Analysis and Prediction of Exon Skipping Events from RNA-Seq with Sequence Information Using Rotation Forest

Abstract
:1. Introduction
2. Results
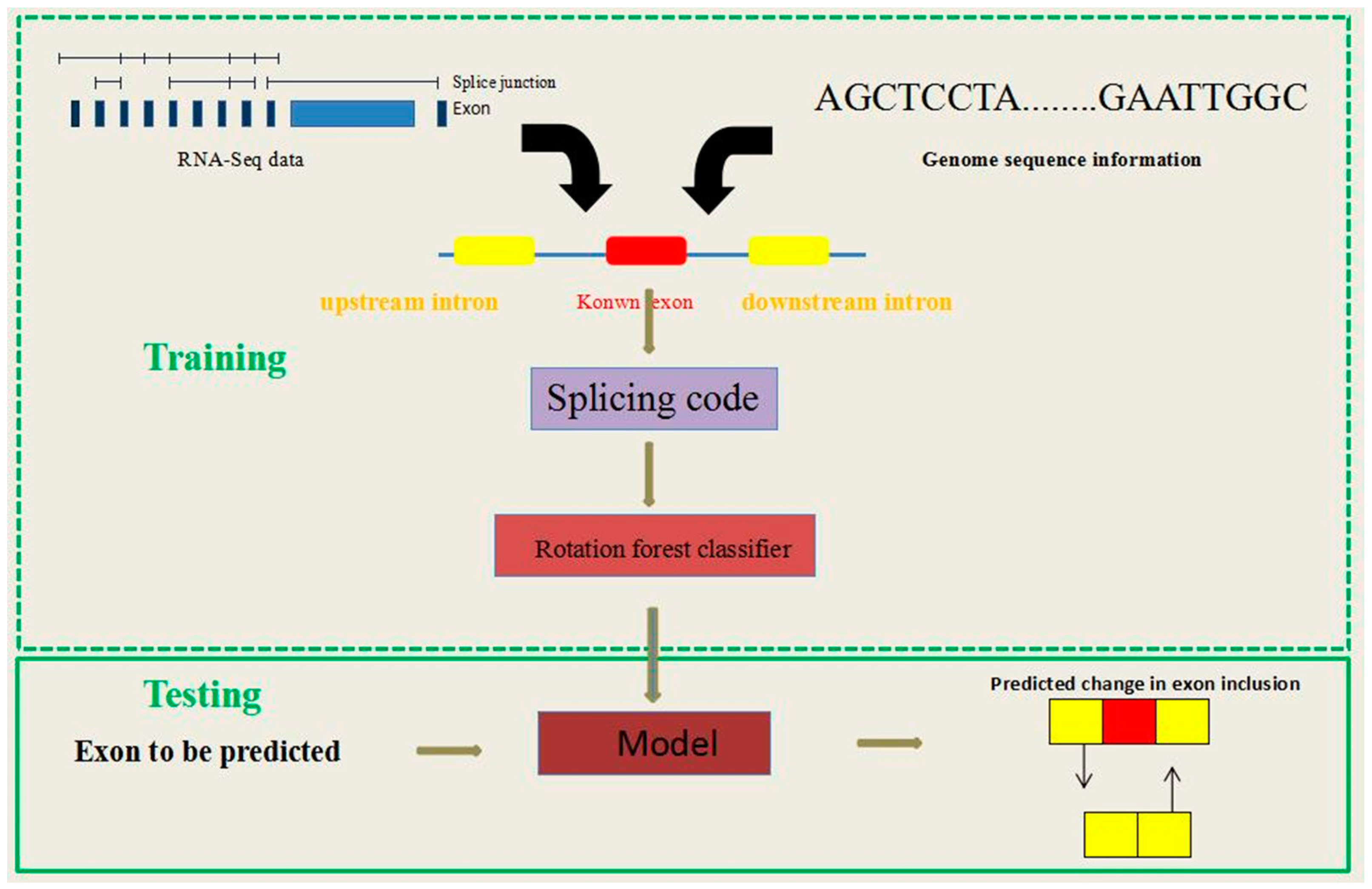
2.1. The RotaF-RSES Framework
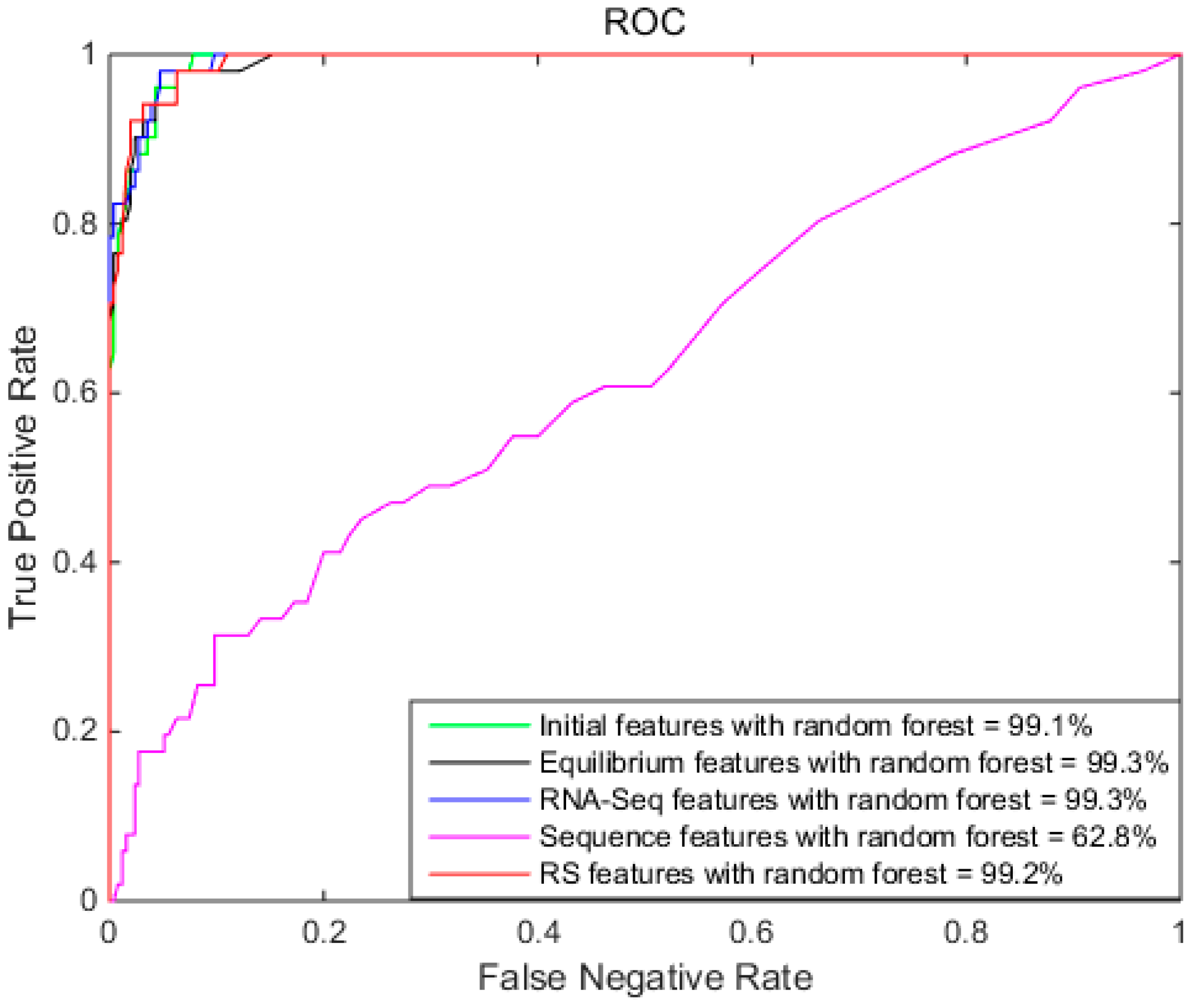
2.2. Comparison of Different Features with Random Forest
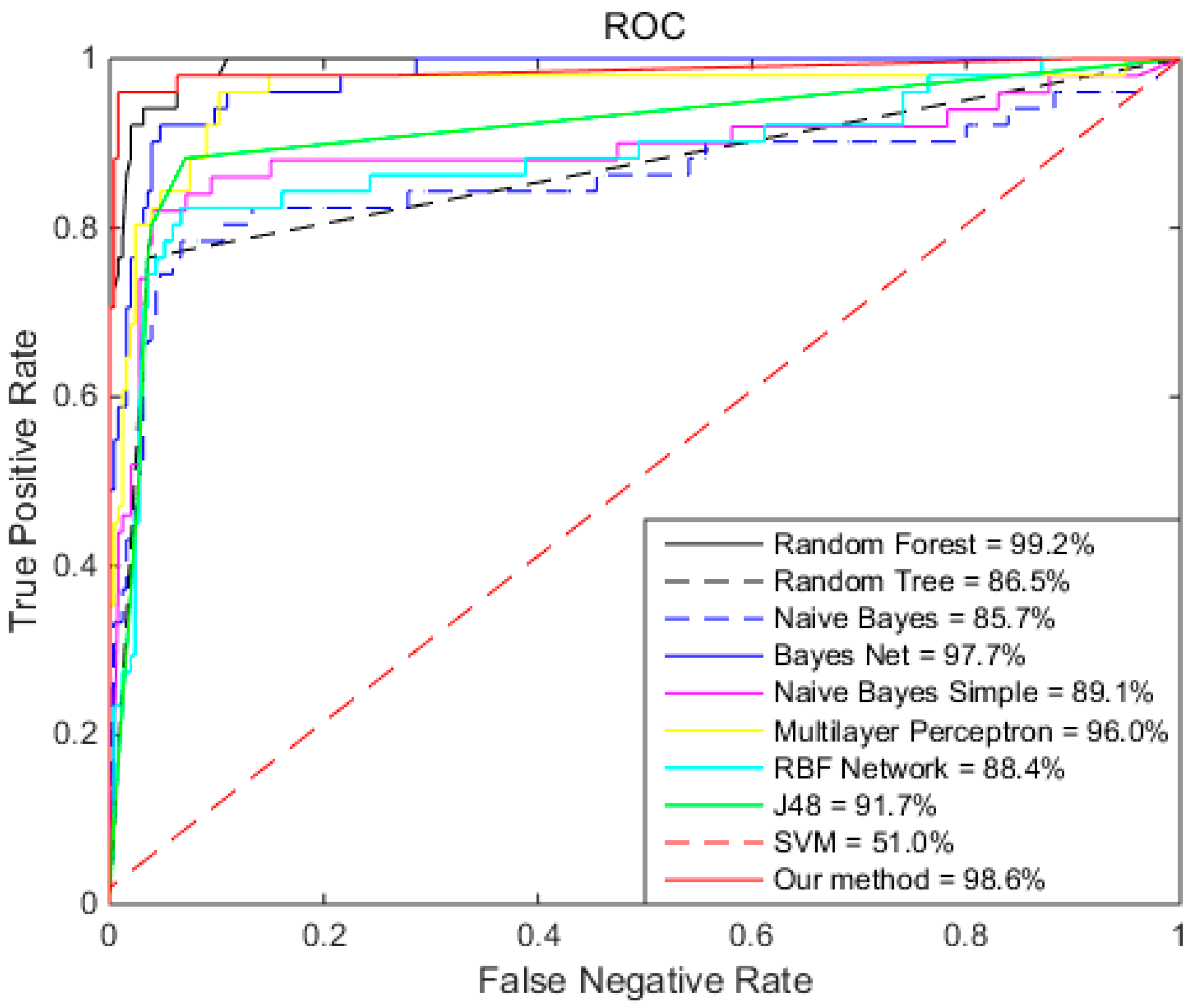
2.3. Comparison of Different Algorithms
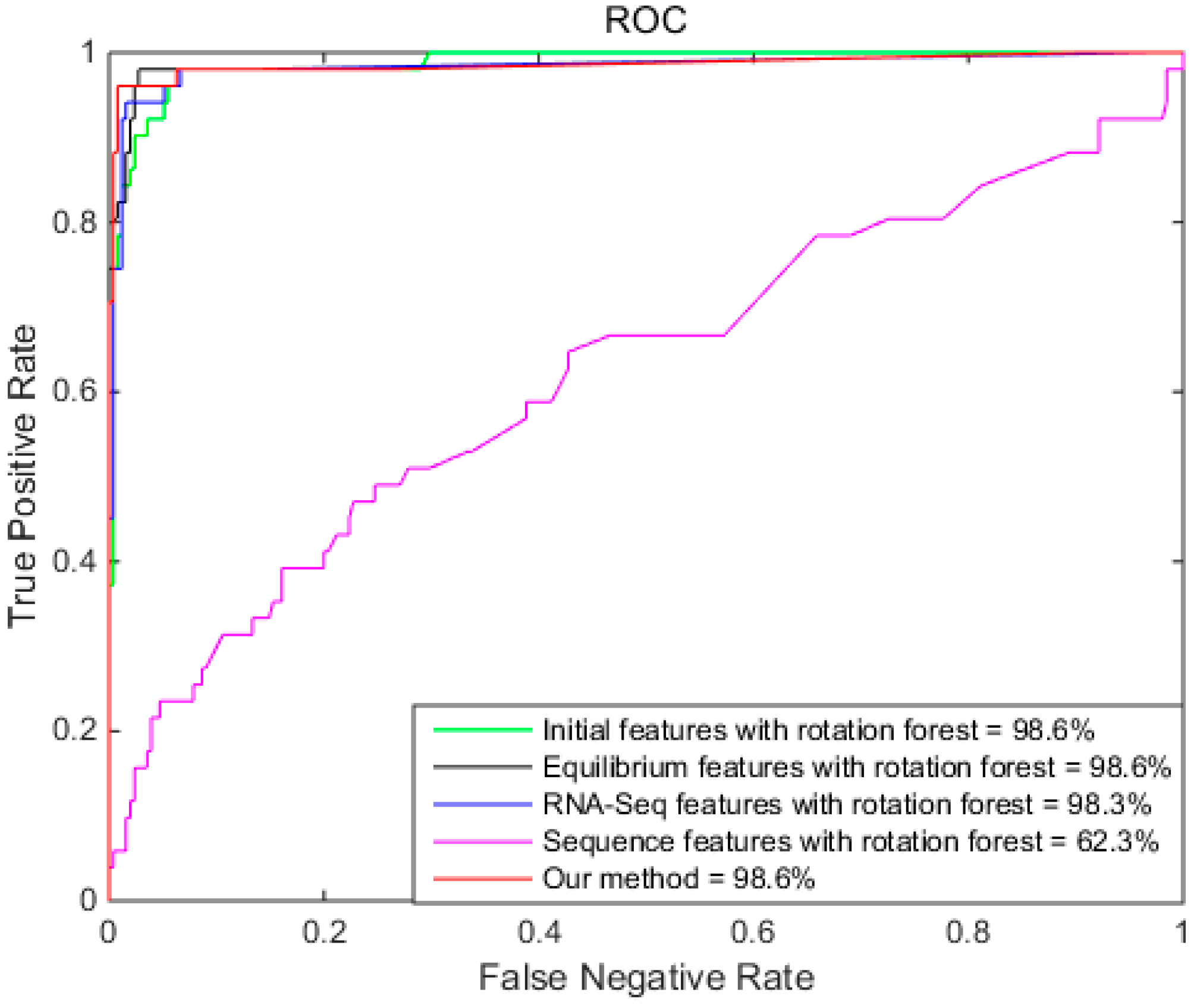
2.4. Comparison of Different Features with Rotation Forest
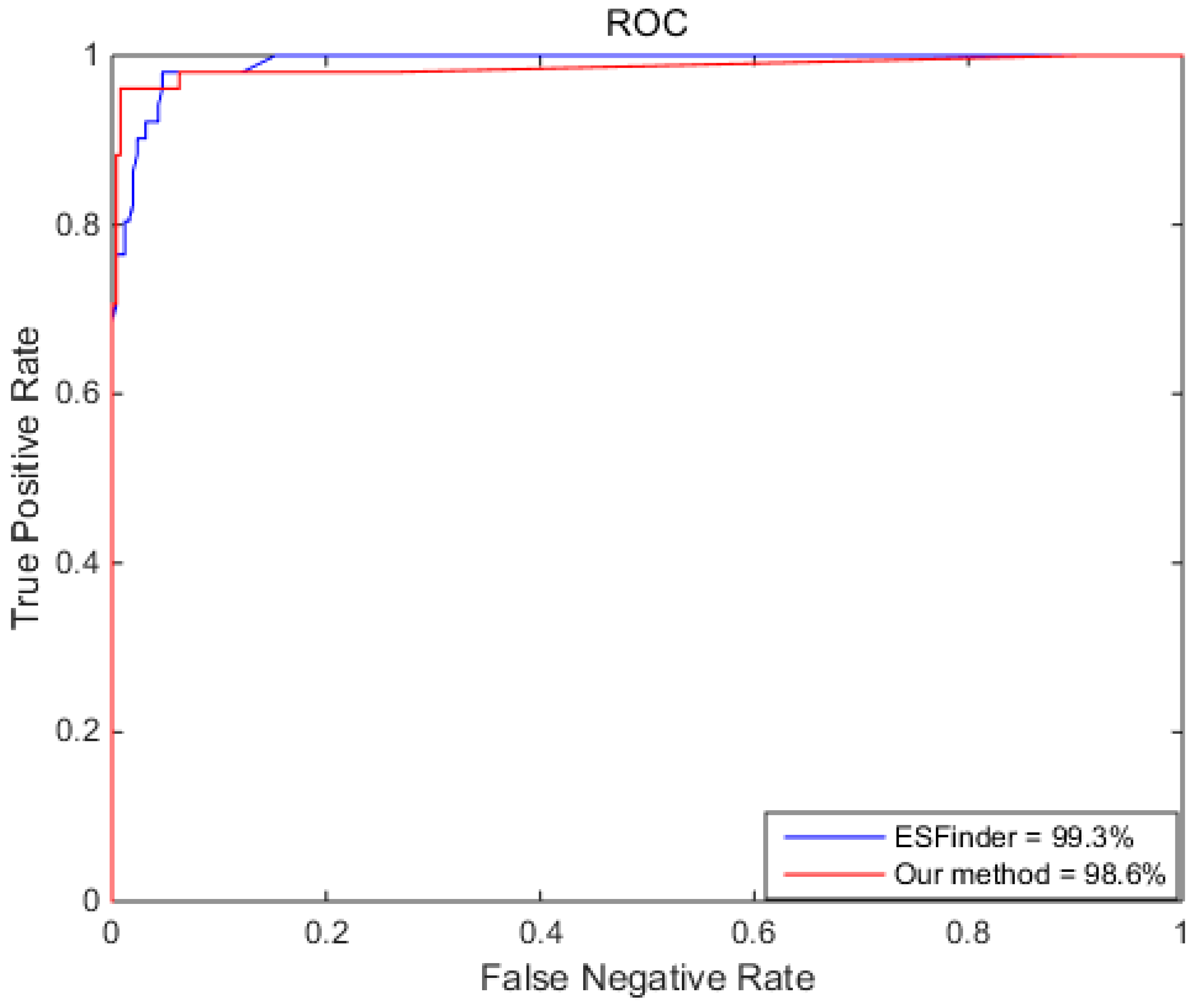
2.5. Comparing RotaF-RSES with Other Methods
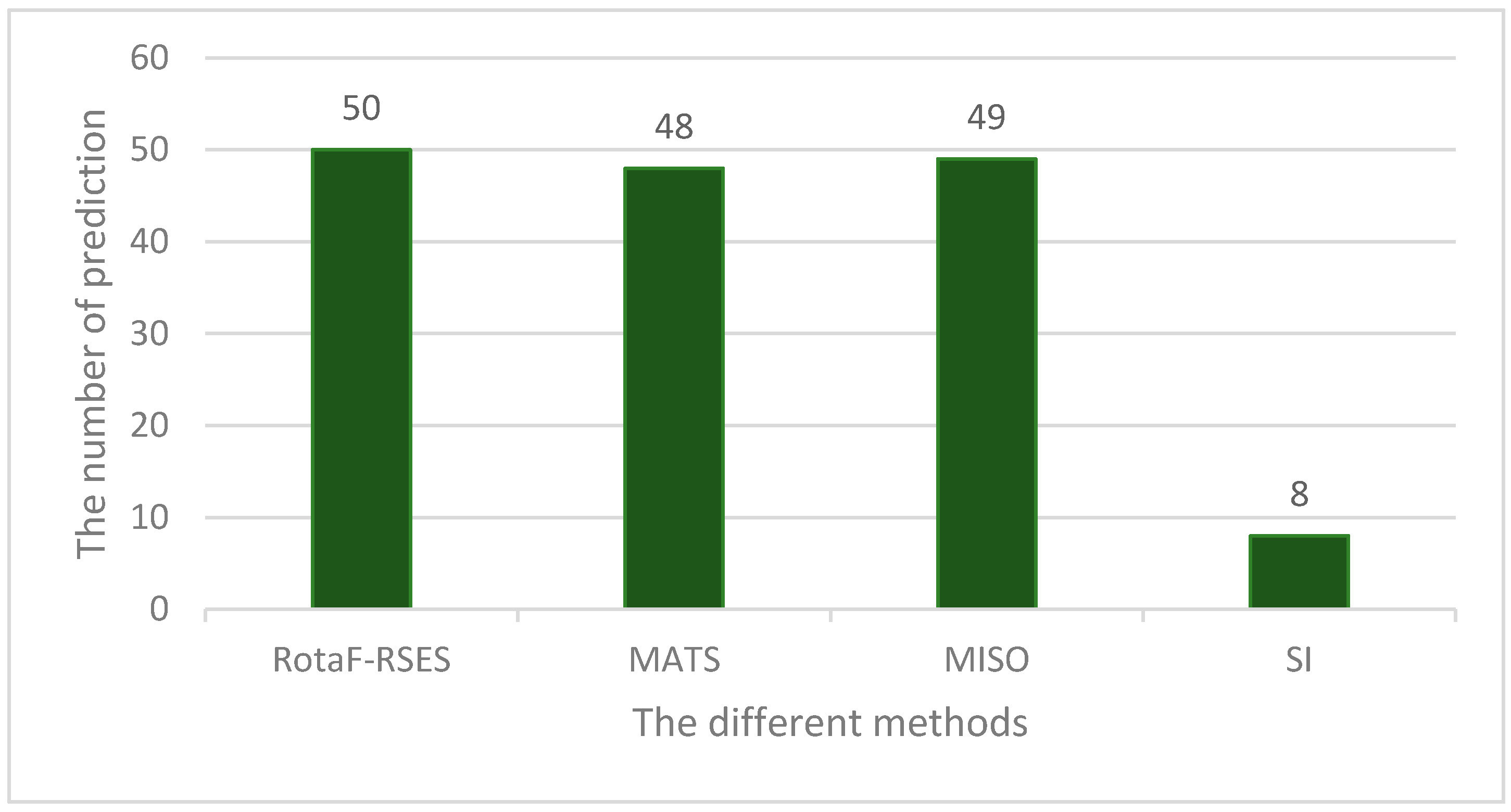
2.6. RotaF-RSES Prediction on Independent Test Data
3. Discussion
4. Materials and Methods
4.1. Dataset
4.2. Feature Extraction
4.3. Sequence Features
4.4. RNA-Seq Features
4.5. Rotation Forest
- (1)
- We split D randomly into K subsets. The feature number of each subset was , which obtains k instances subsets, based on feature subsets .
- (2)
- Using PCA to obtain feature conversion, for example, subset Si and obtain M feature vector, and M’ feature vectors (non-zero) were selected to form a feature vector matrix .
- (3)
- Step (2) is repeated and the result is inputted into a matrix R. We found these features and their initial position in S according to the feature vector in R. Each feature vector was rearranged according to the initial position to obtain a new R*, and a new sample was set up.
