Arachidonic Acid Metabolite as a Novel Therapeutic Target in Breast Cancer Metastasis



Abstract
1. Introduction
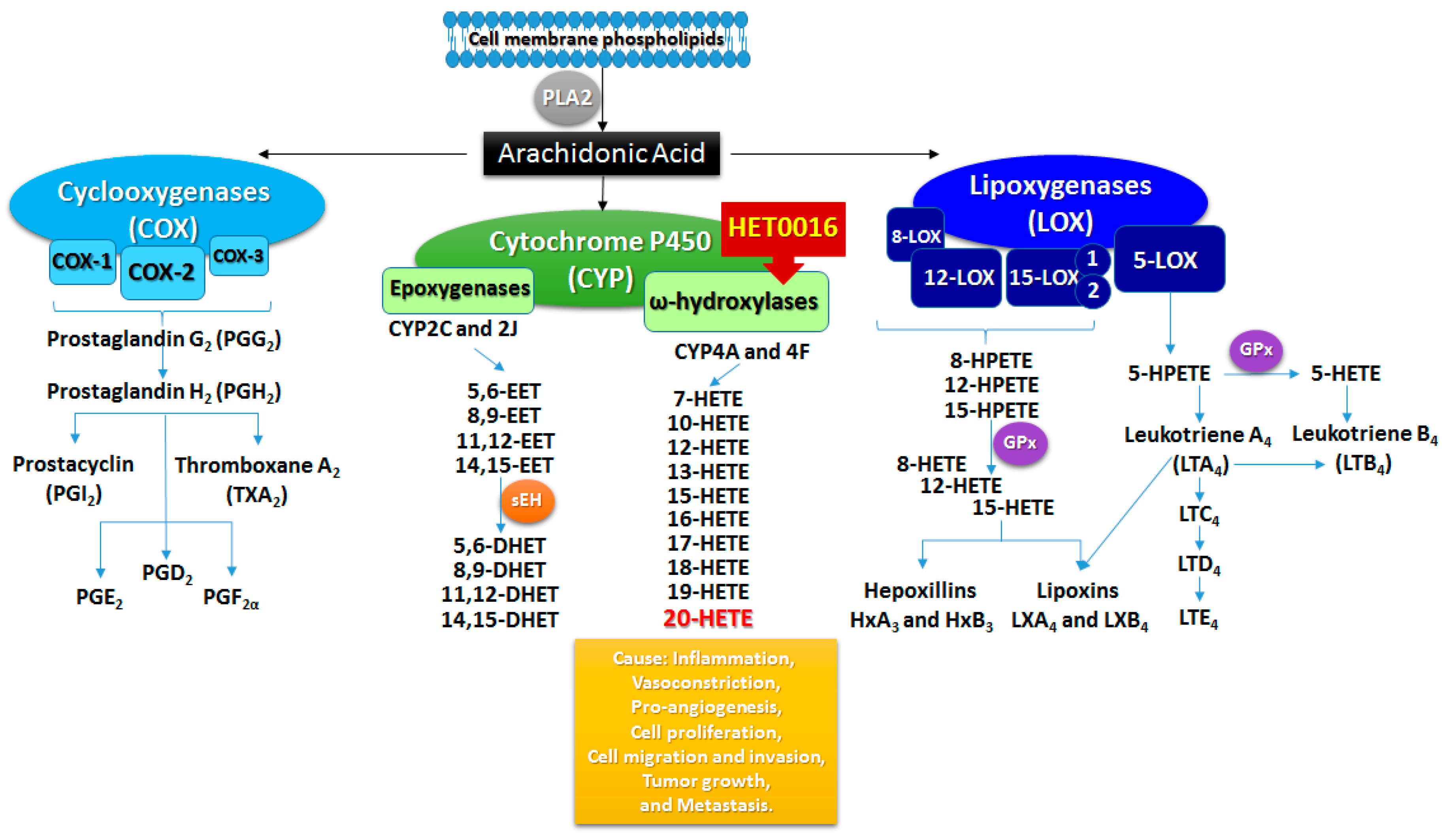
2. Arachidonic Acid Metabolism
3. Cytochrome P450 Mechanisms in Obesity and Breast Cancer
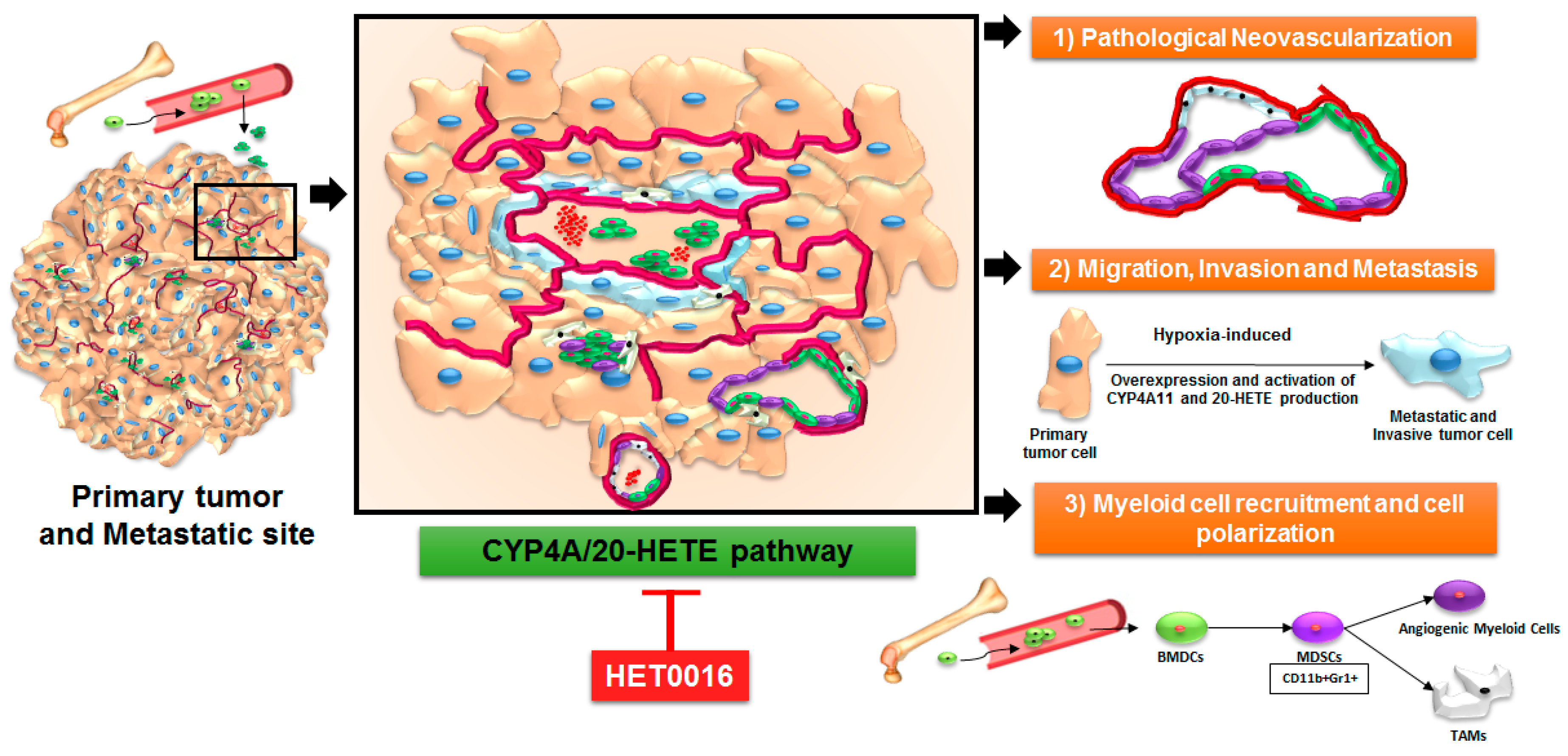
4. Arachidonic Acid Pathway and 20-HETE in Primary Tumors and Metastasis
5. Role of 20-HETE in Stromal Cells and Tumor Cells
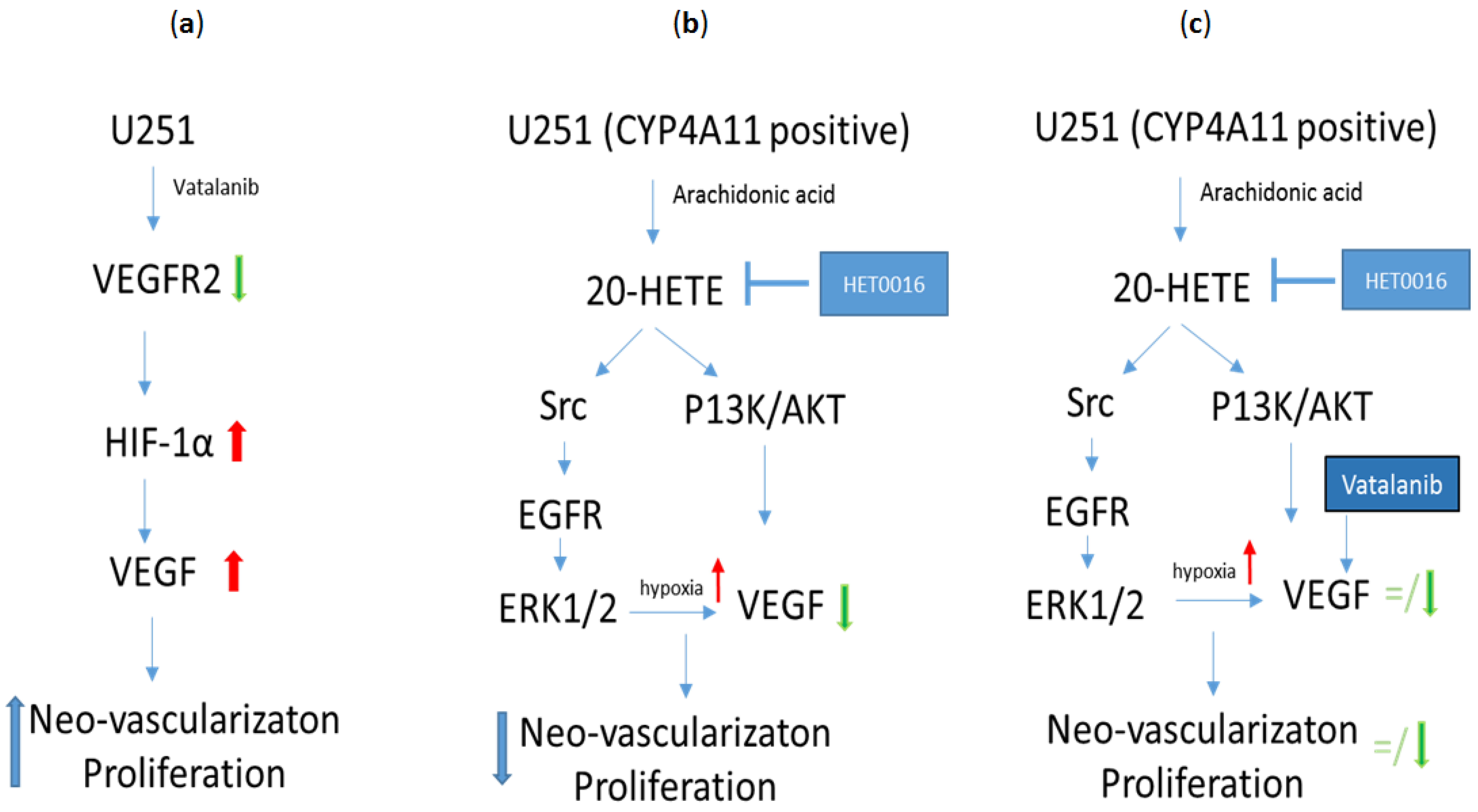
6. HET0016 as a Novel Therapeutic Agent in Treatment of Metastasis
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 20-HETE | 20-Hydroxy-eicosatetraenoic acid |
| 4T1 | Triple negative metastatic murine breast cancer cell line |
| ALX/FPR2 | Lipoxin A4 receptor/formyl peptide receptor |
| AA | Arachidonic acid |
| AAT | Antiangiogenic therapy |
| AKT | Protein kinase B |
| AMPK | 5′ AMP-activated protein kinase |
| BC | Breast cancer |
| BM | Brain metastasis |
| BSO | Buthionine sulfoximine |
| BT-474 | Breast cancer luminal B subtype cell line |
| COX | Cyclooxygenase enzyme |
| CXCR4 | C-X-C chemokine receptor type 4 also known as fusion or CD184; |
| CYP | Cytochrome P450 |
| CYP4A | Cytochrome P450, family 4, subfamily A |
| CYP4F | Cytochrome P450, family 4, subfamily F |
| DHETs | Dihydroxy-eicosatrienoic acids |
| EES | Extravascular extracellular space |
| EETs | Epoxy-eicosatrienoic acids |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| sEH | Epoxide hydrolase |
| EPCs | Endothelial progenitor cells |
| ER | Estrogen receptor |
| ERK | Extracellular signal-regulated kinases |
| Fas | Fas ligand or CD95 ligand |
| FGF-2 | Basic fibroblast growth factor 2 |
| FLA-16 | Novel flavonoid |
| GBM | Glioblastoma |
| GBM811 | Glioblastoma-derived from patient |
| GL261 | Murine glioblastoma cell line |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GSH | Glutathione |
| GPx | Glutathione peroxidase |
| HER2 | Human epidermal growth factor receptor2 |
| HET0016 | N-hydroxy-N′-(4-butyl-2 methyl phenyl) formamidine |
| HF2303 | Glioblastoma-derived from patient |
| HIF-1α | Hypoxia-inducible factor 1 α |
| HO | Hepoxillin |
| HPβCD | 2-Hydroxypropyl β-cyclodextrin |
| HPETE | Hydroperoxy-eicosatetraenoic acid |
| HUVEC | Human umbilical vein endothelial cells |
| IL | Interleukin |
| IV | Intravenous |
| Ki67 | Proliferation marker |
| Ktrans | Forward permeability transfer constant |
| LOX | Lipoxygenase |
| LT | Leukotriene |
| LX | Lipoxin |
| MAPK | Mitogen-Activated Protein Kinase |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| MDA-MB-231 | Triple negative metastatic human breast cancer cell line |
| MDSCs | Myeloid-derived suppressor cells |
| MMPs | Matrix metalloproteinases |
| MRI | Magnetic resonance imaging |
| MVD | Microvessel density |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| eNOS | Endothelial nitric oxide synthase |
| PDX | Patient-derived xenograft |
| PET | Polyethylene terephthalate |
| PG | Prostaglandin |
| PI3K | Phosphatidylinositol-3-kinases |
| PLA2 | Phospholipase A2 |
| PPARs | Peroxisome proliferator-activated receptors |
| PR | Progesterone receptor |
| ROS | Reactive oxygen species |
| SCF | Stem cell factor |
| SDF-1α | Stromal cell-derived factor 1 α |
| STAT1 | Signal transducer and activator of transcription 1 |
| T2DM | Type 2 diabetes mellitus |
| T47D | Breast cancer luminal A subtype cell line |
| TAMs | Tumor-associated macrophages |
| TGF-β | Transforming growth factor β 1 |
| Tie-2 | Transmembrane tyrosine-protein kinase receptor |
| TME | Tumor microenvironment |
| TMZ | Temozolomide |
| TNBC | Triple-negative breast cancer |
| TNFα | Tumor necrosis factor α |
| U251 | Human glioblastoma cell line |
| ve | extracellular space or interstitial volume |
| VE-cadherin | Vascular endothelial cadherin |
| VEGF | Vascular endothelial growth factor |
| VLA-4 | Very late antigen-4 |
| vp | Blood plasma pool |
| α-SMA | α-smooth muscle actin |
References
- Flemban, A.; Qualtrough, D. The potential role of hedgehog signaling in the luminal/basal phenotype of breast epithelia and in breast cancer invasion and metastasis. Cancers 2015, 7, 1863–1884. [Google Scholar] [CrossRef] [PubMed]
- Caan, B.J.; Sweeney, C.; Habel, L.A.; Kwan, M.L.; Kroenke, C.H.; Weltzien, E.K.; Quesenberry, C.P., Jr.; Castillo, A.; Factor, R.E.; Kushi, L.H.; et al. Intrinsic subtypes from the pam50 gene expression assay in a population-based breast cancer survivor cohort: Prognostication of short- and long-term outcomes. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cospons. Am. Soc. Prev. Oncol. 2014, 23, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Sestak, I.; Cuzick, J.; Dowsett, M.; Lopez-Knowles, E.; Filipits, M.; Dubsky, P.; Cowens, J.W.; Ferree, S.; Schaper, C.; Fesl, C.; et al. Prediction of late distant recurrence after 5 years of endocrine treatment: A combined analysis of patients from the austrian breast and colorectal cancer study group 8 and arimidex, tamoxifen alone or in combination randomized trials using the pam50 risk of recurrence score. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 916–922. [Google Scholar]
