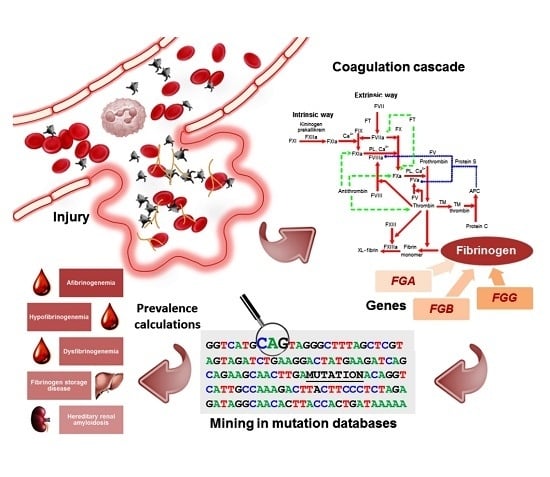
1. Introduction
Fibrinogen is a 340-kDa glycoprotein that plays a crucial role in the hemostatic cascade, being the substrate for fibrin clot formation and the support for platelet aggregation [
1,
2,
3]. It is also essential for several other biological functions, such as wound healing, inflammation, and angiogenesis [
4,
5,
6].
Into the circulation, thrombin cleaves fibrinopeptides A and B to convert fibrinogen into fibrin, which spontaneously polymerizes, and thus forms double-stranded protofibrils, which, in turn, assemble into fibers, ultimately leading to the fibrin clot [
7]. In hepatocytes—the primary site of fibrinogen biosynthesis—the molecule is rapidly assembled in the endoplasmic reticulum (ER), and secreted as a hexamer composed of two sets of three homologous polypeptide chains (called Aα, Bβ, and γ) [
8,
9,
10,
11]. These are encoded by paralogous genes (
FGA,
FGB, and
FGG), which are clustered in a 50-kb region on chromosome 4 (4q31.3–q32.1) [
12].
Monoallelic and biallelic mutations in
FGA,
FGB, and
FGG genes are associated with different inherited conditions, reflecting the pleiotropic function of the fibrinogen protein [
13]. Congenital fibrinogen defects are conventionally classified on the basis of plasma concentration as quantitative (type I) and qualitative (type II) deficiencies [
14,
15,
16]. Quantitative deficiencies include afibrinogenemia/severe hypofibrinogenemia (Online Mendelian Inheritance in Man (OMIM) #202400; [
17]) and hypofibrinogenemia (OMIM +134820, *134830, *134850), which are characterized by the lack/extremely low or by reduced amounts of immunoreactive fibrinogen (<150–160 mg/dL), leading to hemorrhagic manifestations, which can vary from very mild to life threatening. Qualitative deficiencies comprise dysfibrinogenemia (OMIM +134820, *134830, *134850) and hypo-dysfibrinogenemia, and are characterized by a discrepancy between antigen levels and functional (abnormally low) activity. Patients that are diagnosed with these conditions are either asymptomatic, or can suffer from bleeding symptoms, thrombophilia, or even both [
15].
Hypofibrinogenemia and afibrinogenemia have long been considered as different clinical entities. Indeed, they represent the phenotypic expression of the same quantitative trait (i.e., diminished plasma fibrinogen level), which is determined, respectively, by heterozygous or homozygous/combined heterozygous mutations that are affecting one of the fibrinogen genes. As for dysfibrinogenemias and hypodysfibrinogenemias, these disorders are usually inherited as an autosomal dominant trait: they are caused by a single genetic defect ultimately affecting a functional property of the fibrinogen protein, such as the release (impaired or delayed) of fibrinopeptides A and B, defective polymerization, crosslinking, or thrombin binding, as well as delayed plasmin digestion [
16].
Rare hypofibrinogenemic patients can present with liver disease due to the accumulation of mutant fibrinogens within hepatocytes. This condition is called fibrinogen storage disease (FSD), and is generally caused by heterozygous mutations, leading to an impaired secretion of the abnormal fibrinogen, which however maintains its capacity for polymerization and spontaneously aggregates in hepatocellular ER. In the vast majority of cases, mutations leading to FSD are missense variants that are located in a defined region of the C-terminal γ chain (residues 284–375) [
18,
19]. FSD-causing mutation carriers show a great variability in the severity of liver injury, going from the lack of symptoms to severe liver fibrosis/cirrhosis; more severe manifestations can be secondary to xenobiotic intake (e.g., estrogen therapy, alcohol abuse), to viral infections, or even to cancer [
18,
20].