- (4)
- Multiple base classifiers were obtained using the above procedure. The final result was determined by the maximum class confidence.
4.6. Performance Evaluation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Ann. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Ast, G. How did alternative splicing evolve? Nat. Rev. Genet. 2004, 5, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Shemesh, R.; Cohen, Y.; Basechess, O.; Ast, G.; Shamir, R. A Non-EST-Based Method for Exon-Skipping Prediction. Genome Res. 2004, 14, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.W.; Nostrand, E.L.V.; Liang, T.Y. Correction: Discovery and Analysis of Evolutionarily Conserved Intronic Splicing Regulatory Elements. PLoS Genet. 2007, 3, e122. [Google Scholar] [CrossRef]
- Chen, W.; Lin, H.; Feng, P.; Wang, J. Exon skipping event prediction based on histone modifications. Interdiscip. Sci. Comput. Life Sci. 2014, 6, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Dror, G.; Sorek, R.; Shamir, R. Accurate identification of alternatively spliced exons using support vector machine. Bioinformatics 2005, 21, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Richard, H.; Schulz, M.H.; Sultan, M.; Nürnberger, A.; Schrinner, S.; Balzereit, D.; Dagand, E.; Rasche, A.; Lehrach, H.; Vingron, M.; et al. Prediction of alternative isoforms from exon expression levels in RNA-Seq experiments. Nucleic Acids Res. 2010, 38, e112. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Huang, J.; Dittmar, K.A.; Lu, Z.X.; Zhou, Q.; Carstens, R.P.; Xing, Y. MATS: A Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012, 40, e61. [Google Scholar] [CrossRef] [PubMed]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Alamancos, G.P. Methods to Study Splicing from High-Throughput RNA Sequencing Data. Methods Mol. Biol. 2013, 1126, 357–397. [Google Scholar]
- Feng, H.; Qin, Z.; Zhang, X. Opportunities and methods for studying alternative splicing in cancer with RNA-Seq. Cancer Lett. 2013, 340, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lin, L.; Sun, C.; Wang, X.; Wang, X. Combining local graph clustering and similarity measure for complex detection. In Proceedings of the International Conference on Biomedical Engineering and Informatics, Yantai, China, 16–18 October 2010; Volume 5, pp. 2188–2192. [Google Scholar]
- Bai, Y.; Ji, S.; Jiang, Q.; Wang, Y. Identification Exon Skipping Events From High-Throughput RNA Sequencing Data. IEEE Trans. Nanobiosci. 2015, 14, 562–569. [Google Scholar]
- Du, X.; Jing, A.; Hu, X. A novel feature extraction scheme for prediction of protein-protein interaction sites. Mol. Biosyst. 2015, 11, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Heckerman, D.; Wellman, M.P. Bayesian networks. Commun. ACM 1995, 38, 27–30. [Google Scholar] [CrossRef]