- Tamimi, R.M.; Baer, H.J.; Marotti, J.; Galan, M.; Galaburda, L.; Fu, Y.; Deitz, A.C.; Connolly, J.L.; Schnitt, S.J.; Colditz, G.A.; et al. Comparison of molecular phenotypes of ductal carcinoma in situ and invasive breast cancer. Breast Cancer Res. 2008, 10, R67. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.E.; Warwick, J.; Carpenter, R.; Bowen, R.L.; Duffy, S.W.; Jones, J.L. Molecular subtyping of dcis: Heterogeneity of breast cancer reflected in pre-invasive disease. Br. J. Cancer 2011, 104, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Oh, D.S.; Wessels, L.; Weigelt, B.; Nuyten, D.S.; Nobel, A.B.; van’t Veer, L.J.; Perou, C.M. Concordance among gene-expression-based predictors for breast cancer. N. Engl. J. Med. 2006, 355, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Society, A.C. Breast Cancer Facts & Figures 2015–2016; American Cancer Society, Inc.: Atlanta, GA, USA, 2015. [Google Scholar]
- Howlader, N.; Altekruse, S.F.; Li, C.I.; Chen, V.W.; Clarke, C.A.; Ries, L.A.; Cronin, K.A. US incidence of breast cancer subtypes defined by joint hormone receptor and her2 status. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Arvold, N.D.; Taghian, A.G.; Niemierko, A.; Abi Raad, R.F.; Sreedhara, M.; Nguyen, P.L.; Bellon, J.R.; Wong, J.S.; Smith, B.L.; Harris, J.R. Age, breast cancer subtype approximation, and local recurrence after breast-conserving therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 3885–3891. [Google Scholar] [CrossRef] [PubMed]
- Voduc, K.D.; Cheang, M.C.; Tyldesley, S.; Gelmon, K.; Nielsen, T.O.; Kennecke, H. Breast cancer subtypes and the risk of local and regional relapse. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Metzger-Filho, O.; Sun, Z.; Viale, G.; Price, K.N.; Crivellari, D.; Snyder, R.D.; Gelber, R.D.; Castiglione-Gertsch, M.; Coates, A.S.; Goldhirsch, A.; et al. Patterns of recurrence and outcome according to breast cancer subtypes in lymph node-negative disease: Results from international breast cancer study group trials viii and ix. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 3083–3090. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Toft, D.J.; Cryns, V.L. Minireview: Basal-like breast cancer: From molecular profiles to targeted therapies. Mol. Endocrinol. 2011, 25, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, R.; Perou, C.M.; Symmans, W.F.; Ibrahim, N.; Cristofanilli, M.; Anderson, K.; Hess, K.R.; Stec, J.; Ayers, M.; Wagner, P.; et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Cancer Res. 2005, 11, 5678–5685. [Google Scholar] [CrossRef] [PubMed]
- Sihto, H.; Lundin, J.; Lundin, M.; Lehtimaki, T.; Ristimaki, A.; Holli, K.; Sailas, L.; Kataja, V.; Turpeenniemi-Hujanen, T.; Isola, J.; et al. Breast cancer biological subtypes and protein expression predict for the preferential distant metastasis sites: A nationwide cohort study. Breast Cancer Res. 2011, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Yücel, B.; Bahar, S.; Kaçan, T.; Şeker, M.; Celasun, M. Importance of metastasis site in survival of patients with breast cancer. Austin J. Med. Oncol. 2014, 1, 1–7. [Google Scholar]
- Kimbung, S.; Loman, N.; Hedenfalk, I. Clinical and molecular complexity of breast cancer metastases. Semin. Cancer Biol. 2015, 35, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Ruger, R. Molecular basis of lung tropism of metastasis. Cancer Genom. Proteom. 2016, 13, 129–139. [Google Scholar]
- Largillier, R.; Ferrero, J.M.; Doyen, J.; Barriere, J.; Namer, M.; Mari, V.; Courdi, A.; Hannoun-Levi, J.M.; Ettore, F.; Birtwisle-Peyrottes, I.; et al. Prognostic factors in 1,038 women with metastatic breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 2008, 19, 2012–2019. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Ren, Z.; Hameed, O.; Chanda, D.; Morgan, C.J.; Siegal, G.P.; Wei, S. Breast cancer subtypes predispose the site of distant metastases. Am. J. Clin. Pathol. 2015, 143, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Rostami, R.; Mittal, S.; Rostami, P.; Tavassoli, F.; Jabbari, B. Brain metastasis in breast cancer: A comprehensive literature review. J. Neuro-Oncol. 2016, 127, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Witzel, I.; Oliveira-Ferrer, L.; Pantel, K.; Muller, V.; Wikman, H. Breast cancer brain metastases: Biology and new clinical perspectives. Breast Cancer Res. 2016, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.; Foekens, J.A.; Martens, J.W. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Brooks, M.; Wicha, M.S. Epithelial-mesenchymal plasticity of breast cancer stem cells: Implications for metastasis and therapeutic resistance. Curr. Pharm. Des. 2015, 21, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Di, G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin. J. Cancer Res. 2017, 29, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Duechler, M.; Peczek, L.; Zuk, K.; Zalesna, I.; Jeziorski, A.; Czyz, M. The heterogeneous immune microenvironment in breast cancer is affected by hypoxia-related genes. Immunobiology 2014, 219, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Shou, D.; Wen, L.; Song, Z.; Yin, J.; Sun, Q.; Gong, W. Suppressive role of myeloid-derived suppressor cells (mdscs) in the microenvironment of breast cancer and targeted immunotherapies. Oncotarget 2016, 7, 64505–64511. [Google Scholar] [CrossRef] [PubMed]