Heterozygous mutations, which are affecting a small region of the C-terminal portion of the Aα chain, have been described in patients with hereditary renal amyloidosis (HRA) (OMIM +134820). These genetic defects are associated with a mild decrease in fibrinogen levels and are supposed to destabilize the native fold of circulating Aα chain degradation peptides, so that they spontaneously aggregate into amyloid fibrils, prevalently in the glomeruli of the kidneys, and, to a lesser extent, in heart muscle, spleen, and liver. Fibrinogen amyloidosis is the most common form of HRA, and clinical symptoms include hypertension, proteinuria, and azotemia [
21].
The frequency of congenital fibrinogen disorders in the general population is very low. International registries, such as those from the United States, Italy, Iran, and the United Kingdom, suggest that afibrinogenemia is one of the rarest among rare bleeding disorders, with only 1–2 cases per million people [
22]; however, these registries lack prospective and systematic evaluations that could lead to incidence/prevalence determination. In addition, “true” incidence/prevalence estimates for a-, hypo-, and dysfibrinogenemia are made difficult, because many patients are asymptomatic [
15,
22].
With these premises, we here defined the global mutational landscape of
FGA,
FGB, and
FGG, and tried to determine ethnic-specific prevalence of inherited fibrinogen disorders, by analyzing exome and genome data from almost 140,000 individuals available through the publicly available genome Aggregation Database (gnomAD) resource [
23,
24].
3. Discussion
With the exception of the involvement of some peculiar molecular mechanism, such as uniparental isodisomy of the entire chromosome 4 [
30], the genetic bases of fibrinogen disorders are invariably constituted by homozygous/heterozygous mutations within the fibrinogen gene cluster. Here, we took advantage of exome and genome data of ~140,000 individuals to estimate the prevalence of recessively-inherited fibrinogen deficiency, as well as the collective prevalence of all the dominantly-inherited fibrinogen disorders. Our estimates indicated that: (i) the world-wide prevalence for recessively-inherited fibrinogen deficiencies could be 10-fold higher than that reported so far; (ii) prevalence among different populations seems to be extremely different (ranging from 1 in 10
6 in East Asians up to 24.5 in 10