- Titterington, D.M.; Gelpke, G.J. Comparison of discrimination techniques applied to a complex data set of head injured patients. J. R. Stat. Soc. 1981, 144, 145–175. [Google Scholar] [CrossRef]
- Segal, R.; Kothari, M.L.; Madnani, S. Radial basis function (RBF) network adaptive power system stabilizer. IEEE Trans. Power Syst. 2000, 15, 722–727. [Google Scholar] [CrossRef]
- Shalev-Shwartz, S.; Singer, Y.; Srebro, N. Pegasos: Primal Estimated sub-gradient Solver for SVM. Math. Program. 2011, 127, 20–24. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. ACM Sigkdd Explor. Newslett. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Ryan, M.C.; Cleland, J.; Kim, R.; Wong, W.C.; Weinstein, J.N. SpliceSeq: A resource for analysis and visualization of RNA-Seq data on alternative splicing and its functional impacts. Bioinformatics 2012, 28, 2385–2387. [Google Scholar] [CrossRef] [PubMed]
- Barash, Y.; Calarco, J.A.; Gao, W.; Pan, Q.; Wang, X.; Shai, O.; Blencowe, B.J.; Frey, B.J. Deciphering the splicing code. Nature 2010, 465, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [PubMed]
- Iñiguez, L.P.; Ramírez, M.; Barbazuk, W.B.; Hernández, G. Identification and analysis of alternative splicing events in Phaseolus vulgaris and Glycine max. BMC Genom. 2017, 18, 650. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Bollineni, R.C.; Strozynski, M.; Koehler, C.J.; Thiede, B. Identification of alternative splice variants using unique tryptic peptide sequences for database searches. J. Proteome Res. 2017, 16, 2571–2578. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Gao, X.; Wang, J.; Gao, C.; Li, X.; Li, X.; Gong, X.; Zeng, X. Transcriptome Sequencing to Identify Transcription Factor Regulatory Network and Alternative Splicing in Endothelial Cells Under VEGF Stimulation. J. Mol. Neurosci. 2016, 58, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.G.; Xia, J.F.; Li, X.L.; Huang, D.S. Predicting protein–protein interactions from sequence using correlation coefficient and high-quality interaction dataset. Amino Acids 2010, 38, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Shen, H.B. RNA-protein binding motifs mining with a new hybrid deep learning based cross-domain knowledge integration approach. BMC Bioinform. 2017, 18, 136. [Google Scholar] [CrossRef] [PubMed]
- Li, H. The Sequence Alignment-Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Caragea, D.; Brown, S. Exploring Alternative Splicing Features Using Support Vector Machines. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine, Philadelphia, PA, USA, 3–5 November 2008; Volume 5, pp. 231–238. [Google Scholar]
- Busch, A.; Hertel, K.J. Splicing predictions reliably classify different types of alternative splicing. RNA A Publ. RNA Soc. 2015, 21, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goren, A.; Ast, G. Alternative splicing and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 5, 14–26. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Kuncheva, L.I.; Alonso, C.J. Rotation Forest: A New Classifier Ensemble Method. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Akama, Y. Realizability interpretation of PA by iterated limiting PCA. Math. Struct. Comput. Sci. 2014, 24, e240603. [Google Scholar] [CrossRef]
- Zweig, M.H.; Campbell, G. Receiver-operating characteristic (ROC) plots: A fundamental evaluation tool in clinical medicine. Clin. Chem. 1993, 39, 561–577. [Google Scholar] [PubMed]
- Du, X.; Cheng, J.; Zheng, T.; Duan, Z.; Qian, F. A Novel Feature Extraction Scheme with Ensemble Coding for Protein–Protein Interaction Prediction. Int. J. Mol. Sci. 2014, 15, 12731–12749. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Accuracy | Specificity | Sensitivity | AUC |
|---|---|---|---|---|
| Initial | 96.4% | 98.0% | 88.5% | 99.1% |
| Equilibrium | 96.2% | 98.0% | 86.8% | 99.3% |
| RNA-Seq | 96.1% | 97.3% | 90.2% | 99.3% |
| Sequence | 82.7% | 95.7% | 17.6% | 62.8% |
| RS | 96.7% | 97.6% | 92.2% | 99.2% |
| Algorithm | Accuracy | Specificity | Sensitivity | AUC |
|---|---|---|---|---|
| Random Forest | 96.7% | 97.6% | 92.2% | 99.2% |
| Random Tree | 93.1% | 96.5% | 76.5% | 86.5% |