- Filipazzi, P.; Valenti, R.; Huber, V.; Pilla, L.; Canese, P.; Iero, M.; Castelli, C.; Mariani, L.; Parmiani, G.; Rivoltini, L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Ostrand-Rosenberg, S. Reduction of myeloid-derived suppressor cells and induction of m1 macrophages facilitate the rejection of established metastatic disease. J. Immunol. 2005, 174, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.A.; Li, Y.; Chen, S.H. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary t cell activation induced through cd3/cd28 costimulation. J. Immunol. 2000, 165, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Kapoor, V.; Jassar, A.S.; Kaiser, L.R.; Albelda, S.M. Gemcitabine selectively eliminates splenic gr-1+/cd11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Solito, S.; Falisi, E.; Diaz-Montero, C.M.; Doni, A.; Pinton, L.; Rosato, A.; Francescato, S.; Basso, G.; Zanovello, P.; Onicescu, G.; et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood 2011, 118, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Waight, J.D.; Netherby, C.; Hensen, M.L.; Miller, A.; Hu, Q.; Liu, S.; Bogner, P.N.; Farren, M.R.; Lee, K.P.; Liu, K.; et al. Myeloid-derived suppressor cell development is regulated by a stat/irf-8 axis. J. Clin. Investig. 2013, 123, 4464–4478. [Google Scholar] [CrossRef] [PubMed]
- Bergenfelz, C.; Larsson, A.M.; von Stedingk, K.; Gruvberger-Saal, S.; Aaltonen, K.; Jansson, S.; Jernstrom, H.; Janols, H.; Wullt, M.; Bredberg, A.; et al. Systemic monocytic-mdscs are generated from monocytes and correlate with disease progression in breast cancer patients. PLoS ONE 2015, 10, e0127028. [Google Scholar] [CrossRef] [PubMed]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A.; et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef] [PubMed]
- Arvelo, F.; Sojo, F.; Cotte, C. Tumour progression and metastasis. Ecancermedicalscience 2016, 10, 617. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Escribese, M.M.; Casas, M.; Corbi, A.L. Influence of low oxygen tensions on macrophage polarization. Immunobiology 2012, 217, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. Ccl2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Arbab, A.S. Myeloid cell signatures in tumor microenvironment predicts therapeutic response in cancer. OncoTargets Ther. 2016, 9, 1047–1055. [Google Scholar]
- Borin, T.F.; Shankar, A.; Angara, K.; Rashid, M.H.; Jain, M.; Iskander, A.; Ara, R.; Lebedyeva, I.; Korkaya, H.; Achyut, B.R.; et al. HET0016 decreases lung metastasis from breast cancer in immune-competent mouse model. PLoS ONE 2017, 12, e0178830. [Google Scholar] [CrossRef] [PubMed]
- Borin, T.F.; Zuccari, D.A.; Jardim-Perassi, B.V.; Ferreira, L.C.; Iskander, A.S.; Varma, N.R.; Shankar, A.; Guo, A.M.; Scicli, G.; Arbab, A.S. HET0016, a selective inhibitor of 20-hete synthesis, decreases pro-angiogenic factors and inhibits growth of triple negative breast cancer in mice. PLoS ONE 2014, 9, e116247. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Borin, T.F.; Iskander, A.; Varma, N.R.; Achyut, B.R.; Jain, M.; Mikkelsen, T.; Guo, A.M.; Chwang, W.B.; Ewing, J.R.; et al. Combination of vatalanib and a 20-hete synthesis inhibitor results in decreased tumor growth in an animal model of human glioma. OncoTargets Ther. 2016, 9, 1205–1219. [Google Scholar]
- Bosisio, E.; Galli, C.; Galli, G.; Nicosia, S.; Spagnuolo, C.; Tosi, L. Correlation between release of free arachidonic acid and prostaglandin formation in brain cortex and cerebellum. Prostaglandins 1976, 11, 773–781. [Google Scholar] [CrossRef]
- Bergstroem, S.; Danielsson, H.; Samuelsson, B. The enzymatic formation of prostaglandin e2 from arachidonic acid prostaglandins and related factors 32. Biochim. Biophys. Acta 1964, 90, 207–210. [Google Scholar] [CrossRef]
- Rahman, M.; Wright, J.T., Jr.; Douglas, J.G. The role of the cytochrome p450-dependent metabolites of arachidonic acid in blood pressure regulation and renal function: A review. Am. J. Hypertens. 1997, 10, 356–365. [Google Scholar] [CrossRef]
- Bazinet, R.P.; Laye, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Kroetz, D.L.; Xu, F. Regulation and inhibition of arachidonic acid omega-hydroxylases and 20-hete formation. Ann. Rev. Pharmacol. Toxicol. 2005, 45, 413–438. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.F.; Hao, Y.H.; Liu, M.Z.; Yue, J.; Ni, J.; Yuan, B.F.; Feng, Y.Q. Analysis of cytochrome p450 metabolites of arachidonic acid by stable isotope probe labeling coupled with ultra high-performance liquid chromatography/mass spectrometry. J. Chromatogr. A 2015, 1410, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Roman, R.J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 2002, 82, 131–185. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. The factor in edhf: Cytochrome p450 derived lipid mediators and vascular signaling. Vasc. Pharmacol. 2016, 86, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Qi, G.; Han, Y.; Shen, X.; Yao, F.; Xuan, C.; Gu, Y.; Qian, S.Y.; Zeng, Q.; O’Rourke, S.T.; et al. 20-hydroxyeicosatetraenoic acid is a key mediator of angiotensin ii-induced apoptosis in cardiac myocytes. J. Cardiovasc. Pharmacol. 2015, 66, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, M.; Yoshimura, R. Arachidonic acid pathway: A molecular target in human testicular cancer (review). Mol. Med. Rep. 2009, 2, 527–531. [Google Scholar] [PubMed]