6 in non-Finnish Europeans); and, (iii) heterozygous carriers of mutations in the fibrinogen cluster (i.e., individuals possibly at risk to develop a form of fibrinogen disorder, as well as asymptomatic/undiagnosed subjects) should be present in the general world-wide population at a frequency of ~1 every 100 individuals.
Notably, we have to acknowledge that our estimates suffer from some important limitations. First, our prevalence calculations relied on the use of prediction programs aimed at evaluating the deleteriousness of missense/splicing mutations with unknown biological significance. In this respect, we have to notice that, although these algorithms can present limitations [
31,
32], their use currently represents a standard approach, especially if researchers have to deal with data from large-scale sequencing projects. Indeed, it has been demonstrated that different methods can individually show a limited overall predictive value, which, however, increases significantly when considering only concordant outputs from different software (e.g., it has been calculated an encouraging predictive value of ~90% when taking into account concordant results from four different prediction methods) [
31]. We hence based our prevalence calculations on the use of seven different prediction software for missense variants, and of three programs for splicing variants. This choice proved to be quite “conservative”: for instance, excluding from prevalence calculations all of the variants that were never reported in fibrinogen-related databases, we observed a global prevalence rates for recessively-inherited fibrinogen deficiencies of 6.8 in 10
6 individuals (0.033 in 10
6 individuals for the
FGA gene, 0.16 in 10
6 for
FGB, 6.68 in 10
6 for
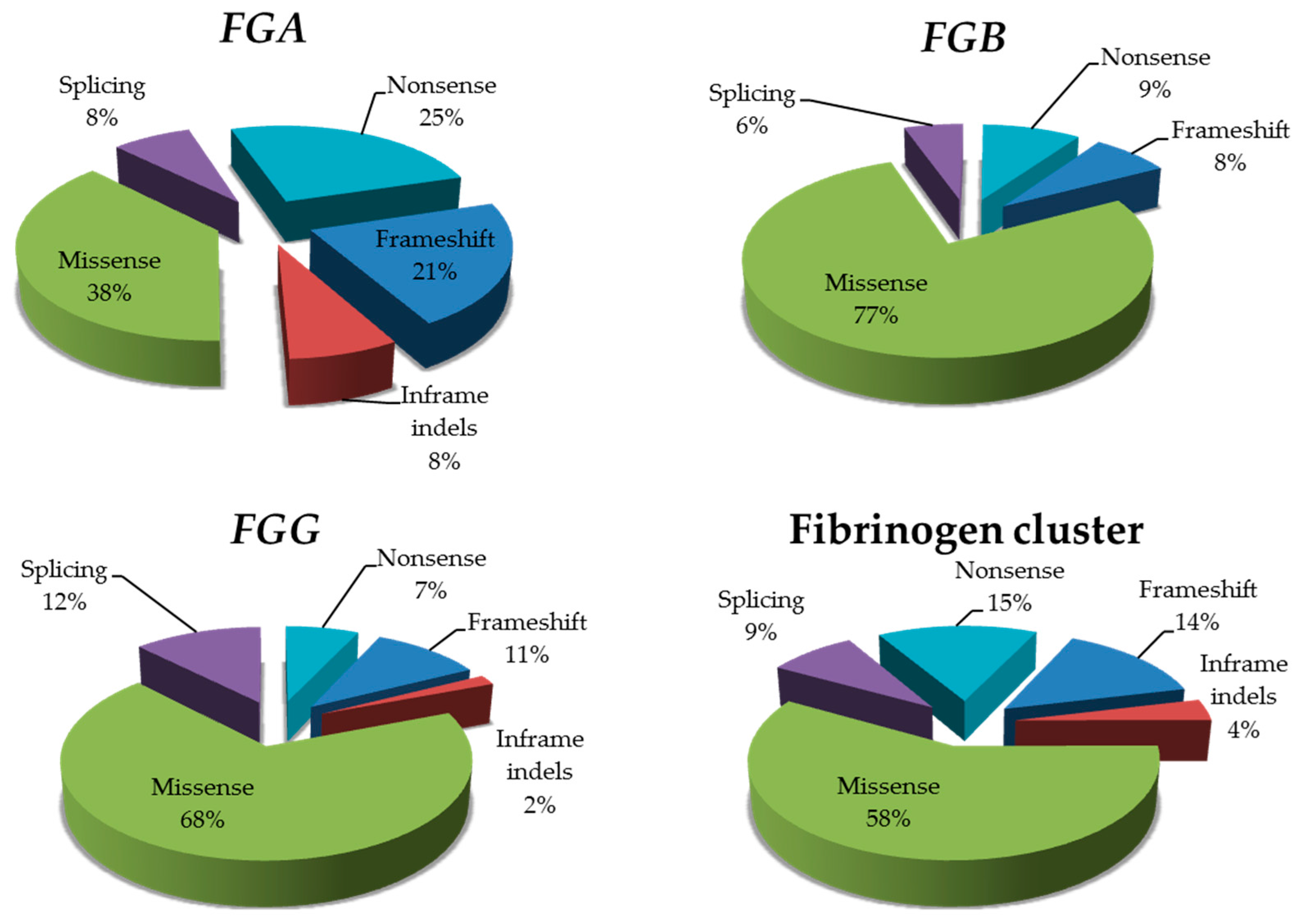
FGG). Importantly, the good performance of our in-silico approach is also testified by predictions performed on fibrinogen missense variants reported as associated with fibrinogen disorders in the databases: we observed an overall prediction rate for the fibrinogen cluster of 78% (if considering all together concordant predictions from seven of seven and six of seven algorithms). The only exception pertained the nine
FGA mutations that were described as cause of amyloidosis: none of them was predicted as damaging neither by 7 of 7 nor by 6 of 7 algorithms. This was probably because all these variants involve a peculiar trait of the Aα chain, i.e., the C-terminal unstructured tail of the protein.
