Heat Shock Proteins in Vascular Diabetic Complications: Review and Future Perspective


 ,
,
Abstract
1. Introduction
2. Diabetes Micro and Macrovascular Complications
3. Heat Shock Proteins
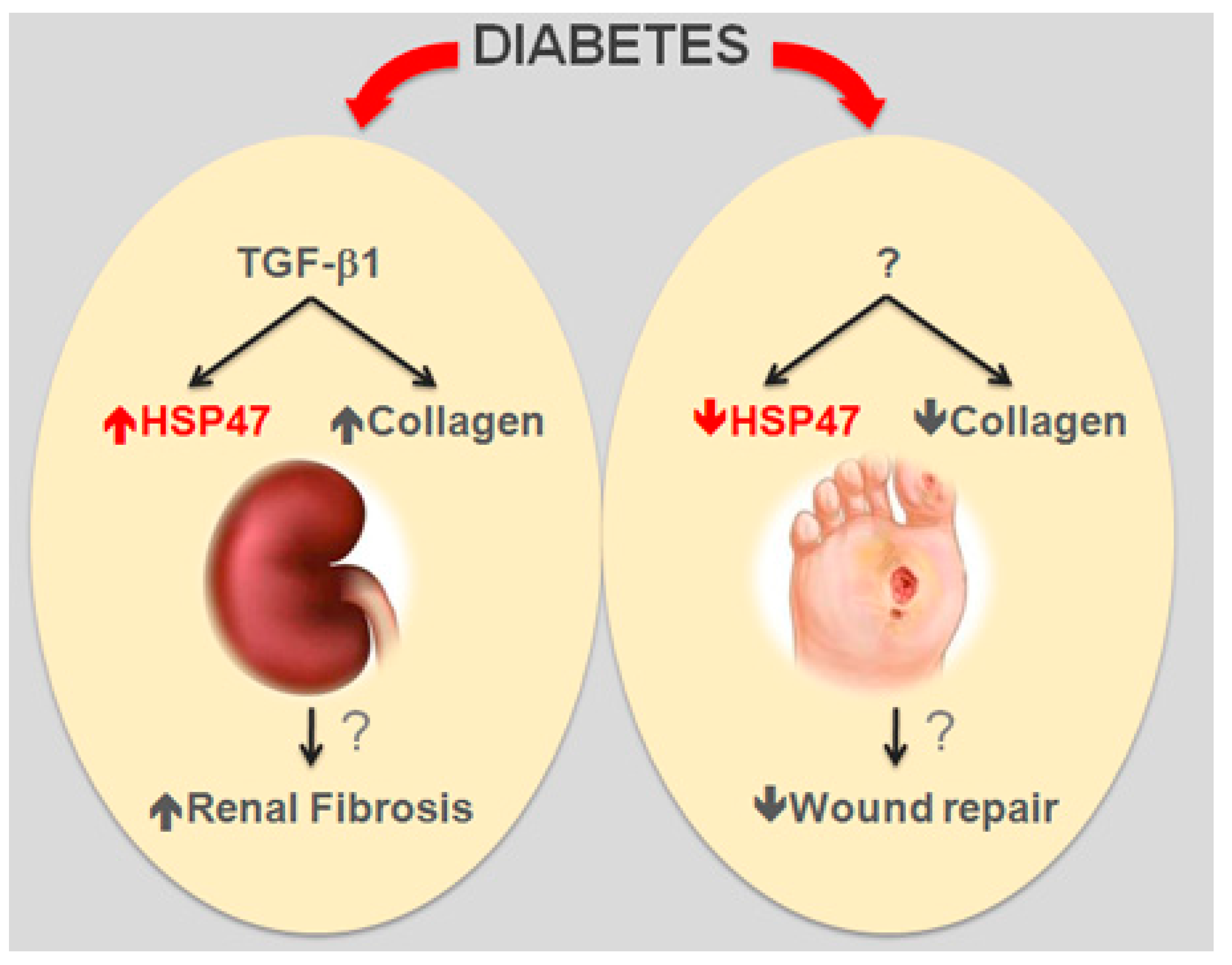
3.1. Heat Shock Protein 47—SerpinH1
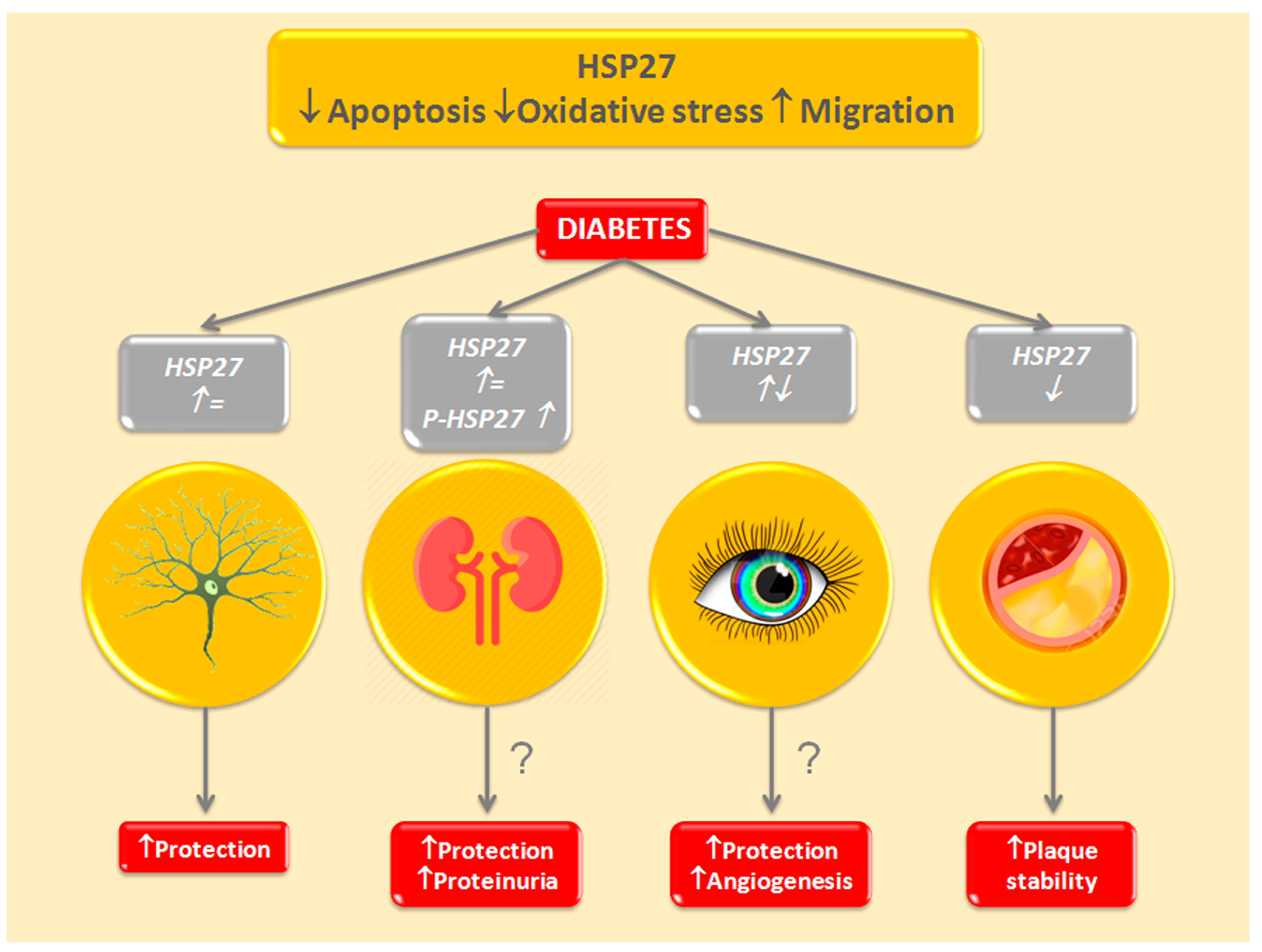
3.2. Heat Shock Protein 27—HSPB1
3.2.1. Diabetic Nephropathy
3.2.2. Diabetic Retinopathy
3.2.3. Diabetic Neuropathy
3.2.4. Diabetic Macrovascular Diseases
3.3. Heat Shock Protein C—HSP90
3.3.1. Diabetic Nephropathy
3.3.2. Diabetic Retinopathy
3.3.3. Diabetic Neuropathy
3.3.4. Diabetes Macrovascular Complications
3.4. Heat Shock Protein A—HSP70
3.4.1. Diabetic Nephropathy
3.4.2. Diabetic Retinopathy
3.4.3. Diabetic Neuropathy
3.4.4. Diabetic Macrovascular Diseases
3.5. Heat Shock Protein D1—HSP60
3.5.1. Microvascular Complications
3.5.2. Macrovascular Complications
4. Summary of Evidence
5. Limits of Current Research and Future Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Low Wang, C.C.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical Update: Cardiovascular Disease in Diabetes Mellitus. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Bakris, G.L.; Bilous, R.W.; Chiang, J.L.; De Boer, I.H.; Goldstein-Fuchs, J.; Hirsch, I.B.; Kalantar-Zadeh, K.; Narva, A.S.; Navaneethan, S.D.; et al. Diabetic Kidney Disease: A Report From an ADA Consensus Conference. Am. J. Kidney Dis. 2014, 64, 510–533. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Singh, R.; Kishore, L.; Kaur, N. Diabetic peripheral neuropathy : Current perspective and future directions. Pharmacol. Res. 2014, 80, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Malone, J.I. Diabetic central neuropathy: CNS damage related to hyperglycemia. Diabetes 2016, 65, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Gong, J.; Murshid, A. Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front. Immunol. 2016, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Nagata, K. Biology of Hsp47 (Serpin H1), a collagen-specific molecular chaperone. Semin. Cell Dev. Biol. 2017, 62, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S.; Kumatori, A.; Harada, T.; Taguchi, T. Coexpression of collagens and collagen-binding heat shock protein 47 in human diabetic nephropathy and IgA nephropathy. Nephron 1998, 80, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Razzaque, M.S.; Cheng, M.; Taguchi, T. The renal expression of heat shock protein 47 and collagens in acute and chronic experimental diabetes in rats. Histochem. J. 2001, 33, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Wolf, G. Epithelial-to-Mesenchymal Transition in Diabetic Nephropathy: Fact or Fiction? Cells 2015, 4, 631–652. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, S.; Abe, H.; Takahashi, T.; Yamamoto, Y.; Takeuchi, M.; Arai, H.; Nagata, K.; Kita, T.; Okamoto, H.; Yamamoto, H.; et al. Advanced Glycation End Products Increase Collagen-specific Chaperone Protein in Mouse Diabetic Nephropathy. J. Biol. Chem. 2004, 279, 19816–19823. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Liu, R.; Ling, G.; Xiao, L.; Xia, Y.; Liu, F.; Li, J.; Liu, Y.; Chen, Q.; Lv, J.; et al. HSP47 regulates ECM accumulation in renal proximal tubular cells induced by TGF-β1 through ERK1/2 and JNK MAPK pathways. Am. J. Physiol. Ren. Physiol. 2012, 303, F757–F765. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Z.; Wang, Y.; Li, L.; Wang, D.; Zhang, W.; Liu, L.; Jiang, H.; Yang, J.; Cheng, J. Overexpression of miR-29b reduces collagen biosynthesis by inhibiting heat shock protein 47 during skin wound healing. Transl. Res. 2016, 178, 38–53.e6. [Google Scholar] [CrossRef] [PubMed]
- Bellaye, P.-S.; Burgy, O.; Causse, S.; Garrido, C.; Bonniaud, P. Heat shock proteins in fibrosis and wound healing: Good or evil? Pharmacol. Ther. 2014, 143, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S.; Taguchi, T. Collagen-binding heat shock protein (HSP) 47 expression in anti-thymocyte serum (ATS)-induced glomerulonephritis. J. Pathol. 1997, 183, 24–29. [Google Scholar] [CrossRef]