| Naïve Bayes | 51.9% | 44.7% | 88.2% | 85.7% |
| Bayes Net | 94.1% | 94.5% | 92.2% | 97.7% |
| Naïve Bayes Simple | 84.2% | 82.8% | 88.0% | 89.1% |
| Multilayer Perceptron | 93.1% | 97.7% | 70.6% | 96.0% |
| RBF network | 86.9% | 99.6% | 23.5% | 88.4% |
| J48 | 93.1% | 96.5% | 76.5% | 91.7% |
| SVM | 83.7% | 100% | 2% | 51.0% |
| Our Method | 98.4% | 99.2% | 94.1% | 98.6% |
| Features | Accuracy | Specificity | Sensitivity | AUC |
|---|---|---|---|---|
| Initial | 95.8% (96.4%)RF | 98.0% (98.0%)RF | 84.3% (88.5%)RF | 98.6% (99.1%)RF |
| Equilibrium | 96.7% (96.2%)RF | 98.0% (98.0%)RF | 90.2% (86.8%)RF | 98.6% (99.3%)RF |
| RNA-Seq | 97.4% (96.1%)RF | 98.0% (97.3%)RF | 92.1% (90.2%)RF | 98.3% (99.3%)RF |
| Sequence | 83.0% (82.7%)RF | 98.4% (95.7%)RF | 7.9% (17.6%)RF | 62.3% (62.8%)RF |
| RS | 98.4% (96.7%)RF | 99.2% (97.6%)RF | 94.1% (92.2%)RF | 98.6% (99.2%)RF |
| Method | Accuracy | Specificity | Sensitivity | AUC |
|---|---|---|---|---|
| ESFinder | 96.2% | 98.0% | 86.8% | 99.3% |
| Our method | 98.4% | 99.2% | 94.1% | 98.6% |
| Different Methods | Our Method | ESFinder | MATS | MISO | SI |
|---|---|---|---|---|---|
| Correct Predictions | 1910 | 1977 | 91 | 140 | 179 |
| Feature | The Description of These Features |
|---|---|
| Read counts on exons | |
| Reads counts on the upstream intron | |
| Reads counts on the downstream intron | |
| Reads counts supporting the inclusive exon | |