- Koontongkaew, S.; Leelahavanichkul, K. Arachidonic acid metabolism and its implication on head and neck cancer. In Head and Neck Cancer; Agulnik, D.M., Ed.; InTech: London, UK, 2012. [Google Scholar]
- Greene, E.R.; Huang, S.; Serhan, C.N.; Panigrahy, D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011, 96, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. Cox-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proc. Natl. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.M.; Schluesener, H.J.; Meyermann, R.; Serhan, C.N. Cox-3 the enzyme and the concept: Steps towards highly specialized pathways and precision therapeutics? Prostaglandins Leukot. Essent. Fat. Acids 2003, 69, 339–343. [Google Scholar] [CrossRef]
- Willoughby, D.A.; Moore, A.R.; Colville-Nash, P.R. Cox-1, cox-2, and cox-3 and the future treatment of chronic inflammatory disease. Lancet 2000, 355, 646–648. [Google Scholar] [CrossRef]
- Moore, G.Y.; Pidgeon, G.P. Cross-talk between cancer cells and the tumour microenvironment: The role of the 5-lipoxygenase pathway. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Z.; Hennig, R.; Adrian, T.E. Lipoxygenase and cyclooxygenase metabolism: New insights in treatment and chemoprevention of pancreatic cancer. Mol. Cancer 2003, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Larre, S.; Tran, N.; Fan, C.; Hamadeh, H.; Champigneulles, J.; Azzouzi, R.; Cussenot, O.; Mangin, P.; Olivier, J.L. Pge2 and ltb4 tissue levels in benign and cancerous prostates. Prostaglandins Other Lipid Mediat. 2008, 87, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Venerito, M.; Kuester, D.; Harms, C.; Schubert, D.; Wex, T.; Malfertheiner, P. Upregulation of leukotriene receptors in gastric cancer. Cancers 2011, 3, 3156–3168. [Google Scholar] [CrossRef] [PubMed]
- Hennig, R.; Ventura, J.; Segersvard, R.; Ward, E.; Ding, X.Z.; Rao, S.M.; Jovanovic, B.D.; Iwamura, T.; Talamonti, M.S.; Bell, R.H., Jr.; et al. Ly293111 improves efficacy of gemcitabine therapy on pancreatic cancer in a fluorescent orthotopic model in athymic mice. Neoplasia 2005, 7, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Adrian, T.E.; Hennig, R.; Friess, H.; Ding, X. The role of ppargamma receptors and leukotriene b(4) receptors in mediating the effects of ly293111 in pancreatic cancer. PPAR Res. 2008, 2008, 827096. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Honn, K.V. 12(s)-hete in cancer metastasis. Adv. Exp. Med. Biol. 1999, 447, 181–191. [Google Scholar] [PubMed]
- Steele, V.E.; Holmes, C.A.; Hawk, E.T.; Kopelovich, L.; Lubet, R.A.; Crowell, J.A.; Sigman, C.C.; Kelloff, G.J. Lipoxygenase inhibitors as potential cancer chemopreventives. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cospons. Am. Soc. Prev. Oncol. 1999, 8, 467–483. [Google Scholar]
- Wong, B.C.; Wang, W.P.; Cho, C.H.; Fan, X.M.; Lin, M.C.; Kung, H.F.; Lam, S.K. 12-lipoxygenase inhibition induced apoptosis in human gastric cancer cells. Carcinogenesis 2001, 22, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G.; Honn, K.V. Apoptosis of w256 carcinosarcoma cells of the monocytoid origin induced by ndga involves lipid peroxidation and depletion of gsh: Role of 12-lipoxygenase in regulating tumor cell survival. J. Cell. Physiol. 1997, 172, 155–170. [Google Scholar] [CrossRef]
- Nigam, S.; Patabhiraman, S.; Ciccoli, R.; Ishdorj, G.; Schwarz, K.; Petrucev, B.; Kuhn, H.; Haeggstrom, J.Z. The rat leukocyte-type 12-lipoxygenase exhibits an intrinsic hepoxilin a3 synthase activity. J. Biol. Chem. 2004, 279, 29023–29030. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, N.B.; Mohammed, A.; Rao, C.V. Role of lipoxins, resolvins, and other bioactive lipids in colon and pancreatic cancer. Cancer Metastasis Rev. 2011, 30, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, J.A.; Sharma-Walia, N. Lipoxins: Nature’s way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192. [Google Scholar] [PubMed]
- Pidgeon, G.P.; Lysaght, J.; Krishnamoorthy, S.; Reynolds, J.V.; O’Byrne, K.; Nie, D.; Honn, K.V. Lipoxygenase metabolism: Roles in tumor progression and survival. Cancer Metastasis Rev. 2007, 26, 503–524. [Google Scholar] [CrossRef] [PubMed]
- Curzio, M.; Esterbauer, H.; Poli, G.; Biasi, F.; Cecchini, G.; Di Mauro, C.; Cappello, N.; Dianzani, M.U. Possible role of aldehydic lipid peroxidation products as chemoattractants. Int. J. Tissue React. 1987, 9, 295–306. [Google Scholar] [PubMed]
- Wan, Y.Y.; Cai, Y.; Li, J.; Yuan, Q.; French, B.; Gonzalez, F.J.; French, S. Regulation of peroxisome proliferator activated receptor alpha-mediated pathways in alcohol fed cytochrome p450 2e1 deficient mice. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2001, 19, 117–130. [Google Scholar] [CrossRef]