- Sunamoto, M.; Kuze, K.; Tsuji, H.; Ohishi, N.; Yagi, K.; Nagata, K.; Kita, T.; Doi, T. Antisense oligonucleotides against collagen-binding stress protein HSP47 suppress collagen accumulation in experimental glomerulonephritis. Lab. Investig. 1998, 78, 967–972. [Google Scholar] [PubMed]
- Xia, Z.; Abe, K.; Furusu, A.; Miyazaki, M.; Obata, Y.; Tabata, Y.; Koji, T.; Kohno, S. Suppression of renal tubulointerstitial fibrosis by small interfering RNA targeting heat shock protein 47. Am. J. Nephrol. 2008, 28, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Ushioda, R.; Ito, S.; Ikeda, K.; Masago, Y.; Nagata, K. Deletion of the collagen-specific molecular chaperone Hsp47 causes endoplasmic reticulum stress-mediated apoptosis of hepatic stellate cells. J. Biol. Chem. 2015, 290, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Ogawa, K.; Takeuchi, K.; Takagi, M.; Yoshida, M.; Hirokawa, T.; Hirayama, S.; Shin-Ya, K.; Shimada, I.; Doi, T.; et al. A small-molecule compound inhibits a collagen-specific molecular chaperone and could represent a potential remedy for fibrosis. J. Biol. Chem. 2017, 292, 20076–20085. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, L. The plasmid encoding HSP47 enhances collagen expression and promotes skin wound healing in an alloxan-induced diabetic model. Cell Biol. Int. 2009, 33, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Agrawal, N.K.; Gupta, S.K.; Mohan, G.; Chaturvedi, S.; Singh, K. Decreased expression of heat shock proteins may lead to compromised wound healing in type 2 diabetes mellitus patients. J. Diabetes Complicat. 2015, 29, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.-P. Mammalian HspB1 (Hsp27) is a molecular sensor linked to the physiology and environment of the cell. Cell Stress Chaperones 2017, 22, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Batulan, Z.; Pulakazhi Venu, V.K.; Li, Y.; Koumbadinga, G.; Alvarez-Olmedo, D.G.; Shi, C.; O’Brien, E.R. Extracellular Release and Signaling by Heat Shock Protein 27: Role in Modifying Vascular Inflammation. Front. Immunol. 2016, 7, 285. [Google Scholar] [CrossRef] [PubMed]
- Barutta, F.; Pinach, S.; Giunti, S.; Vittone, F.; Forbes, J.M.; Chiarle, R.; Arnstein, M.; Perin, P.C.; Camussi, G.; Cooper, M.E.; et al. Heat shock protein expression in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2008, 295, F1817–F1824. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Sanz, A.B.; Sanchez-Lopez, E.; Ruiz-Ortega, M.; Benito-Martin, A.; Saleem, M.A.; Mathieson, P.W.; Mezzano, S.; Egido, J.; Ortiz, A. HSP27/HSPB1 as an adaptive podocyte antiapoptotic protein activated by high glucose and angiotensin II. Lab. Investig. 2012, 92, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-K.; Ronkina, N.; Höft, A.; Prohl, C.; Menne, J.; Gaestel, M.; Haller, H.; Meier, M. Deletion of MK2 signalling in vivo inhibits small Hsp phosphorylation but not diabetic nephropathy. Nephrol. Dial. Transpl. 2008, 23, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Tikoo, K.; Meena, R.L.; Kabra, D.G.; Gaikwad, A.B. Change in post-translational modifications of histone H3, heat-shock protein-27 and MAP kinase p38 expression by curcumin in streptozotocin-induced type I diabetic nephropathy. Br. J. Pharmacol. 2008, 153, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, M.E.; Muggli, E.E. Small heat shock protein alteration provides a mechanism to reduce mesangial cell contractility in diabetes and oxidative stress. Kidney Int. 2000, 57, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Smoyer, W.E.; Gupta, A.; Mundel, P.; Ballew, J.D.; Welsh, M.J. Altered expression of glomerular heat shock protein 27 in experimental nephrotic syndrome. J. Clin. Investig. 1996, 97, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Jakhotia, S.; Sivaprasad, M.; Shalini, T.; Reddy, P.Y.; Viswanath, K.; Jakhotia, K.; Sahay, R.; Sahay, M.; Reddy, G.B. Circulating Levels of Hsp27 in Microvascular Complications of Diabetes: Prospects as a Biomarker of Diabetic Nephropathy. Available online: http://www.webcitation.org/6uqzjNQLb (accessed on 9 November 2017).