| Reads counts supporting the exclusive exon | |
| Reads counts on the corresponding gene |
| Skeletal Muscle (S) | Brain (B) |
|---|---|
| Feature | The Definition of These Features |
|---|---|
| Skeletal Muscle (S) | Brain (B) |
|---|---|
| Skeletal Muscle (S) | Brain (B) | Divergence |
|---|---|---|
| SP | BP |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, X.; Hu, C.; Yao, Y.; Sun, S.; Zhang, Y. Analysis and Prediction of Exon Skipping Events from RNA-Seq with Sequence Information Using Rotation Forest. Int. J. Mol. Sci. 2017, 18, 2691. https://doi.org/10.3390/ijms18122691
Du X, Hu C, Yao Y, Sun S, Zhang Y. Analysis and Prediction of Exon Skipping Events from RNA-Seq with Sequence Information Using Rotation Forest. International Journal of Molecular Sciences. 2017; 18(12):2691. https://doi.org/10.3390/ijms18122691
Chicago/Turabian StyleDu, Xiuquan, Changlin Hu, Yu Yao, Shiwei Sun, and Yanping Zhang. 2017. "Analysis and Prediction of Exon Skipping Events from RNA-Seq with Sequence Information Using Rotation Forest" International Journal of Molecular Sciences 18, no. 12: 2691. https://doi.org/10.3390/ijms18122691
APA StyleDu, X., Hu, C., Yao, Y., Sun, S., & Zhang, Y. (2017). Analysis and Prediction of Exon Skipping Events from RNA-Seq with Sequence Information Using Rotation Forest. International Journal of Molecular Sciences, 18(12), 2691. https://doi.org/10.3390/ijms18122691