- Robertson, G.; Leclercq, I.; Farrell, G.C. Nonalcoholic steatosis and steatohepatitis. Ii. Cytochrome p-450 enzymes and oxidative stress. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1135–G1139. [Google Scholar] [PubMed]
- Bell, L.C.; Guengerich, F.P. Oxidation kinetics of ethanol by human cytochrome p450 2e1. Rate-limiting product release accounts for effects of isotopic hydrogen substitution and cytochrome b5 on steady-state kinetics. J. Biol. Chem. 1997, 272, 29643–29651. [Google Scholar] [CrossRef] [PubMed]
- Gorsky, L.D.; Koop, D.R.; Coon, M.J. On the stoichiometry of the oxidase and monooxygenase reactions catalyzed by liver microsomal cytochrome p-450. Products of oxygen reduction. J. Biol. Chem. 1984, 259, 6812–6817. [Google Scholar] [PubMed]
- Ekstrom, G.; Ingelman-Sundberg, M. Rat liver microsomal nadph-supported oxidase activity and lipid peroxidation dependent on ethanol-inducible cytochrome p-450 (p-450iie1). Biochem. Pharmacol. 1989, 38, 1313–1319. [Google Scholar] [CrossRef]
- Wu, D.; Cederbaum, A.I. Removal of glutathione produces apoptosis and necrosis in hepg2 cells overexpressing cyp2e1. Alcohol. Clin. Exp. Res. 2001, 25, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Cederbaum, A.I. Cyp2e1 overexpression in hepg2 cells induces glutathione synthesis by transcriptional activation of gamma-glutamylcysteine synthetase. J. Biol. Chem. 2000, 275, 15563–15571. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Cederbaum, A.I. Induction of catalase, alpha, and microsomal glutathione s-transferase in cyp2e1 overexpressing hepg2 cells and protection against short-term oxidative stress. Hepatology 2001, 33, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Kaipainen, A.; Greene, E.R.; Huang, S. Cytochrome p450-derived eicosanoids: The neglected pathway in cancer. Cancer Metastasis Rev. 2010, 29, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes p450 in health and disease. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef] [PubMed]
- Nie, D.; Che, M.; Zacharek, A.; Qiao, Y.; Li, L.; Li, X.; Lamberti, M.; Tang, K.; Cai, Y.; Guo, Y.; et al. Differential expression of thromboxane synthase in prostate carcinoma: Role in tumor cell motility. Am. J. Pathol. 2004, 164, 429–439. [Google Scholar] [CrossRef]
- Medhora, M.; Dhanasekaran, A.; Gruenloh, S.K.; Dunn, L.K.; Gabrilovich, M.; Falck, J.R.; Harder, D.R.; Jacobs, E.R.; Pratt, P.F. Emerging mechanisms for growth and protection of the vasculature by cytochrome p450-derived products of arachidonic acid and other eicosanoids. Prostaglandins Other Lipid Mediat. 2007, 82, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Zhang, F.; Jiang, M.; Wang, M.H.; Zand, B.A.; Abraham, N.G.; Nasjletti, A.; Laniado-Schwartzman, M. Transfection and functional expression of cyp4a1 and cyp4a2 using bicistronic vectors in vascular cells and tissues. J. Pharmacol. Exp. Ther. 2004, 311, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Al-Shabrawey, M.; Wang, M.H. Cyclooxygenase- and cytochrome p450-derived eicosanoids in stroke. Prostaglandins Other Lipid Mediat. 2016, 122, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.; Wang, M.H.; Reddy, K.M.; Falck, J.R.; Schwartzman, M.L. Kinetic profile of the rat cyp4a isoforms: Arachidonic acid metabolism and isoform-specific inhibitors. Am. J. Physiol. 1999, 276, R1691–R1700. [Google Scholar] [PubMed]
- Muller, D.N.; Schmidt, C.; Barbosa-Sicard, E.; Wellner, M.; Gross, V.; Hercule, H.; Markovic, M.; Honeck, H.; Luft, F.C.; Schunck, W.H. Mouse cyp4a isoforms: Enzymatic properties, gender- and strain-specific expression, and role in renal 20-hydroxyeicosatetraenoic acid formation. Biochem. J. 2007, 403, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hoopes, S.L.; Garcia, V.; Edin, M.L.; Schwartzman, M.L.; Zeldin, D.C. Vascular actions of 20-hete. Prostaglandins Other Lipid Mediat. 2015, 120, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Joseph, G.; Zhang, F.F.; Nguyen, H.; Jiang, H.; Gotlinger, K.H.; Falck, J.R.; Yang, J.; Schwartzman, M.L.; Guo, A.M. 20-hete contributes to ischemia-induced angiogenesis. Vasc. Pharmacol. 2016, 83, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Joseph, G.; Shkolnik, B.; Ding, Y.; Zhang, F.F.; Gotlinger, K.; Falck, J.R.; Dakarapu, R.; Capdevila, J.H.; Bernstein, K.E.; et al. Angiotensin ii receptor blockade or deletion of vascular endothelial ace does not prevent vascular dysfunction and remodeling in 20-hete-dependent hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R71–R78. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Wang, M.H.; Miyata, N.; Laniado-Schwartzman, M. Cytochrome p450 4a isoform inhibitory profile of N-hydroxy-N′-(4-butyl-2-methylphenyl)-formamidine (het0016), a selective inhibitor of 20-hete synthesis. Biol. Pharm. Bull. 2005, 28, 1651–1654. [Google Scholar] [CrossRef] [PubMed]
- Muthalif, M.M.; Benter, I.F.; Karzoun, N.; Fatima, S.; Harper, J.; Uddin, M.R.; Malik, K.U. 20-hydroxyeicosatetraenoic acid mediates calcium/calmodulin-dependent protein kinase ii-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA 1998, 95, 12701–12706. [Google Scholar] [CrossRef] [PubMed]