- Joussen, A.M.; Huang, S.; Poulaki, V.; Camphausen, K.; Beecken, W.D.; Kirchhof, B.; Adamis, A.P. In vivo retinal gene expression in early diabetes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3047–3057. [Google Scholar]
- Brucklacher, R.M.; Patel, K.M.; VanGuilder, H.D.; Bixler, G.V.; Barber, A.J.; Antonetti, D.A.; Lin, C.-M.; LaNoue, K.F.; Gardner, T.W.; Bronson, S.K.; et al. Whole genome assessment of the retinal response to diabetes reveals a progressive neurovascular inflammatory response. BMC Med. Genom. 2008, 1, 26. [Google Scholar] [CrossRef] [PubMed]
- Kandpal, R.P.; Rajasimha, H.K.; Brooks, M.J.; Nellissery, J.; Wan, J.; Qian, J.; Kern, T.S.; Swaroop, A. Transcriptome analysis using next generation sequencing reveals molecular signatures of diabetic retinopathy and efficacy of candidate drugs. Mol. Vis. 2012, 18, 1123–1146. [Google Scholar] [PubMed]
- Nahomi, R.B.; Palmer, A.; Green, K.M.; Fort, P.E.; Nagaraj, R.H. Pro-inflammatory cytokines downregulate Hsp27 and cause apoptosis of human retinal capillary endothelial cells. Biochim. Biophys. Acta 2014, 1842, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Tang, J.; Li, G.; Berti-Mattera, L.; Lee, C.A.; Bartkowski, D.; Gale, D.; Monahan, J.; Niesman, M.R.; Alton, G.; et al. Effects of p38 MAPK inhibition on early stages of diabetic retinopathy and sensory nerve function. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2158–2164. [Google Scholar] [CrossRef] [PubMed]
- Pinach, S.; Burt, D.; Berrone, E.; Barutta, F.; Bruno, G.; Porta, M.; Perin, P.C.; Gruden, G. Retinal heat shock protein 25 in early experimental diabetes. Acta Diabetol. 2013, 50, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Losiewicz, M.K.; Fort, P.E. Diabetes Impairs the Neuroprotective Properties of Retinal Alpha-crystallins. Investig. Opthalmol. Vis. Sci. 2011, 52, 5034–5042. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.S.; Raghu, G.; Reddy, S.S.; Pasupulati, A.K.; Suryanarayana, P.; Reddy, G.B. Response of small heat shock proteins in diabetic rat retina. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7674–7682. [Google Scholar] [CrossRef] [PubMed]
- Krueger-Naug, A.M.R.; Emsley, J.G.; Myers, T.L.; Currie, R.W.; Clarke, D.B. Injury to retinal ganglion cells induces expression of the small heat shock protein Hsp27 in the rat visual system. Neuroscience 2002, 110, 653–665. [Google Scholar] [CrossRef]
- Sawada, J.; Li, F.; Komatsu, M. R-Ras Inhibits VEGF-Induced p38MAPK Activation and HSP27 Phosphorylation in Endothelial Cells. J. Vasc. Res. 2016, 52, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Lee, H.-J.; Choi, S.; Jin, Y.B.; An, H.J.; Kang, J.-H.; Yoon, S.S.; Lee, Y.-S. Soluble HSPB1 regulates VEGF-mediated angiogenesis through their direct interaction. Angiogenesis 2012, 15, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Jego, G.; Wettstein, G.; Terrier, O.; Cronier, L.; Yousfi, N.; Hébrard, S.; Bouchot, A.; Hazoumé, A.; Joly, A.-L.; et al. Extracellular HSP27 mediates angiogenesis through Toll-like receptor 3. FASEB J. 2013, 27, 4169–4183. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Geuens, T.; Petiot, P.; Péréon, Y.; Adriaenssens, E.; Haidar, M.; Capponi, S.; Maisonobe, T.; Fournier, E.; Dubourg, O.; et al. Axonal Neuropathies due to Mutations in Small Heat Shock Proteins: Clinical, Genetic, and Functional Insights into Novel Mutations. Hum. Mutat. 2017, 38, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi, J.; Van Den Bosch, L.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; Van Impe, K.; Fedotov, V.; et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Benn, S.C.; Perrelet, D.; Kato, A.C.; Scholz, J.; Decosterd, I.; Mannion, R.J.; Bakowska, J.C.; Woolf, C.J. Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron 2002, 36, 45–56. [Google Scholar] [CrossRef]
- Ma, C.H.E.; Omura, T.; Cobos, E.J.; Latrémolière, A.; Ghasemlou, N.; Brenner, G.J.; Van Veen, E.; Barrett, L.; Sawada, T.; Gao, F.; et al. Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J. Clin. Investig. 2011, 121, 4332–4347. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H.; Zhang, W.; Sima, A.A.F. Degeneration of the Golgi and neuronal loss in dorsal root ganglia in diabetic BioBreeding/Worcester rats. Diabetologia 2006, 49, 2763–2774. [Google Scholar] [CrossRef] [PubMed]
- Zochodne, D.W.; Verge, V.M.; Cheng, C.; Sun, H.; Johnston, J. Does diabetes target ganglion neurones? Progressive sensory neurone involvement in long-term experimental diabetes. Brain 2001, 124, 2319–2334. [Google Scholar] [CrossRef] [PubMed]
- Korngut, L.; Ma, C.H.E.; Martinez, J.A.; Toth, C.C.; Guo, G.F.; Singh, V.; Woolf, C.J.; Zochodne, D.W. Overexpression of human HSP27 protects sensory neurons from diabetes. Neurobiol. Dis. 2012, 47, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Barutta, F.; Pinach, S.; Bruno, G.; Perin, P.C.; Gruden, G. Hippocampal heat shock protein 25 expression in streptozotocin-induced diabetic mice. Neuroscience 2012, 227, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Gruden, G.; Bruno, G.; Chaturvedi, N.; Burt, D.; Schalkwijk, C.; Pinach, S.; Stehouwer, C.D.; Witte, D.R.; Fuller, J.H.; Perin, P.C.; et al. Serum heat shock protein 27 and diabetes complications in the EURODIAB prospective complications study: A novel circulating marker for diabetic neuropathy. Diabetes 2008, 57, 1966–1970. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.; Rolandsson Enes, S.; Skärstrand, H.; Pourhamidi, K.; Gottsäter, A.; Wollmer, P.; Rolandsson, O.; Westergren-Thorsson, G.; Dahlin, L.B. Temporal trend of autonomic nerve function and HSP27, MIF and PAI-1 in type 1 diabetes. J. Clin. Transl. Endocrinol. 2017, 8, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Pourhamidi, K.; Dahlin, L.B.; Boman, K.; Rolandsson, O. Heat shock protein 27 is associated with better nerve function and fewer signs of neuropathy. Diabetologia 2011, 54, 3143–3149. [Google Scholar] [CrossRef] [PubMed]
