Drug Resistance Driven by Cancer Stem Cells and Their Niche

Abstract
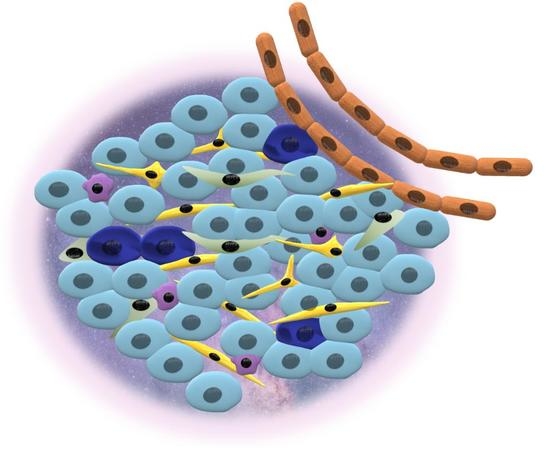
1. Cancer Stem Cells
1.1. Introduction
1.2. CSC History and Origin
1.3. Regulators in CSC Phenotype
1.4. Epithelial-to-Mesenchymal Transition and CSC Phenotype
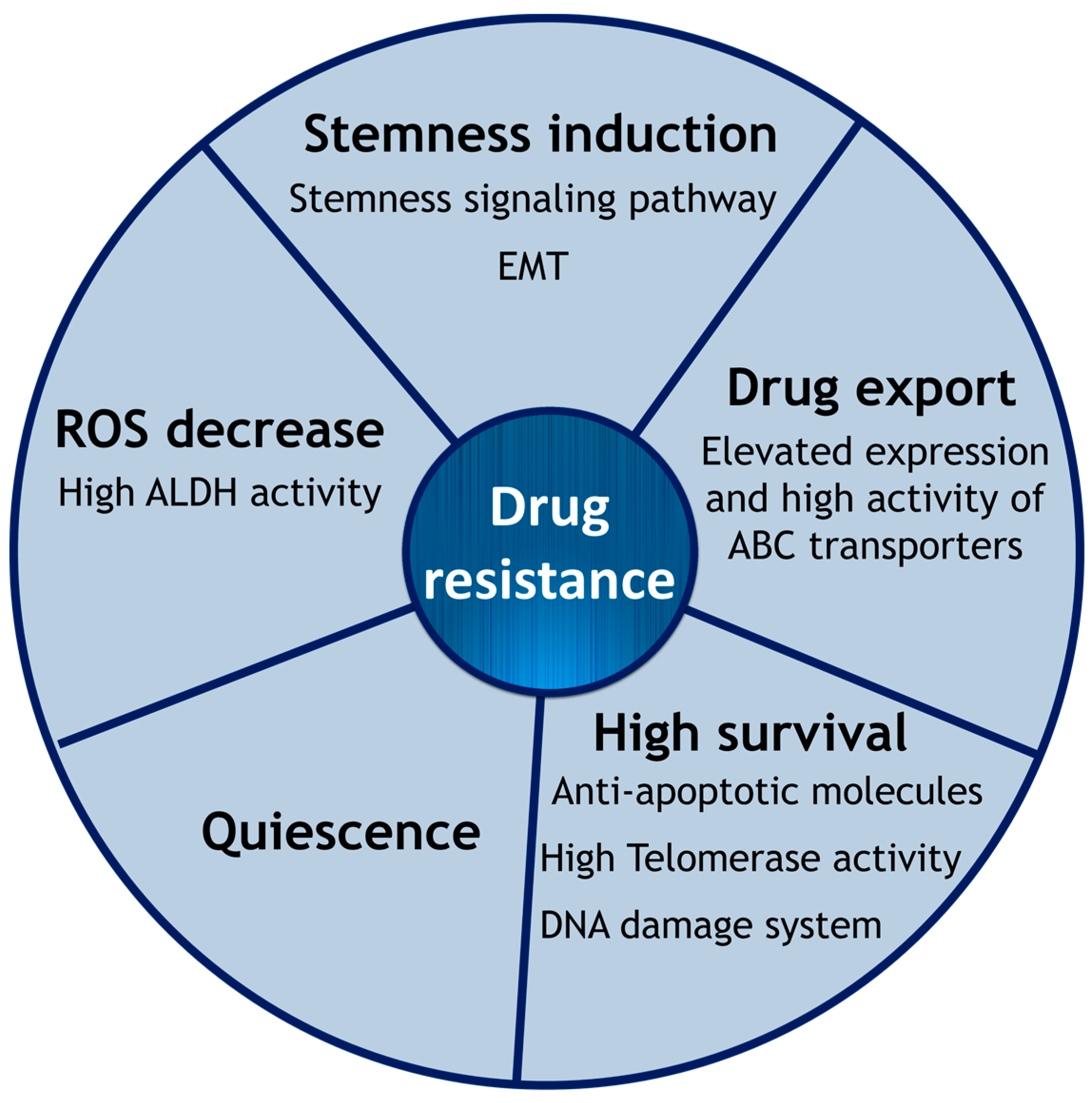
2. Intrinsic Drug Resistance of CSCs
2.1. Epithelial-to-Mesenchymal Transition
2.2. Cell Membrane Transporters: ABC Family
2.3. Hypoxia and ROS
2.4. High Survival Capacity of CSCs
2.5. Effect of MicroRNAs in CSC Phenotype Acquisition
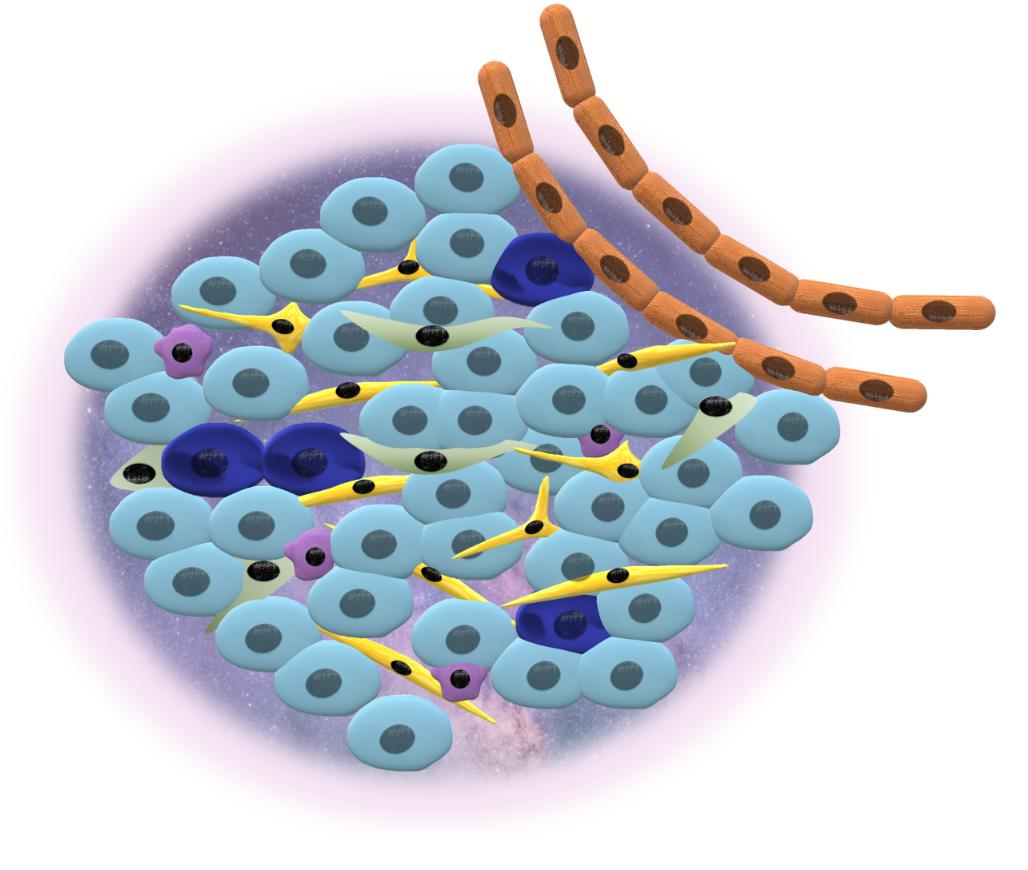
3. Tumor Heterogeneity and Cancer Niche
3.1. Tumor Heterogeneity
3.2. Cancer Niche
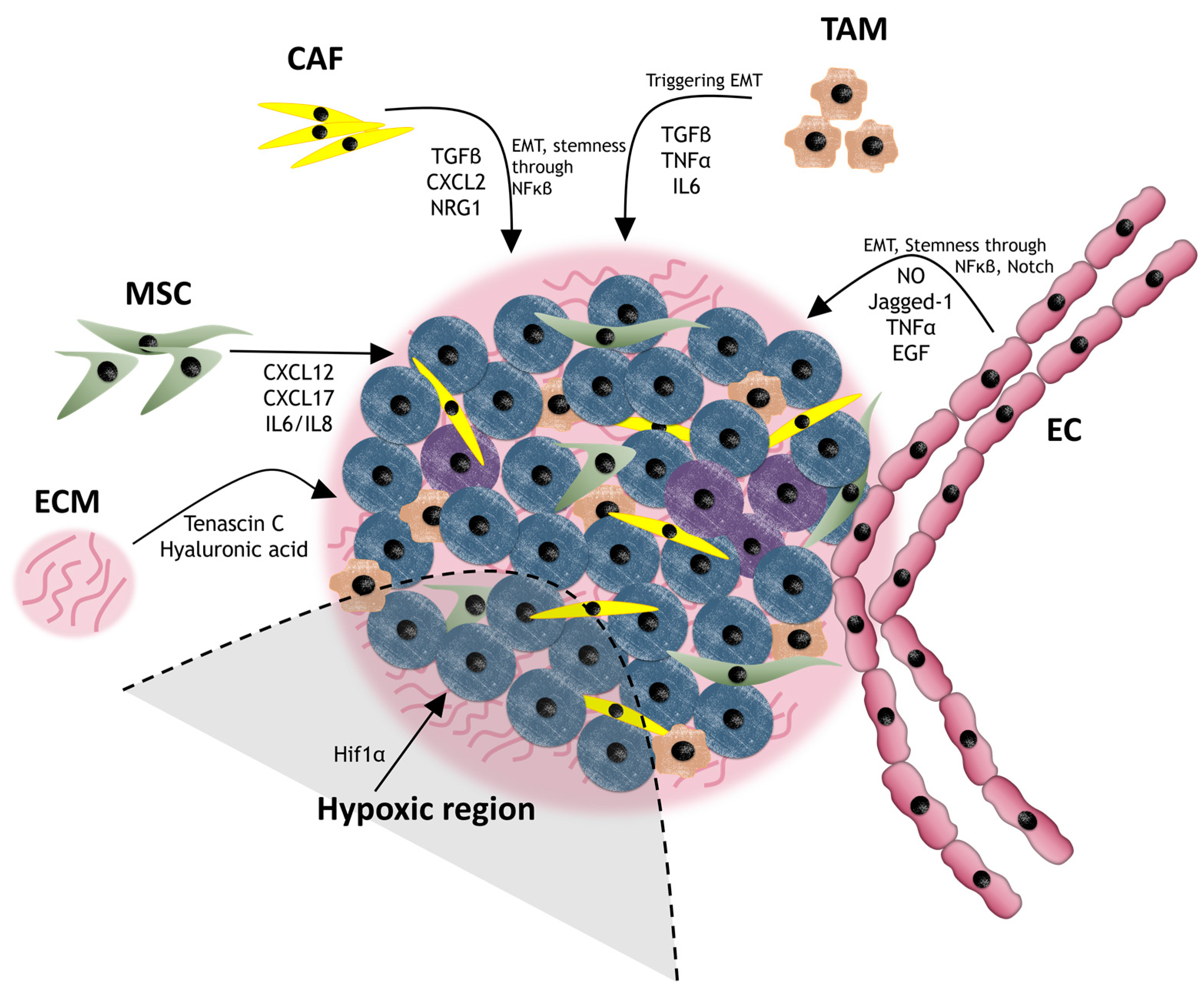
3.3. Cancer Niche and Therapy Resistance
3.3.1. Cancer-Associated Fibroblasts (CAFs)
3.3.2. Mesenchymal Stem Cells (MSCs)
3.3.3. Endothelial Cells (ECs)
3.3.4. Tumor-Associated Macrophages (TAMs)
3.3.5. Extracellular Matrix (ECM)
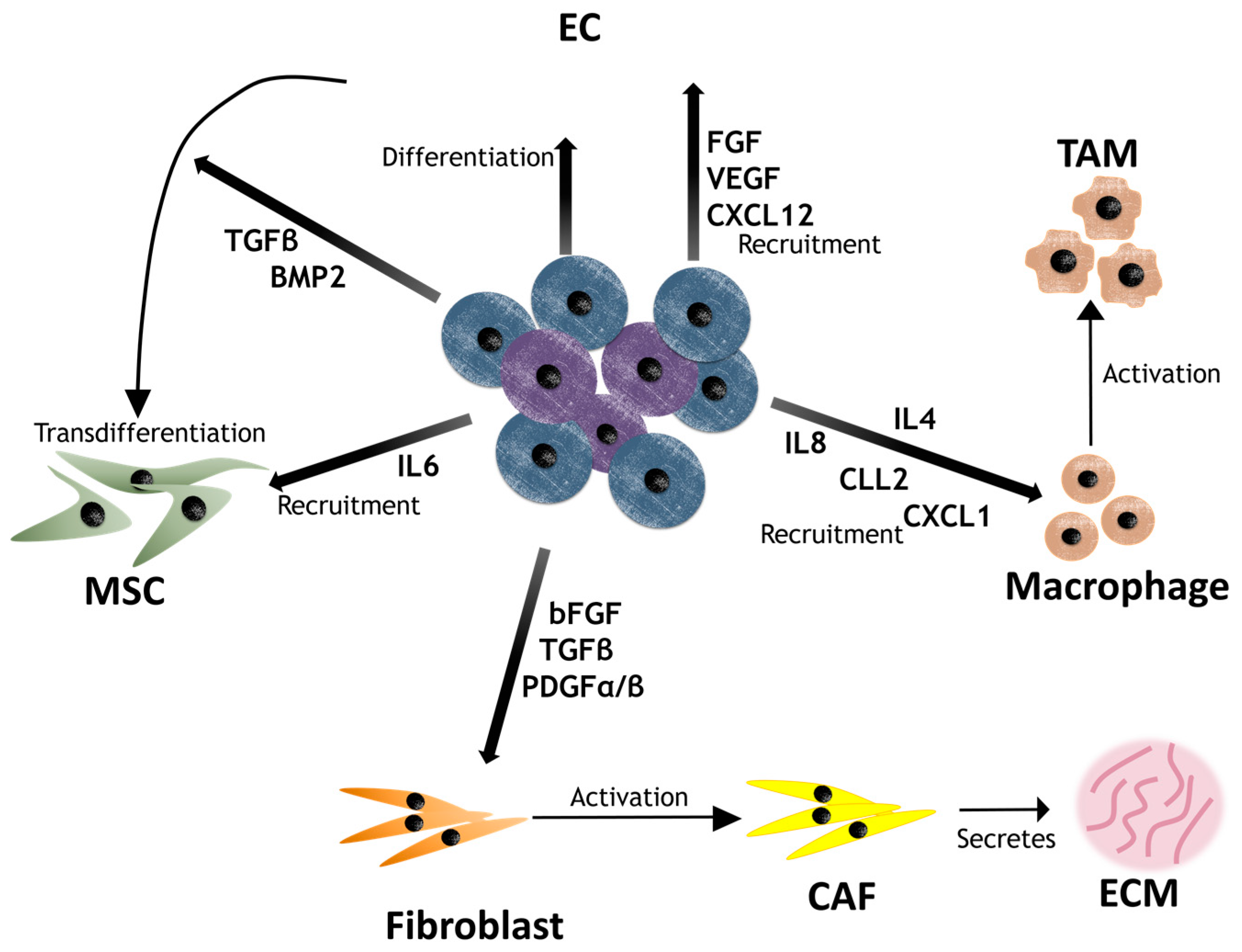
4. Contribution of CSCs to the Niche
5. Treatments and Approaches
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CSC | Cancer stem cell |
| SC | Stem cell |
| Wnt | Wingless |
| EMT | Epithelial-to-mesenchymal transition |
| ROS | Reactive oxygen species |
| ECM | Extracellular matrix |
| CAF | Cancer-associated fibroblast |
| MSC | Mesenchymal stem cell |
| EC | Endothelial cell |