- Kaduce, T.L.; Fang, X.; Harmon, S.D.; Oltman, C.L.; Dellsperger, K.C.; Teesch, L.M.; Gopal, V.R.; Falck, J.R.; Campbell, W.B.; Weintraub, N.L.; et al. 20-hydroxyeicosatetraenoic acid (20-hete) metabolism in coronary endothelial cells. J. Biol. Chem. 2004, 279, 2648–2656. [Google Scholar] [CrossRef] [PubMed]
- Schwartzman, M.L.; Falck, J.R.; Yadagiri, P.; Escalante, B. Metabolism of 20-hydroxyeicosatetraenoic acid by cyclooxygenase. Formation and identification of novel endothelium-dependent vasoconstrictor metabolites. J. Biol. Chem. 1989, 264, 11658–11662. [Google Scholar] [PubMed]
- Garcia, V.; Shkolnik, B.; Milhau, L.; Falck, J.R.; Schwartzman, M.L. 20-hete activates the transcription of angiotensin-converting enzyme via nuclear factor-kappab translocation and promoter binding. J. Pharmacol. Exp. Ther. 2016, 356, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Zahid, H.; Simpson, E.R.; Brown, K.A. Inflammation, dysregulated metabolism and aromatase in obesity and breast cancer. Curr. Opin. Pharmacol. 2016, 31, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Chen, D.; Moy, I.; Brooks, D.C.; Zhao, H. Aromatase, breast cancer and obesity: A complex interaction. Trends Endocrinol. Metab. 2012, 23, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Leonel, C.; Ferreira, L.C.; Borin, T.F.; Moschetta, M.G.; Freitas, G.S.; Haddad, M.R.; de Camargos Pinto Robles, J.A.; Aparecida Pires de Campos Zuccari, D. Inhibition of epithelial-mesenchymal transition in response to treatment with metformin and y27632 in breast cancer cell lines. Anticancer Agents Med. Chem. 2017, 17, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Hunger, N.I.; Docanto, M.; Simpson, E.R. Metformin inhibits aromatase expression in human breast adipose stromal cells via stimulation of amp-activated protein kinase. Breast Cancer Res. Treat. 2010, 123, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated fatty acids. J. Nutr. Metab. 2012, 2012, 539426. [Google Scholar] [CrossRef] [PubMed]
- Horrillo, R.; Gonzalez-Periz, A.; Martinez-Clemente, M.; Lopez-Parra, M.; Ferre, N.; Titos, E.; Moran-Salvador, E.; Deulofeu, R.; Arroyo, V.; Claria, J. 5-lipoxygenase activating protein signals adipose tissue inflammation and lipid dysfunction in experimental obesity. J. Immunol. 2010, 184, 3978–3987. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Wada, K.; Murata, Y.; Nakajima, A.; Yamashiro, T.; Kamisaki, Y. Critical role of leukotriene b4 receptor signaling in mouse 3t3-l1 preadipocyte differentiation. Lipids Health Dis. 2013, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Oh, D.Y.; Bandyopadhyay, G.; Lagakos, W.S.; Talukdar, S.; Osborn, O.; Johnson, A.; Chung, H.; Maris, M.; Ofrecio, J.M.; et al. Ltb4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med. 2015, 21, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Curat, C.A.; Miranville, A.; Sengenes, C.; Diehl, M.; Tonus, C.; Busse, R.; Bouloumie, A. From blood monocytes to adipose tissue-resident macrophages: Induction of diapedesis by human mature adipocytes. Diabetes 2004, 53, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [PubMed]
- Kaaman, M.; Ryden, M.; Axelsson, T.; Nordstrom, E.; Sicard, A.; Bouloumie, A.; Langin, D.; Arner, P.; Dahlman, I. Alox5ap expression, but not gene haplotypes, is associated with obesity and insulin resistance. Int. J. Obes. 2006, 30, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Park, E.C.; Kim, S.I.; Hong, Y.; Hwang, J.W.; Cho, G.S.; Cha, H.N.; Han, J.K.; Yun, C.H.; Park, S.Y.; Jang, I.S.; et al. Inhibition of cyp4a reduces hepatic endoplasmic reticulum stress and features of diabetes in mice. Gastroenterology 2014, 147, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ackerman, R.; Saleh, M.; Gotlinger, K.H.; Kessler, M.; Mendelowitz, L.G.; Falck, J.R.; Arbab, A.S.; Scicli, A.G.; Schwartzman, M.L.; et al. 20-hete regulates the angiogenic functions of human endothelial progenitor cells and contributes to angiogenesis in vivo. J. Pharmacol. Exp. Ther. 2014, 348, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.M.; Janic, B.; Sheng, J.; Falck, J.R.; Roman, R.J.; Edwards, P.A.; Arbab, A.S.; Scicli, A.G. The cytochrome p450 4a/f-20-hydroxyeicosatetraenoic acid system: A regulator of endothelial precursor cells derived from human umbilical cord blood. J. Pharmacol. Exp. Ther. 2011, 338, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.M.; Arbab, A.S.; Falck, J.R.; Chen, P.; Edwards, P.A.; Roman, R.J.; Scicli, A.G. Activation of vascular endothelial growth factor through reactive oxygen species mediates 20-hydroxyeicosatetraenoic acid-induced endothelial cell proliferation. J. Pharmacol. Exp. Ther. 2007, 321, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.M.; Sheng, J.; Scicli, G.M.; Arbab, A.S.; Lehman, N.L.; Edwards, P.A.; Falck, J.R.; Roman, R.J.; Scicli, A.G. Expression of cyp4a1 in u251 human glioma cell induces hyperproliferative phenotype in vitro and rapidly growing tumors in vivo. J. Pharmacol. Exp. Ther. 2008, 327, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ackerman, R.; Guo, A.M. 20-hete in neovascularization. Prostaglandins Other Lipid Mediat. 2012, 98, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.M.; Scicli, G.; Sheng, J.; Falck, J.C.; Edwards, P.A.; Scicli, A.G. 20-hete can act as a nonhypoxic regulator of hif-1{alpha} in human microvascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H602–H613. [Google Scholar] [CrossRef] [PubMed]