- Lepedda, A.J.; Cigliano, A.; Cherchi, G.M.; Spirito, R.; Maggioni, M.; Carta, F.; Turrini, F.; Edelstein, C.; Scanu, A.M.; Formato, M. A proteomic approach to differentiate histologically classified stable and unstable plaques from human carotid arteries. Atherosclerosis 2009, 203, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ventura, J.L.; Duran, M.C.; Blanco-Colio, L.M.; Meilhac, O.; Leclercq, A.; Michel, J.-B.; Jensen, O.N.; Hernandez-Merida, S.; Tuñón, J.; Vivanco, F.; et al. Identification by a differential proteomic approach of heat shock protein 27 as a potential marker of atherosclerosis. Circulation 2004, 110, 2216–2219. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ventura, J.L.; Nicolas, V.; Houard, X.; Blanco-Colio, L.M.; Leclercq, A.; Egido, J.; Vranckx, R.; Michel, J.-B.; Meilhac, O. Biological significance of decreased HSP27 in human atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Park, E.-C.; Bae, S.W.; Park, M.Y.; Kim, S.W.; Yoo, H.S.; Tudev, M.; Ko, Y.H.; Choi, Y.-H.; Kim, S.; et al. Expression of Heat Shock Protein 27 in Human Atherosclerotic Plaques and Increased Plasma Level of Heat Shock Protein 27 in Patients With Acute Coronary Syndrome. Circulation 2006, 114, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Cuerrier, C.M.; Chen, Y.-X.; Tremblay, D.; Rayner, K.; McNulty, M.; Zhao, X.; Kennedy, C.R.J.; De BelleRoche, J.; Pelling, A.E.; O’Brien, E.R. Chronic Over-Expression of Heat Shock Protein 27 Attenuates Atherogenesis and Enhances Plaque Remodeling: A Combined Histological and Mechanical Assessment of Aortic Lesions. PLoS ONE 2013, 8, e55867. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-F.; Liu, S.-J.; Chen, G. Heat shock protein 27 phosphorylation in the proliferation and apoptosis of human umbilical vein endothelial cells induced by high glucose through the phosphoinositide 3-kinase/Akt and extracellular signal-regulated kinase 1/2 pathways. Mol. Med. Rep. 2015, 11, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.S.; Brownlee, M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ. Res. 2016, 118, 1808–1829. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.; Chen, Y.-X.; McNulty, M.; Simard, T.; Zhao, X.; Wells, D.J.; De Belleroche, J.; O’Brien, E.R. Extracellular release of the atheroprotective heat shock protein 27 is mediated by estrogen and competitively inhibits acLDL binding to scavenger receptor-A. Circ. Res. 2008, 103, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Raizman, J.E.; Chen, Y.-X.; Seibert, T.; Hibbert, B.; Cuerrier, C.M.; Salari, S.; Zhao, X.; Hu, T.; Shi, C.; Ma, X.; et al. Heat shock protein-27 attenuates foam cell formation and atherogenesis by down-regulating scavenger receptor-A expression via NF-κB signaling. Biochim. Biophys. Acta 2013, 1831, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Pulakazhi Venu, V.K.; Adijiang, A.; Seibert, T.; Chen, Y.-X.; Shi, C.; Batulan, Z.; O’Brien, E.R. Heat shock protein 27–derived atheroprotection involves reverse cholesterol transport that is dependent on GM-CSF to maintain ABCA1 and ABCG1 expression in ApoE−/− mice. FASEB J. 2017, 31, 2364–2379. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Cleveland, J.C.; Ao, L.; Li, J.; Zeng, Q.; Fullerton, D.A.; Meng, X. Human myocardium releases heat shock protein 27 (HSP27) after global ischemia: The proinflammatory effect of extracellular HSP27 through toll-like receptor (TLR)-2 and TLR4. Mol. Med. 2014, 20, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, H.; Kuroyanagi, G.; Tsujimoto, M.; Enomoto, Y.; Matsushima-Nishiwaki, R.; Onuma, T.; Kojima, A.; Doi, T.; Tanabe, K.; Akamatsu, S.; et al. Release of Phosphorylated HSP27 (HSPB1) from Platelets Is Accompanied with the Acceleration of Aggregation in Diabetic Patients. PLoS ONE 2015, 10, e0128977. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, H.; Kuroyanagi, G.; Tsujimoto, M.; Matsushima-Nishiwaki, R.; Akamatsu, S.; Enomoto, Y.; Iida, H.; Otsuka, T.; Ogura, S.; Iwama, T.; et al. Thrombin Receptor-Activating Protein (TRAP)-Activated Akt Is Involved in the Release of Phosphorylated-HSP27 (HSPB1) from Platelets in DM Patients. Int. J. Mol. Sci. 2016, 17, 737. [Google Scholar] [CrossRef] [PubMed]
- Gruden, G.; Barutta, F.; Catto, I.; Bosco, G.; Caprioli, M.G.; Pinach, S.; Fornengo, P.; Cavallo-Perin, P.; Davini, O.; Cerrato, P.; et al. Serum levels of heat shock protein 27 in patients with acute ischemic stroke. Cell Stress Chaperones 2013, 18, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Phillips, V.L.; Williams, M.J.; Van Rij, A.M.; Jones, G.T. Plasma heat shock protein 27 is associated with coronary artery disease, abdominal aortic aneurysm and peripheral artery disease. Springerplus 2014, 3, 635. [Google Scholar] [CrossRef] [PubMed]
- Seibert, T.A.; Hibbert, B.; Chen, Y.-X.; Rayner, K.; Simard, T.; Hu, T.; Cuerrier, C.M.; Zhao, X.; De Belleroche, J.; Chow, B.J.W.; et al. Serum heat shock protein 27 levels represent a potential therapeutic target for atherosclerosis: Observations from a human cohort and treatment of female mice. J. Am. Coll. Cardiol. 2013, 62, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Kardys, I.; Rifai, N.; Meilhac, O.; Michel, J.-B.; Martin-Ventura, J.L.; Buring, J.E.; Libby, P.; Ridker, P.M. Plasma concentration of heat shock protein 27 and risk of cardiovascular disease: A prospective, nested case-control study. Clin. Chem. 2008, 54, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Burt, D.; Bruno, G.; Chaturvedi, N.; Schalkwijk, C.; Stehouwer, C.D.; Witte, D.R.; Fuller, J.H.; Pinach, S.; Cavallo Perin, P.; Gruden, G. Anti-Heat Shock Protein 27 Antibody Levels and Diabetes Complications in the EURODIAB Study. Diabetes Care 2009, 32, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Karagöz, G.E.; Rüdiger, S.G.D. Hsp90 interaction with clients. Trends Biochem. Sci. 2015, 40, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Sidera, K.; Patsavoudi, E. HSP90 inhibitors: Current development and potential in cancer therapy. Recent Pat. Anticancer Drug Discov. 2014, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Sohn, E.; Jung, D.H.; Lee, Y.M.; Kim, C.S.; Kim, J.; Kim, J.S. Expression of heat shock protein 90 in the kidneys of diabetic db/db mice. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2198–2204. [Google Scholar] [PubMed]
- Zhang, H.-M.; Dang, H.; Kamat, A.; Yeh, C.-K.; Zhang, B.-X. Geldanamycin Derivative Ameliorates High Fat Diet-Induced Renal Failure in Diabetes. PLoS ONE 2012, 7, e32746. [Google Scholar] [CrossRef] [PubMed]
- Kuan, Y.-C.; Hashidume, T.; Shibata, T.; Uchida, K.; Shimizu, M.; Inoue, J.; Sato, R. Heat Shock Protein 90 Modulates Lipid Homeostasis by Regulating the Stability and Function of Sterol Regulatory Element-binding Protein (SREBP) and SREBP Cleavage-activating Protein. J. Biol. Chem. 2017, 292, 3016–3028. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, I.; Oguiza, A.; Recio, C.; Mallavia, B.; Madrigal-Matute, J.; Blanco, J.; Egido, J.; Martin-Ventura, J.-L.; Gomez-Guerrero, C. Targeting HSP90 Ameliorates Nephropathy and Atherosclerosis through Suppression of NF-κB and STAT Signaling Pathways in Diabetic Mice. Diabetes 2015, 64, 3600–3613. [Google Scholar] [CrossRef] [PubMed]