| TAM | Tumor-associated macrophage |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.; Yaswen, P.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.G.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Kawasaki, B.T.; Klarmann, G.J.; Thomas, S.B.; Farrar, W.L. CD44+ CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br. J. Cancer 2008, 98, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; de Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cells heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer Stem Cells: Impact, Heterogeneity, and Uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Brooks, M.D.; Burness, M.L.; Wicha, M.S. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell Stem Cell 2015, 17, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Fukunaga-kalabis, M.; Schmidt, E.C.; Susan, E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Garcia-Mayea, Y.; Mir, C.; Lorente, J.; Rubio, I.T.; LLeonart, M.E. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat. Rev. 2016, 49, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Radulescu, S.; Ridgway, R.A.; Cordero, J.; Athineos, D.; Salgueiro, P.; Poulsom, R.; Neumann, J.; Jung, A.; Patel, S.; Woodgett, J.; et al. Acute WNT signalling activation perturbs differentiation within the adult stomach and rapidly leads to tumour formation. Oncogene 2013, 32, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Warrier, S.; Bhuvanalakshmi, G.; Arfuso, F.; Rajan, G.; Millward, M.; Dharmarajan, A. Cancer stem-like cells from head and neck cancers are chemosensitized by the Wnt antagonist, sFRP4, by inducing