- Angara, K.; Rashid, M.H.; Shankar, A.; Ara, R.; Iskander, A.; Borin, T.F.; Jain, M.; Achyut, B.R.; Arbab, A.S. Vascular mimicry in glioblastoma following anti-angiogenic and anti-20-hete therapies. Histol. Histopathol. 2017, 32, 917–928. [Google Scholar] [PubMed]
- Jain, M.; Gamage, N.H.; Alsulami, M.; Shankar, A.; Achyut, B.R.; Angara, K.; Rashid, M.H.; Iskander, A.; Borin, T.F.; Wenbo, Z.; et al. Intravenous formulation of het0016 decreased human glioblastoma growth and implicated survival benefit in rat xenograft models. Sci. Rep. 2017, 7, 41809. [Google Scholar] [CrossRef] [PubMed]
- Angara, K.; Borin, T.F.; Arbab, A.S. Vascular mimicry: A novel neovascularization mechanism driving anti-angiogenic therapy (aat) resistance in glioblastoma. Transl. Oncol. 2017, 10, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. 1990, 82, 4–6. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.S.; Weinberg, R.A. Tumor-host interactions: A far-reaching relationship. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 4022–4028. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Huang, S.; Kieran, M.W.; Kaipainen, A. Ppargamma as a therapeutic target for tumor angiogenesis and metastasis. Cancer Biol. Ther. 2005, 4, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated sdf-1/cxcl12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Lin, E.Y.; Pollard, J.W. Role of infiltrated leucocytes in tumour growth and spread. Br. J. Cancer 2004, 90, 2053–2058. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef] [PubMed]
- Sa, G.; Murugesan, G.; Jaye, M.; Ivashchenko, Y.; Fox, P.L. Activation of cytosolic phospholipase a2 by basic fibroblast growth factor via a p42 mitogen-activated protein kinase-dependent phosphorylation pathway in endothelial cells. J. Biol. Chem. 1995, 270, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, A.; Bodiga, S.; Gruenloh, S.; Gao, Y.; Dunn, L.; Falck, J.R.; Buonaccorsi, J.N.; Medhora, M.; Jacobs, E.R. 20-hete increases survival and decreases apoptosis in pulmonary arteries and pulmonary artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H777–H786. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chai, H.; Li, Y.; Zhao, H.; Xie, X.; Zheng, H.; Wang, C.; Wang, X.; Yang, G.; Cai, X.; et al. Increased expression of cyp4z1 promotes tumor angiogenesis and growth in human breast cancer. Toxicol. Appl. Pharmacol. 2012, 264, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chen, L.; Yang, Y.Q.; Falck, J.R.; Guo, A.M.; Li, Y.; Yang, J. Cytochrome p450 ω-hydroxylase promotes angiogenesis and metastasis by upregulation of vegf and mmp-9 in non-small cell lung cancer. Cancer Chemother. Pharmacol. 2011, 68, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Folberg, R.; Hendrix, M.J.; Maniotis, A.J. Vasculogenic mimicry and tumor angiogenesis. Am. J. Pathol. 2000, 156, 361–381. [Google Scholar] [CrossRef]
- Folberg, R.; Maniotis, A.J. Vasculogenic mimicry. Acta Pathol. Microbiol. Immunol. Scand. 2004, 112, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.C.; Ma, G.; Chen, S.H.; Pan, P.Y. Polarization and reprogramming of myeloid-derived suppressor cells. J. Mol. Cell Biol. 2013, 5, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.W.; Yu, T.J.; Zhang, J.; Li, Y.; Chen, H.L.; Yang, G.F.; Yu, W.; Liu, Y.Z.; Liu, X.X.; Duan, C.F.; et al. Cyp4a in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene 2017, 36, 5045–5057. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Y.; Chen, H.; Zhang, J.; Zhang, J.; Qin, T.; Duan, C.; Chen, X.; Liu, Y.; Zhou, X.; et al. Inhibition of cyp4a by a novel flavonoid fla-16 prolongs survival and normalizes tumor vasculature in glioma. Cancer Lett. 2017, 402, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Nithipatikom, K.; Isbell, M.A.; See, W.A.; Campbell, W.B. Elevated 12- and 20-hydroxyeicosatetraenoic acid in urine of patients with prostatic diseases. Cancer Lett. 2006, 233, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Alexanian, A.; Miller, B.; Roman, R.J.; Sorokin, A. 20-hete-producing enzymes are up-regulated in human cancers. Cancer Genom. Proteom. 2012, 9, 163–169. [Google Scholar]