- García, R.; Merino, D.; Gómez, J.M.; Nistal, J.F.; Hurlé, M.A.; Cortajarena, A.L.; Villar, A.V. Extracellular heat shock protein 90 binding to TGFβ receptor I participates in TGFβ-mediated collagen production in myocardial fibroblasts. Cell. Signal. 2016, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Kim, H.J.; Yu, M.R.; Kim, W.-Y.; Kim, J.; Ryu, J.H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Ziyadeh, F. Heat shock protein 90 inhibitor attenuates renal fibrosis through degradation of transforming growth factor-β type II receptor. Lab. Investig. 2012, 92, 1583–1596. [Google Scholar] [CrossRef] [PubMed]
- Sontake, V.; Wang, Y.; Kasam, R.K.; Sinner, D.; Reddy, G.B.; Naren, A.P.; McCormack, F.X.; White, E.S.; Jegga, A.G.; Madala, S.K. Hsp90 regulation of fibroblast activation in pulmonary fibrosis. JCI Insight 2017, 2, e91454. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hoshimaru, M.; Aoki, T.; Takahashi, J.B.; Ohtsuka, T.; Asahi, M.; Matsuura, N.; Kikuchi, H. Expression of heat shock proteins in the developing rat retina. Neurosci. Lett. 1996, 205, 215–217. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Kim, Y.-S.; Park, C.-H.; Chung, I.-Y.; Yoo, J.-M.; Kim, J.-G.; Lee, B.-J.; Kang, S.-S.; Cho, G.-J.; Choi, W.-S. Protein kinase C-delta mediates neuronal apoptosis in the retinas of diabetic rats via the Akt signaling pathway. Diabetes 2008, 57, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Zhang, X.M.; Wang, X.D.; Wang, B.J.; Wang, W. 17-AAG, a Hsp90 inhibitor, attenuates the hypoxia-induced expression of SDF-1alpha and ILK in mouse RPE cells. Mol. Biol. Rep. 2010, 37, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-C.; Kao, Y.-H.; Hu, P.-S.; Chen, J.-H. Geldanamycin, a HSP90 inhibitor, attenuates the hypoxia-induced vascular endothelial growth factor expression in retinal pigment epithelium cells in vitro. Exp. Eye Res. 2007, 85, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.H.; An, H.; Chang, D.-J.; Baek, Y.-Y.; Cho, C.S.; Jun, H.O.; Park, S.-J.; Kim, J.H.; Lee, H.-Y.; Kim, K.-W.; et al. Hypoxia-mediated retinal neovascularization and vascular leakage in diabetic retina is suppressed by HIF-1α destabilization by SH-1242 and SH-1280, novel hsp90 inhibitors. J. Mol. Med. 2014, 92, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.B.; Blagg, B.S. To fold or not to fold: Modulation and consequences of Hsp90 inhibition. Future Med. Chem. 2009, 1, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.J.; Li, C.; Yu, C.; Lu, Y.; Krise, J.M.; McIntosh, M.P.; Rajewski, R.A.; Blagg, B.S.J.; Dobrowsky, R.T. Inhibiting heat-shock protein 90 reverses sensory hypoalgesia in diabetic mice. ASN Neuro 2010, 2, e00040. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.J.; Pan, P.; Farmer, K.L.; Zhao, H.; Blagg, B.S.J.; Dobrowsky, R.T. Modulating molecular chaperones improves sensory fiber recovery and mitochondrial function in diabetic peripheral neuropathy. Exp. Neurol. 2012, 235, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ma, J.; Zhao, H.; Blagg, B.S.; Dobrowsky, R.T. Induction of Heat Shock Protein 70 (Hsp70) prevents Neuregulin-Induced Demyelination by Enhancing the Proteasomal Clearance of c-Jun. ASN Neuro 2012, 4, e00102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, H.; Blagg, B.S.J.; Dobrowsky, R.T. C-terminal heat shock protein 90 inhibitor decreases hyperglycemia-induced oxidative stress and improves mitochondrial bioenergetics in sensory neurons. J. Proteome Res. 2012, 11, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Pan, P.; Anyika, M.; Blagg, B.S.J.; Dobrowsky, R.T. Modulating Molecular Chaperones Improves Mitochondrial Bioenergetics and Decreases the Inflammatory Transcriptome in Diabetic Sensory Neurons. ACS Chem. Neurosci. 2015, 6, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.; Warnholtz, A.; et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001, 88, E14–E22. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Venkatakrishnan, A.; Yu, S.; Kazlauskas, A. Protein kinase A-dependent translocation of Hsp90 alpha impairs endothelial nitric-oxide synthase activity in high glucose and diabetes. J. Biol. Chem. 2007, 282, 9364–9371. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Konopinski, R.; Yan, B.; Centonze, V.E.; Natarajan, M. High glucose-induced IKK-Hsp-90 interaction contributes to endothelial dysfunction. Am. J. Physiol. Cell Physiol. 2009, 296, C182–C192. [Google Scholar] [CrossRef] [PubMed]
- Presley, T.; Vedam, K.; Druhan, L.J.; Ilangovan, G. Hyperthermia-induced Hsp90·eNOS preserves mitochondrial respiration in hyperglycemic endothelial cells by down-regulating Glut-1 and up-regulating G6PD activity. J. Biol. Chem. 2010, 285, 38194–38203. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J.; Xie, Z.; Viollet, B.; Zou, M.-H. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes 2006, 55, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Businaro, R.; Profumo, E.; Tagliani, A.; Buttari, B.; Leone, S.; D’Amati, G.; Ippoliti, F.; Leopizzi, M.; D’Arcangelo, D.; Capoano, R.; et al. Heat-shock protein 90: A novel autoantigen in human carotid atherosclerosis. Atherosclerosis 2009, 207, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; López-Franco, O.; Blanco-Colio, L.M.; Muñoz-García, B.; Ramos-Mozo, P.; Ortega, L.; Egido, J.; Martín-Ventura, J.L. Heat shock protein 90 inhibitors attenuate inflammatory responses in atherosclerosis. Cardiovasc. Res. 2010, 86, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, I.; Oguiza, A.; Recio, C.; Lopez-Sanz, L.; Bernal, S.; Egido, J.; Gomez-Guerrero, C. Interplay between HSP90 and Nrf2 pathways in diabetes-associated atherosclerosis. Clínica e Investigación en Arteriosclerosis 2017, 29, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-F.; Sahu, D.; Tsen, F.; Zhao, Z.; Fan, J.; Kim, R.; Wang, X.; O’Brien, K.; Li, Y.; Kuang, Y.; et al. A fragment of secreted Hsp90α carries properties that enable it to accelerate effectively both acute and diabetic wound healing in mice. J. Clin. Investig. 2011, 121, 4348–4361. [Google Scholar] [CrossRef] [PubMed]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef] [PubMed]