Second, we could in theory have underestimated the total number of causative mutations for systematic bias still characterizing exome-sequencing data. For instance, promoters and intronic regions are not included by design in exome sequencing, so that variants located in such regions can easily go undetected. Insertions and deletions are not always correctly recognized by variant-calling programs, so they can go unnoticed as well. More importantly, gross deletions, and rearrangements may be not detected at all. This represents a substantial problem in the calculation of prevalence for fibrinogen deficiencies, since one of the most recurrent mutation for these disorders is the well-known 11-kb deletion, eliminating the majority of the
FGA gene [
16,
33]. This deletion has been reported in at least eight afibrinogenemic patients [
34], and, together with other two gross deletions that are described in the
FGA gene (i.e., a 15-kb and a 4.1-kb deletion) [
30,
35], account for ~9% of all cases of afibrinogenemia characterized by mutations in
FGA [
34].
The high prevalence rates that we estimated from the gnomAD repository are indeed not completely unexpected. The most recent World Federation of Hemophilia (WFH) annual global survey, which was conducted in 2015, found that inherited fibrinogen deficiencies on a global scale account for 1777 of 304,362 inherited bleeding disorders (0.6%) [
36], confirming fibrinogen deficiencies as quite rare disorders, and approximately 85 times less common than hemophilia A (these data are based on questionnaires sent to national hemophilia associations linked with the Federation). However, digging deeper in the WFH data, it becomes clear that prevalence data on fibrinogen deficiencies are somehow underestimated, at least in some populations. For instance, though it is not clear if data reported concern autosomal-recessive, autosomal-dominant, or both forms of the fibrinogen deficiency, it is possible to calculate exceptional prevalence rates of 8.7 and 14.3 per million people for the United Kingdom and the Slovak Republic, respectively. Concerning Italy, a country with a population that is comparable to that of the United Kingdom (~60 million people), no data are reported on fibrinogen deficiencies in the WFH 2015 survey. However, in the “Human fibrinogen database”, a total of 104 fibrinogen-deficient Italian cases are described (54 coming from our center). This figure alone is sufficient to suggest higher prevalence rates for fibrinogen-related disorders than those reported so far, especially if considering that the database has a clear bias towards published data. To have a better idea of the Italian situation, we took advantage of our whole-exome dataset [
37], which was composed of exomes of 1750 healthy controls (80% males, age < 45 years, no history of thromboembolic disease). Sequence coverage of fibrinogen cluster was optimal in all individuals, and allowed us to retrieve a total of 24 variants, corresponding to eight different mutations (one frameshift mutation and seven missense damaging variants; distribution: one variant in
FGA, two in
FGB, 5 in
FGG). Once again, the burden of mutations affecting the
FGG gene appears to be the highest, with just 1 mutation (p.Ala108Gly) considerably driving the prevalence rates for recessively-inherited fibrinogen deficiencies (0.08 per 10
6 individuals for the
FGA gene, 0.33 per 10
6 individuals for
FGB, and an exceptional 36 per 10
6 individuals for the
FGG gene) (
Supplementary Table S3). Interestingly, the “driving effect” that was exerted by the p.Ala108Gly mutation could also be at the basis of the marked differences in prevalence rates that were observed in different populations (from 1 in 10
6 in East Asians to 24.5 in 10
6 in non-Finnish Europeans). In particular, Ivaskevicius and colleagues [
38] reported that the p.Ala108Gly mutation is associated with a specific haplotype, hence denoting a single, ancestral event (founder effect). Given the observed frequencies of the mutation in the different ethnic groups (
Table 4), one could speculate that the ancestral mutation event could have originated in Africans (before the major divergence of non-African populations), and that, subsequently, the mutation could have spread towards Europe and Asia following human migrations [
39]. However, with the last divergence between South and East Asians, it is conceivable that the p.Ala108Gly mutation could have not reached the Far East.