apoptosis, decreasing stemness, drug resistance and epithelial to mesenchymal transition. Cancer Gene Ther. 2014, 21, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ishii, H.; Nishida, N.; Takemasa, I.; Mizushima, T.; Ikeda, M.; Yokobori, T.; Mimori, K.; Yamamoto, H.; Sekimoto, M.; et al. Significance of Lgr5+ve cancer stem cells in the colon and rectum. Ann. Surg. Oncol. 2011, 18, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W. Self-renewal of the gastric epithelium from stem and progenitor cells. Front. Biosci. (Schol. Ed.) 2013, 5, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog signaling in the maintenance of cancer stem cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Walsh, M.P.; Ali, S.A.; Thompson, E.A.; Murray, N.R.; Flieds, A.P. The PRKCI and SOX2 oncogenes are co-amplified and cooperate to activate hedgehog signaling in lung squamous cell carcinoma. Cancer Cell 2014, 25, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Iliou, M.S.; Esteller, M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol. Oncol. 2012, 6, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.W.; Rueger, M.A.; Bae, S.-K.; Kittappa, R.; McKay, R.D.G. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Concise review: Notch signaling in stem cell systems. Stem Cells 2006, 24, 2437–2447. [Google Scholar] [CrossRef] [PubMed]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Warren, R.Q.; Ivy, S.P. Breast cancer growth and metastasis: Interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 