- Beck, F.X.; Neuhofer, W.; Müller, E. Molecular chaperones in the kidney: Distribution, putative roles, and regulation. Am. J. Physiol. Ren. Physiol. 2000, 279, F203–F215. [Google Scholar]
- Calabrese, V.; Mancuso, C.; Sapienza, M.; Puleo, E.; Calafato, S.; Cornelius, C.; Finocchiaro, M.; Mangiameli, A.; Di Mauro, M.; Stella, A.M.G.; et al. Oxidative stress and cellular stress response in diabetic nephropathy. Cell Stress Chaperones 2007, 12, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-C.; Guh, J.-Y.; Chen, H.-C.; Yang, Y.-L.; Huang, J.-S.; Chuang, L.-Y. Advanced glycation end-product-induced mitogenesis is dependent on Janus kinase 2-induced heat shock protein 70 in normal rat kidney interstitial fibroblast cells. Transl. Res. 2007, 149, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Gellai, R.; Hodrea, J.; Lenart, L.; Hosszu, A.; Koszegi, S.; Balogh, D.; Ver, A.; Banki, N.F.; Fulop, N.; Molnar, A.; et al. Role of O-linked N-acetylglucosamine modification in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2016, 311, F1172–F1181. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Yiu, W.H.; Wu, H.J.; Chan, L.Y.Y.; Leung, J.C.K.; Au, W.S.; Chan, K.W.; Lai, K.N.; Tang, S.C.W. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Chen, X.; Gilbert, R.E.; Zhang, Y.; Waltham, M.; Schache, M.; Kelly, D.J.; Pollock, C.A. High Glucose-Induced Thioredoxin-Interacting Protein in Renal Proximal Tubule Cells Is Independent of Transforming Growth Factor-β1. Am. J. Pathol. 2007, 171, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.-F.; Tsai, P.-J.; Chuang, Y.-L.; Shen, Y.-T.; Tai, T.-A.; Chen, W.-C.; Chou, C.-K.; Ho, L.-C.; Tang, M.-J.; Lai, K.-T.A.; et al. Albumin stimulates renal tubular inflammation through an HSP70-TLR4 axis in mice with early diabetic nephropathy. Dis. Model. Mech. 2015, 8, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- El-Horany, H.E.-S.; Abd-Ellatif, R.N.; Watany, M.; Hafez, Y.M.; Okda, H.I. NLRP3 expression and urinary HSP72 in relation to biomarkers of inflammation and oxidative stress in diabetic nephropathy patients. IUBMB Life 2017, 69, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Morteza, A.; Nakhjavani, M.; Larry, M.; Nargesi, A.A.; Esteghamati, A. Heat shock protein 70 and albuminuria in patients with type 2 diabetes: A matched case control study. Cell Stress Chaperones 2013, 18, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Buraczynska, M.; Swatowski, A.; Buraczynska, K.; Dragan, M.; Ksiazek, A. Heat-shock protein gene polymorphisms and the risk of nephropathy in patients with Type 2 diabetes. Clin. Sci. 2009, 116, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mohammad, G.; dos Santos, J.M.; Zhong, Q. Abrogation of MMP-9 Gene Protects Against the Development of Retinopathy in Diabetic Mice by Preventing Mitochondrial Damage. Diabetes 2011, 60, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Sayed, K.M.; Mahmoud, A.A. Heat shock protein-70 and hypoxia inducible factor-1α in type 2 diabetes mellitus patients complicated with retinopathy. Acta Ophthalmol. 2016, 94, e361–e366. [Google Scholar] [CrossRef] [PubMed]
- Gruden, G.; Bruno, G.; Chaturvedi, N.; Burt, D.; Pinach, S.; Schalkwijk, C.; Stehouwer, C.D.; Witte, D.R.; Fuller, J.H.; Cavallo-Perin, P.; et al. ANTI-HSP60 and ANTI-HSP70 antibody levels and micro/ macrovascular complications in type 1 diabetes: The EURODIAB Study. J. Intern. Med. 2009, 266, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Dobrowsky, R.T. Targeting the Diabetic Chaperome to Improve Peripheral Neuropathy. Curr. Diabetes Rep. 2016, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Jia, Y.; Liu, Y.; Wang, H.; Ren, G.; Wang, H. The detection and role of heat shock protein 70 in various nondisease conditions and disease conditions: A literature review. Cell Stress Chaperones 2015, 20, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Karpe, P.A.; Tikoo, K. Heat shock prevents insulin resistance-induced vascular complications by augmenting angiotensin-(1-7) signaling. Diabetes 2014, 63, 1124–1139. [Google Scholar] [CrossRef] [PubMed]
- Giacconi, R.; Caruso, C.; Lio, D.; Muti, E.; Cipriano, C.; Saba, V.; Boccoli, G.; Gasparini, N.; Malavolta, M.; Mocchegiani, E. 1267 HSP70-2 polymorphism as a risk factor for carotid plaque rupture and cerebral ischaemia in old type 2 diabetes-atherosclerotic patients. Mech. Ageing Dev. 2005, 126, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Mambula, S.S.; Gray, P.J.; Theriault, J.R. Extracellular heat shock proteins in cell signaling. FEBS Lett. 2007, 581, 3689–3694. [Google Scholar] [CrossRef] [PubMed]
- Nakhjavani, M.; Morteza, A.; Asgarani, F.; Khalilzadeh, O.; Ghazizadeh, Z.; Bathaie, S.Z.; Esteghamati, A. The dual behavior of heat shock protein 70 and asymmetric dimethylarginine in relation to serum CRP levels in type 2 diabetes. Gene 2012, 498, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Borges, T.J.; Wieten, L.; Van Herwijnen, M.J.C.; Broere, F.; Van Der Zee, R.; Bonorino, C.; Van Eden, W. The anti-inflammatory mechanisms of Hsp70. Front. Immunol. 2012, 3, 95. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-Y.; Brown, N.K.; Wu, W.; Khedri, Z.; Yu, H.; Chen, X.; Van De Vlekkert, D.; D’Azzo, A.; Zheng, P.; Liu, Y. Broad and direct interaction between TLR and Siglec families of pattern recognition receptors and its regulation by Neu1. eLife 2014, 3, e04066. [Google Scholar] [CrossRef] [PubMed]
- Dulin, E.; García-Barreno, P.; Guisasola, M.C. Extracellular heat shock protein 70 (HSPA1A) and classical vascular risk factors in a general population. Cell Stress Chaperones 2010, 15, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Herz, I.; Rosso, R.; Roth, A.; Keren, G.; George, J. Serum levels of anti heat shock protein 70 antibodies in patients with stable and unstable angina pectoris. Acute Card. Care 2006, 8, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ventura, J.L.; Leclercq, A.; Blanco-Colio, L.M.; Egido, J.; Rossignol, P.; Meilhac, O.; Michel, J.-B. Low plasma levels of HSP70 in patients with carotid atherosclerosis are associated with increased levels of proteolytic markers of neutrophil activation. Atherosclerosis 2007, 194, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. Increased Serum Levels of Heat Shock Protein 70 Are Associated With Low Risk of Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Pockley, A.G.; Georgiades, A.; Thulin, T.; de Faire, U.; Frostegård, J. Serum heat shock protein 70 levels predict the development of atherosclerosis in subjects with established hypertension. Hypertension 2003, 42, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Chen, X.; Middleditch, M.; Huang, L.; Vazhoor Amarsingh, G.; Reddy, S.; Lu, J.; Zhang, S.; Ruggiero, K.; Phillips, A.R.J.; et al. Quantitative proteomic profiling identifies new renal targets of copper(II)-selective chelation in the reversal of diabetic nephropathy in rats. Proteomics 2009, 9, 4309–4320. [Google Scholar] [CrossRef] [PubMed]