- Guo, M.; Roman, R.J.; Fenstermacher, J.D.; Brown, S.L.; Falck, J.R.; Arbab, A.S.; Edwards, P.A.; Scicli, A.G. 9l gliosarcoma cell proliferation and tumor growth in rats are suppressed by N-hydroxy-N′-(4-butyl-2-methylphenol) formamidine (het0016), a selective inhibitor of cyp4a. J. Pharmacol. Exp. Ther. 2006, 317, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Roman, R.J.; Falck, J.R.; Edwards, P.A.; Scicli, A.G. Human u251 glioma cell proliferation is suppressed by HET0016 [N-hydroxy-N′-(4-butyl-2-methylphenyl)formamidine], a selective inhibitor of cyp4a. J. Pharmacol. Exp. Ther. 2005, 315, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Li, Y.; Wang, Y.; Zhao, H.; Zhang, J.; Chai, H.; Tang, T.; Yue, J.; Guo, A.M.; Yang, J. Downregulation of cox-2 and cyp 4a signaling by isoliquiritigenin inhibits human breast cancer metastasis through preventing anoikis resistance, migration and invasion. Toxicol. Appl. Pharmacol. 2014, 280, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.G.; Ning, Y.G.; Chen, C.; Ma, D.; Liu, Z.J.; Yang, S.; Zhou, J.; Xiao, X.; Zhang, X.A.; Edin, M.L.; et al. Cytochrome p450 epoxygenase promotes human cancer metastasis. Cancer Res. 2007, 67, 6665–6674. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Edin, M.L.; Lee, C.R.; Huang, S.; Bielenberg, D.R.; Butterfield, C.E.; Barnes, C.M.; Mammoto, A.; Mammoto, T.; Luria, A.; et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Investig. 2012, 122, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Liu, L.Z. Akt signaling in regulating angiogenesis. Curr. Cancer Drug Targets 2008, 8, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.; Sheng, S.; Pardee, A.B. Metastasis and akt activation. Cell Cycle 2008, 7, 2991–2996. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.B.; Nabha, S.M.; Atanaskova, N. Role of map kinase in tumor progression and invasion. Cancer Metastasis Rev. 2003, 22, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.H.; Beury, D.W.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: Critical cells driving immune suppression in the tumor microenvironment. Adv. Cancer Res. 2015, 128, 95–139. [Google Scholar] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The role of myeloid-derived suppressor cells (mdsc) in cancer progression. Vaccines 2016, 4. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Subtypes | Molecular Characterization | Prevalence | Prognosis | Treatment |
|---|---|---|---|---|
| Luminal A | Estrogen receptor (ER)-positive, Progesterone receptor (PR)-positive or negative, Human epidermal growth factor receptor 2 (HER2)-negative | 30–70% [2,3,4,5,6,7] | Best prognosis, high survival rates, and low recurrence rates [3,4,5,8] | Treatment for these tumors often includes chemotherapy and anti-hormone therapy |
| Luminal B | ER-positive, PR-positive or negative, HER2-positive | 10–20% [2,3,4,5,6,7] Luminal B tumors are often diagnosed at a younger age than luminal A tumors [7,8,9] | Luminal B tumors tend to have factors that lead to a poorer prognosis, compared to luminal A tumors, including poorer tumor grade, larger tumor size and lymph node-positivity [3,4,5,8,9,10,11] Patients with luminal B tumors tend to have fairly high survival rates, although not as high as those with luminal A tumors [4,8] | The treatment for luminal B tumors includes anti-hormone therapy, anti-HER2 therapies and radiation, depending on tumor grade and lymph nodes status |
| HER2-enriched | ER-negative, PR-negative, HER2-positive | 5–15% [3,5,7] HER2-type tumors may be diagnosed at a younger age than luminal A and luminal B tumors [8] | HER2-type tumors tend to have lymph node-positivity and poorer tumor grade [3,4,5,8,10] | HER2-type breast cancers can be treated with anti-HER2 drugs such as trastuzumab (Herceptin), lapatinib, capecitabine. Before these drugs were available, HER2-type tumors had a fairly poor prognosis [3,12] |
| Basal-like or Triple-negative breast cancer | ER-negative, PR-negative, HER2-negative | 15–20% [2,3,4,5,6,7] These tumors tend to occur more often in younger women [5,9] | Triple-negative/basal-like tumors are often aggressive and have a poorer prognosis compared to ER-positive subtypes (luminal A and luminal B tumors) [3,5] | Triple-negative tumors can be treated successfully with chemotherapy and radiation, depending on tumor grade, lymph nodes status and disease stage |
| Species | 20-HETE Production |
|---|---|
| Mouse | CYP4A10; CYP4A12a; CYP4A12b; CYP4A14 |
| Rat | CYP4A1; CYP4A2; CYP4A3 |
| Rabbit | CYP4A4; CYP4A6; CYP4A7 |
| Human | CYP4A11; CYP4A22; CYP4F2; CYP4F3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borin, T.F.; Angara, K.; Rashid, M.H.; Achyut, B.R.; Arbab, A.S. Arachidonic Acid Metabolite as a Novel Therapeutic Target in Breast Cancer Metastasis. Int. J. Mol. Sci. 2017, 18, 2661. https://doi.org/10.3390/ijms18122661
Borin TF, Angara K, Rashid MH, Achyut BR, Arbab AS. Arachidonic Acid Metabolite as a Novel Therapeutic Target in Breast Cancer Metastasis. International Journal of Molecular Sciences. 2017; 18(12):2661. https://doi.org/10.3390/ijms18122661
Chicago/Turabian StyleBorin, Thaiz F., Kartik Angara, Mohammad H. Rashid, Bhagelu R. Achyut, and Ali S. Arbab. 2017. "Arachidonic Acid Metabolite as a Novel Therapeutic Target in Breast Cancer Metastasis" International Journal of Molecular Sciences 18, no. 12: 2661. https://doi.org/10.3390/ijms18122661
APA StyleBorin, T. F., Angara, K., Rashid, M. H., Achyut, B. R., & Arbab, A. S. (2017). Arachidonic Acid Metabolite as a Novel Therapeutic Target in Breast Cancer Metastasis. International Journal of Molecular Sciences, 18(12), 2661. https://doi.org/10.3390/ijms18122661