The issue related to the fundamental contribution of the p.Ala108Gly mutation to prevalence estimates should be carefully kept in mind. In fact, if we do not consider this variant at all, the prevalence rates for recessively-inherited fibrinogen disorders would dramatically drop (on a global scale prevalence would be 3.2 per million people), with highest values that would be registered in Africans/Africans Americans (i.e., 4 per million people) and the lowest in Ashkenazi Jews (for this population, recessively-inherited fibrinogen deficiencies would be virtually absent). The same dramatic drop would be registered for autosomal-dominant fibrinogen disorders (
Supplementary Table S4). The p.Ala108Gly mutation (legacy name γAla82Gly) was repeatedly reported as being associated with moderately-decreased fibrinogen levels and with mild bleeding tendency [
25,
26,
34]. In addition, in a recent meta-analysis aimed at identifying loci for fibrinogen concentration, the p.Ala108Gly allele clearly emerged among the strongest predictors of decreased fibrinogen levels (β = −0.2179;
p = 4.0 × 10
−82) [
40]. Importantly, Ivaskevicius and colleagues [
38], by screening 616 blood donors, already observed the p.Ala108Gly as a common
FGG variant in Caucasians, thus calculating an allele frequency of 0.0032 and a frequency for homozygous individuals of 1 in 95,000. These data well reconcile with those calculated using the gnomAD database.
Our data raise the problem of why such remarkably high prevalence can be calculated for recessively-inherited fibrinogen deficiencies. The most likely explanation could rely on the relatively-low fibrinogen levels that are associated with the highly-prevalent p.Ala108Gly mutation, and the consequent mild/absent symptomatology characterizing many carrier/homozygous individuals (which can go unnoticed) [
38]. Alternatively, we can hypothesize two additional explanations.
A first possibility could be that the p.Ala108Gly allele, at the heterozygous state, confers a selective advantage, and thus, has spread throughout the gene pool. This hypothesis strongly emerges among other genetic explanations, such as a possible genetic drift (which can be excluded due to the presence of the p.Ala108Gly allele world-wide), a potential transmission distortion (which can be left out since the Hardy-Weinberg equilibrium is perfectly respected), or a high mutation rate. This last possibility can be discounted on the basis of the above-mentioned observation that the p.Ala108Gly mutation is associated with a founder effect [
38]. It remains to understand why a pro-hemorrhagic allele could represent a selective advantage; in this respect, it is worth noticing that a high fibrinogen concentration has long been recognized as a strong and established predictor of cardiovascular disease outcomes (myocardial infarction, stroke, venous thromboembolism), autoimmune disorders with an inflammatory component, as well as cancer [
5,
41,
42,
43,
44]. However, most of these phenotypes are late onset and therefore are predicted to have a limited effect on natural selection; moreover, the heterozygous advantage hypothesized to explain the frequency of the factor V Leiden mutation points to the opposite direction, being related to moderate hypercoagulability [
45].
Conversely, it could be plausible that we do not detect in the general population the high rate of predicted homozygous/compound heterozygous individuals because of problems that are associated with pregnancy (defects in fetal implantation in afibrinogenemic/hypofibrinogenemic women and/or embryos). It is indeed well recognized that fibrinogen has a critical role in maintaining pregnancy: at six weeks of gestation, maternal endothelial cells are replaced by cytotrophoblasts, starting a remodeling process of vessels that involves an active bleeding near the cytotrophoblastic shell, followed by the formation of the Nitabuch’s layer (a fibrinoid layer). This process is highly compromised in patients with quantitative fibrinogen defects, ultimately leading to spontaneous miscarriages [
46]. This notion is well supported by different studies analyzing series of fibrinogen-deficient women, all reporting miscarriage as a frequent complication [
47,
48,
49,
50]. Further corroborating this observation, a recent study—aimed at elucidating by whole-exome sequencing the genetic etiology of recurrent pregnancy loss—reported the identification of mutations in the
FGA gene as having a potential role in implantation/pregnancy biology [
51]. Hence, when considering the high frequency of heterozygous carriers of fibrinogen defects in the general population, we can hypothesize the fibrinogen cluster as a future “biomarker” to be screened also for recurrent pregnancy loss.
In conclusion, in this work, we have exploited the enormous potential that is provided by public-available repositories to paint a more clear landscape of the genetic burden associated with fibrinogen genes and their related disorders. From our analysis, it clearly emerges that putative disease alleles are much more frequent than expected, a trend already observed for other genes/disorders [
52,
53]. Caution should be placed to interpret these data, since some of the identified variants could be non-pathogenic and some others could be not fully penetrant. Nonetheless, our analysis represents the first attempt to evaluate the prevalence rates of fibrinogen disorders in populations other than those that are coming from North America and Europe, also indicating the mutations/genes to be prioritized for genetic screenings.





{kind=link}
{kind=link}