- Aluksanasuwan, S.; Sueksakit, K.; Fong-ngern, K.; Thongboonkerd, V. Role of HSP60 (HSPD1) in diabetes-induced renal tubular dysfunction: Regulation of intracellular protein aggregation, ATP production, and oxidative stress. FASEB J. 2017, 31, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Kowluru, R.A. Novel Role of Mitochondrial Matrix Metalloproteinase-2 in the Development of Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2011, 52, 3832–3841. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Young, B.J.; Martinus, R.D. Expression of chaperonin 60 in the hippocampus of the streptozotocin diabetic rat. Neuroreport 2006, 17, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, R.K.; Kanwar, J.R.; Wang, D.; Ormrod, D.J.; Krissansen, G.W. Temporal expression of heat shock proteins 60 and 70 at lesion-prone sites during atherogenesis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Kleindienst, R.; Xu, Q.; Willeit, J.; Waldenberger, F.R.; Weimann, S.; Wick, G. Immunology of atherosclerosis. Demonstration of heat shock protein 60 expression and T lymphocytes bearing alpha/beta or gamma/delta receptor in human atherosclerotic lesions. Am. J. Pathol. 1993, 142, 1927–1937. [Google Scholar] [PubMed]
- Wick, G.; Jakic, B.; Buszko, M.; Wick, M.C.; Grundtman, C. The role of heat shock proteins in atherosclerosis. Nat. Rev. Cardiol. 2014, 11, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, C.; Wei, X.; Li, P.; Cui, Y.; Qin, Y.; Wei, X.; Jin, M.; Kohama, K.; Gao, Y. Heat shock protein 60 stimulates the migration of vascular smooth muscle cells via Toll-like receptor 4 and ERK MAPK activation. Sci. Rep. 2015, 5, 15352. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q. Role of heat shock proteins in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Afek, A.; George, J.; Gilburd, B.; Rauova, L.; Goldberg, I.; Kopolovic, J.; Harats, D.; Shoenfeld, Y. Immunization of Low-density Lipoprotein Receptor Deficient (LDL-RD) Mice with Heat Shock Protein 65 (HSP-65) Promotes Early Atherosclerosis. J. Autoimmun. 2000, 14, 115–121. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Afek, A.; Gilburd, B.; Shoenfeld, Y.; Harats, D. Cellular and humoral immune responses to heat shock protein 65 are both involved in promoting fatty-streak formation in LDL-receptor deficient mice. J. Am. Coll. Cardiol. 2001, 38, 900–905. [Google Scholar] [CrossRef]
- George, J.; Greenberg, S.; Barshack, I.; Goldberg, I.; Keren, G.; Roth, A. Immunity to heat shock protein 65--an additional determinant in intimal thickening. Atherosclerosis 2003, 168, 33–38. [Google Scholar] [CrossRef]
- Knoflach, M.; Kiechl, S.; Kind, M.; Said, M.; Sief, R.; Gisinger, M.; Van Der Zee, R.; Gaston, H.; Jarosch, E.; Willeit, J.; et al. Cardiovascular risk factors and atherosclerosis in young males: ARMY study (Atherosclerosis Risk-Factors in Male Youngsters). Circulation 2003, 108, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Knoflach, M.; Kiechl, S.; Penz, D.; Zangerle, A.; Schmidauer, C.; Rossmann, A.; Shingh, M.; Spallek, R.; Griesmacher, A.; Bernhard, D.; et al. Cardiovascular Risk Factors and Atherosclerosis in Young Women: Atherosclerosis Risk Factors in Female Youngsters (ARFY Study). Stroke 2009, 40, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Halcox, J.P.J.; Deanfield, J.; Shamaei-Tousi, A.; Henderson, B.; Steptoe, A.; Coates, A.R.M.; Singhal, A.; Lucas, A. Circulating Human Heat Shock Protein 60 in the Blood of Healthy Teenagers: A Novel Determinant of Endothelial Dysfunction and Early Vascular Injury? Arterioscler. Thromb. Vasc. Biol. 2005, 25, e141–e142. [Google Scholar] [CrossRef] [PubMed]
- Hoppichler, F.; Koch, T.; Dzien, A.; Gschwandtner, G.; Lechleitner, M. Prognostic Value of Antibody Titre to Heat-Shock Protein 65 on Cardiovascular Events. Cardiology 2000, 94, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Mandal, K.; Schett, G.; Mayr, M.; Wick, G.; Oberhollenzer, F.; Willeit, J.; Kiechl, S.; Xu, Q. Association of serum-soluble heat shock protein 60 with carotid atherosclerosis: Clinical significance determined in a follow-up study. Stroke 2005, 36, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Foteinos, G.; Afzal, A.R.; Mandal, K.; Jahangiri, M.; Xu, Q. Anti-heat shock protein 60 autoantibodies induce atherosclerosis in apolipoprotein E-deficient mice via endothelial damage. Circulation 2005, 112, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Tse, K.; Sette, A.; Ley, K. Vaccination to modulate atherosclerosis. Autoimmunity 2015, 48, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Shamaei-Tousi, A.; Stephens, J.W.; Bin, R.; Cooper, J.A.; Steptoe, A.; Coates, A.R.M.; Henderson, B.; Humphries, S.E. Association between plasma levels of heat shock protein 60 and cardiovascular disease in patients with diabetes mellitus. Eur. Heart J. 2006, 27, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Rabczynski, M.; Fiodorenko-Dumas, Z.; Adamiec, R.; Paprocka-Borowicz, M.; Dumas, I. Role of anti-HSP 60/65 antibodies in atherogenesis in patients with type 2 diabetes and lower limb ischemia. J. Physiol. Pharmacol. 2012, 63, 691–696. [Google Scholar] [PubMed]
- De Maio, A. Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: A form of communication during injury, infection, and cell damage. Cell Stress Chaperones 2011, 16, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Vega, V.L.; Rodríguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gu, H.; Huang, W.; Peng, J.; Li, Y.; Yang, L.; Qin, D.; Essandoh, K.; Wang, Y.; Peng, T.; et al. Hsp20-Mediated Activation of Exosome Biogenesis in Cardiomyocytes Improves Cardiac Function and Angiogenesis in Diabetic Mice. Diabetes 2016, 65, 3111–3128. [Google Scholar] [CrossRef] [PubMed]