2011, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- Counter, C.M.; Avilion, A.A.; Lefeuvrel, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchettil, S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar] [PubMed]
- Allsopp, R.C.; Morin, G.B.; DePinho, R.; Harley, C.B.; Weissman, I.L. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood 2003, 102, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.T.; Beauchamp, A.D.; Calado, R.T.; Bradford, J.W.; Regal, J.A.; Shenoy, A.; Liang, Y.; Lansdorp, P.M.; Young, N.S.; Ly, H. Functional characterization of natural telomerase mutations found in patients with hematologic disorders. Blood 2007, 109, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Makki, J.; Myint, O.; Wynn, A.A.; Samsudin, A.T.; John, D.V. Expression distribution of cancer stem cells, epithelial to mesenchymal transition, and telomerase activity in breast cancer and their association with clinicopathologic characteristics. Clin. Med. Insights Pathol. 2015, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial–mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Shook, D.; Keller, R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech. Dev. 2003, 120, 1351–1383. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Portillo, F.; Cano, A. Transcriptional regulation of cadherins during development and carcinogenesis. Int. J. Dev. Biol. 2004, 48, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.; Eaton, E.N.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Campbell, L.L.; Polyak, K.; et al. The epithelial-mesenchymal transition generates cekks with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Hollier, B.G.; Tinnirello, A.A.; Werden, S.J.; Evans, K.W.; Taube, J.H.; Sarkar, T.R.; Sphyris, N.; Shariati, M.; Kumar, S.V.; Battula, V.L.; et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013, 73, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Yoshioka, Y.; Minoura, K.; Takahashi, R.U.; Takeshita, F.; Taya, T.; Horii, R.; Fukuoka, Y.; Kato, T.; Kosaka, N.; et al. An integrative genomic analysis revealed the relevance of microRNA and gene expression for drug-resistance in human breast cancer cells. Mol. Cancer 2011, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Efferth, T. Tumor heterogeneity, single-cell sequencing, and drug resistance. Pharmaceuticals 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Konkimalla, V.B.; Wang, Y.F.; Sauerbrey, A.; Meinhardt, S.; Zintl, F.; Mattern, J.; Volm, M. Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin. Cancer Res. 2008, 14, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.P.; Das, D.N.; Panda, P.K.; Mukhopadhyay, S.; Sinha, N.; Praharaj, P.P.; Agarwal, R.; Bhutia, S.K. Implications of cancer stem cells in developing therapeutic resistance in oral cancer. Oral Oncol. 2016, 62, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Dallas, N.A.; Xia, L.; Fan, F.; Gray, M.J.; Gaur, P.; van Buren, G.; Samuel, S.; Kim, M.P.; Lim, S.J.; Ellis, L.M. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insuline-like growth factor-1 receptor inhibitor. Cancer Res. 2009, 69, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef] [PubMed]
