Roberts, D.J.; Seiden, M.V.; Scadden, D.T.; Rueda, B.R.; et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, K.; Yang, Z.; Han, N.; Quan, X.; Guo, X.; Li, C. Bladder cancer stem cells: Clonal origin and therapeutic perspectives. Oncotarget 2017, 8, 66668–66679. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Ohkuma, M.; Kim, H.M.; Akita, H.; Takiuchi, D.; Hatano, H.; Nagano, H.; et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Investig. 2010, 120, 3326–3339. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Manyam, G.C.; Mmeje, C.O.; Baggerly, K.A.; Kamat, A.M. Surface PD-L1, E-cadherin, CD24, and VEGFR2 as markers of epithelial cancer stem cells associated with rapid tumorigenesis. Sci. Rep. 2017, 7, 9602. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Grogan, T.R.; Hieronymus, H.; Hashimoto, T.; Mottahedeh, J.; Cheng, D.; Zhang, L.; Huang, K.; Stoyanova, T.; Park, J.W.; et al. Low CD38 Identifies Progenitor-like Inflammation-Associated Luminal Cells that Can Initiate Human Prostate Cancer and Predict Poor Outcome. Cell Rep. 2016, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Hao, X.; Yan, M.; Yao, M.; Ge, C.; Gu, J.; Li, J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int. J. Cancer 2010, 126, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Hosen, N.; Park, C.Y.; Tatsumi, N.; Oji, Y.; Sugiyama, H.; Gramatzki, M.; Krensky, A.M.; Weissman, I.L. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 11008–11013. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Rijavec, M.; Kern, I.; Sodja, E.; Korosec, P.; Cufer, T. BMI1, ALDH1A1, and CD133 Transcripts Connect Epithelial-Mesenchymal Transition to Cancer Stem Cells in Lung Carcinoma. Stem Cells Int. 2016, 2016, 9714315. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.S.; Hur, W.; Kim, T.K.; Hong, S.W.; Kim, S.W.; Choi, J.E.; Sung, P.S.; Song, M.J.; Lee, B.C.; Hwang, D.; et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. 2012, 315, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.P.; Bristow, R.G.; Kapoor, A. Prostate cancer stem cells: Deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget 2015, 6, 1900–1919. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Perrin, R.; Hill, R.P.; Dubrovska, A.; Kurth, I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int. J. Radiat. Biol. 2014, 90, 636–652. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- White, P.T.; Subramanian, C.; Zhu, Q.; Zhang, H.; Zhao, H.; Gallagher, R.; Timmermann, B.N.; Blagg, B.S.; Cohen, M.S. Novel HSP90 inhibitors effectively target functions of thyroid cancer stem cell preventing migration and invasion. Surgery 2016, 159, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, H.Y.; Park, K.K.; Choi, Y.K.; Nam, J.S.; Hong, I.S. CWP232228 targets liver cancer stem cells through Wnt/beta-catenin signaling: A novel therapeutic approach for liver cancer treatment. Oncotarget 2016, 7, 20395–20409. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.B.; Hong, I.S.; Kim, R.J.; Lee, S.Y.; Park, S.J.; Lee, E.S.; Park, J.H.; Yun, C.H.; Chung, J.U.; Lee, K.J.; et al. Wnt/beta-Catenin Small-Molecule Inhibitor CWP232228 Preferentially Inhibits the Growth of Breast Cancer Stem-like Cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Hur, W.; Choi, J.E.; Kim, J.H.; Hwang, D.; Yoon, S.K. Role of ADAM17 in invasion and migration of CD133-expressing liver cancer stem cells after irradiation. Oncotarget 2016, 7, 23482–23497. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.A.; Caudell, J.J.; El-Haddad, G.; Berglund, A.E.; Welsh, E.A.; Yue, B.; Hoffe, S.E.; Naghavi, A.O.; Abuodeh, Y.A.; Frakes, J.M.; et al. Radiosensitivity Differences Between Liver Metastases Based on Primary Histology Suggest Implications for Clinical Outcomes After Stereotactic Body Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, A.J.; Jones, I.M. Second cancers after radiotherapy: Any evidence for radiation-induced genomic instability? Oncogene 2003, 22, 7018–7027. [Google Scholar] [CrossRef] [PubMed]
- Hegemann, N.S.; Schlesinger-Raab, A.; Ganswindt, U.; Horl, C.; Combs, S.E.; Holzel, D.; Gschwend, J.E.; Stief, C.; Belka, C.; Engel, J. Risk of second cancer following radiotherapy for prostate cancer: A population-based analysis. Radiat. Oncol. 2017, 12, 2. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hur, W.; Yoon, S.K. Molecular Pathogenesis of Radiation-Induced Cell Toxicity in Stem Cells. Int. J. Mol. Sci. 2017, 18, 2749. https://doi.org/10.3390/ijms18122749
Hur W, Yoon SK. Molecular Pathogenesis of Radiation-Induced Cell Toxicity in Stem Cells. International Journal of Molecular Sciences. 2017; 18(12):2749. https://doi.org/10.3390/ijms18122749
Chicago/Turabian StyleHur, Wonhee, and Seung Kew Yoon. 2017. "Molecular Pathogenesis of Radiation-Induced Cell Toxicity in Stem Cells" International Journal of Molecular Sciences 18, no. 12: 2749. https://doi.org/10.3390/ijms18122749
APA StyleHur, W., & Yoon, S. K. (2017). Molecular Pathogenesis of Radiation-Induced Cell Toxicity in Stem Cells. International Journal of Molecular Sciences, 18(12), 2749. https://doi.org/10.3390/ijms18122749