- Geiger, A.; Walker, A.; Nissen, E. Human fibrocyte-derived exosomes accelerate wound healing in genetically diabetic mice. Biochem. Biophys. Res. Commun. 2015, 467, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Spertini, F.; Leimgruber, A.; Morel, B.; Khazaeli, M.B.; Yamamoto, K.; Dayer, J.M.; Weisbart, R.H.; Lee, M.L. Idiotypic vaccination with a murine anti-dsDNA antibody: Phase I study in patients with nonactive systemic lupus erythematosus with nephritis. J. Rheumatol. 1999, 26, 2602–2608. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| HSP Names | Old Names |
|---|---|
| HSPB | |
| HSPB1 | HSP27; CMT2F; HMN2B; HSP28; HSP25; HS.76067; DKFZp586P1322 |
| HSPB2 | HSP27; MKBP; Hs.78846; LOH11CR1K; MGC133245 |
| HSPD1 | HSP60; GroEL |
| HSPA | |
| HSPA1A | HSP70-1; HSP72; HSPA1 |
| HSPA1B | HSP70-2 |
| HSPA2 | Heat-shock 70kD protein-2 |
| HSPA5 | BIP; GRP78; MIF2 |
| HSPA8 | HSC70; HSC71; HSP71; HSP73 |
| HSPA9 | GRP75; HSPA9B; MOT; MOT2; PBP74; mot-2 |
| HSPC | |
| HSPC1 | HSP90AA1; HSPN; LAP2; HSP86; HSPC1; HSPCA; HSP89; HSP90; HSP90A; HSP90N; HSPCAL1; HSPCAL4; FLJ31884 |
| HSPC2 | HSP90AA2; HSPCA; HSPCAL3; HSP90 |
| HSPC3 | HSP90AB1; HSPC2; HSPCB; D6S182; HSP90B; FLJ26984; HSP90 |
| HSPC4 | HSP90B1; ECGP; GP96; TRA1; GRP94; endoplasmin |
| HSPC5 | TRAP1; HSP75; HSP90L |
| HSP | Action | Strategy | Specificity | Complication | Animal Model | Effects | Reference |
|---|---|---|---|---|---|---|---|
| HSP27 | Inhibition of phosphorylation | Genetic deletion of MK-2 | +/− | Diabetic nephropathy | STZ-induced diabetic MK-2−/− mice | None | [29] |
| Inhibition of phosphorylation | PHA666859 (p38 inhibitor) | - | Diabetic retinopathy | STZ-induced diabetic rats | Amelioration of retinal vascular injury | [38] | |
| Induction | Genetic overexpression | + | Diabetic neuropathy | STZ-induced diabetic hHSP27 transgenic mice | Amelioration of DSP | [52] | |
| HSP90 | Inhibition | 17-DMAG (HSP90 inhibitor) | + | Diabetic nephropathy | db/db mice HFD | Reduced kidney damage | [78] |
| Inhibition | 17-DMAG (HSP90 inhibitor) | + | Diabetic nephropathy | STZ-induced diabetic ApoE−/− mice | Reduced albuminuria and mesangial expansion | [80] | |
| Inhibition | SH-1242/SH-1280 (HSP90 inhibitors) | + | Diabetic retinopathy | STZ-induced diabetic mice | Reduced retinal vascular leakage | [88] | |
| Inhibition | KU-32 (C-terminal HSP90 inhibitor) | + | Diabetic neuropathy | STZ-induced diabetic mice | Amelioration of DSP | [90] | |
| Inhibition | 17-DMAG (HSP90 inhibitor) | + | Diabetic macrovascular disease | STZ-induced diabetic ApoE−/− mice | Reduced number of atherosclerotic lesions and more stable plaques | [80] | |
| Inhibition | 17-DMAG (HSP90 inhibitor) | + | Diabetic macrovascular disease | STZ-induced diabetic mice | Reduced lesion size and inflammation | [102] | |
| HSP70 | Inhibition | PFTμ/ VER (intracellular HSP70 inhibitors) | + | Diabetic nephropathy | STZ-induced diabetic mice | Reduced albuminuria, tubular injury | [111] |
| Inhibition | KNK437 (HSF-1 inhibitor) | +/− | Diabetic nephropathy | STZ-induced diabetic mice | Reduced albuminuria tubular injury | [111] | |
| Inhibition | HSP70 neutralizing Ab (blockade of eHSP70) | + | Diabetic nephropathy | STZ-induced diabetic mice | Reduce albuminuria | [111] |
| Biomarker | Study Design | Study Population | N | Results | Adjustments | Reference |
|---|---|---|---|---|---|---|
| HSP27 | Hospital-based case-control study | DM2 with microvascular complications vs. C | C = 247 DM2 = 195 (DR = 123, DN = 80, DNu = 109) | HSP27 higher in DM2-DN vs. other groups | Gender, age, BMI | [33] |
| Nested case-control study EURODIAB PCS | DM1 with and without complications | Controls = 168 Cases = 363 | Direct, independent association with DSP OR 2.41 (1.11–5.24) | Conventional risk factors, markers of inflammation, AER | [54] | |
| Case-control study | Subjects with NGT, IGT and DM2 | NGT = 39 IGT = 29 DM2 = 51 | Inverse association with nerve function OR 2.51 (1.25, 5.05) | Age, sex, statin, HbA1c | [56] | |
| Anti-HSP27 | Nested case-control study EURODIAB PCS | DM1 with and without complications | Controls = 168 Cases = 363 | No association with DM1 complications | Age, DM duration, hypertension, HbA1C, smoking, TNF-α | [74] |
| Urinary HSP70 | Case-control study | DM2 (Normo, Micro Macro) vs. C | C = 15 DM2 = 45 (Normo = 15 Micro = 15, Macro = 15) | Urinary HSP70 higher in Micro/Macro than in Normo DM2 | None | [112] |
| HSP70 | Case-control study | DM2 with and without albuminuria | DM2-Normo = 40 DM2-Alb = 40 | HSP70 higher in Alb than in Normo DM2 | None | [113] |
| Case-control study | DM2 with and without DR vs. C | C = 70 DM2 without DR = 50 DM2 with DR = 50 | HSP70 higher in DM2 with DR | None | [116] | |
| Anti-HSP70 | Nested case-control study EURODIAB PCS | DM1 with and without complications | Controls = 168 Cases = 363 | Independent, inverse association with DR [OR 0.35 (0.15–0.80)] and CVD [OR 0.39 (0.17–0.87)] | Age, DM duration, hypertension, HbA1c, smoking, TNFα, homocysteine, AER | [117] |
| HSP60 | Cross-sectional UDACS Study | DM patients with and without CVD | DM1 = 147 DM2 = 708 DM without CVD = 607 DM with CVD = 241 | HSP60 detectable more frequently in subjects with CVD and MI | Age, sex, ethnic group, smoking | [151] |
| Anti-HSP60 | Nested case-control study EURODIAB PCS | DM1 with and without complications | Controls = 168 Cases = 363 | No associations | - | [117] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellini, S.; Barutta, F.; Mastrocola, R.; Imperatore, L.; Bruno, G.; Gruden, G. Heat Shock Proteins in Vascular Diabetic Complications: Review and Future Perspective. Int. J. Mol. Sci. 2017, 18, 2709. https://doi.org/10.3390/ijms18122709
Bellini S, Barutta F, Mastrocola R, Imperatore L, Bruno G, Gruden G. Heat Shock Proteins in Vascular Diabetic Complications: Review and Future Perspective. International Journal of Molecular Sciences. 2017; 18(12):2709. https://doi.org/10.3390/ijms18122709
Chicago/Turabian StyleBellini, Stefania, Federica Barutta, Raffaella Mastrocola, Luigi Imperatore, Graziella Bruno, and Gabriella Gruden. 2017. "Heat Shock Proteins in Vascular Diabetic Complications: Review and Future Perspective" International Journal of Molecular Sciences 18, no. 12: 2709. https://doi.org/10.3390/ijms18122709
APA StyleBellini, S., Barutta, F., Mastrocola, R., Imperatore, L., Bruno, G., & Gruden, G. (2017). Heat Shock Proteins in Vascular Diabetic Complications: Review and Future Perspective. International Journal of Molecular Sciences, 18(12), 2709. https://doi.org/10.3390/ijms18122709