- Sahlberg, S.H.; Spiegelberg, D.; Glimelius, B.; Stenerlöw, B.; Nestor, M. Evaluation of cancer stem cell markers CD133, CD44, CD24: Association with AKT isoforms and radiation resistance in colon cancer cells. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.D.; Hier, M.; Mlynarek, A.; Kowalski, L.P.; Alaoui-Jamali, M.A. Recurrent oral cancer: Current and emerging therapeutic approaches. Front. Pharmacol. 2012, 3, 149. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Peng, Y.; Liu, Y.; Xin, H.; Zhan, X.; Tan, W. Twist1 and Snail link Hedgehog signaling to tumor-initiating cell-like properties and acquired chemoresistance independently of ABC transporters. Stem Cells 2015, 33, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Franklin, C.K.; Mackay, A.; Reis-Filho, J.S.; Isacke, C.M. Epithelial and mesenchymal subpopulations within normal basal breast cell lines exhibit distinct stem cell/progenitor properties. Stem Cells 2012, 30, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Bradford, G.B.; Williams, B.; Rossi, R.; Bertoncello, I. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp. Hematol. 1997, 25, 445–453. [Google Scholar] [PubMed]
- Zhu, L.-F.; Hu, Y.; Yang, C.-C.; Xu, X.-H.; Ning, T.-Y.; Wang, Z.-L.; Ye, J.-H.; Liu, L.-K. Snail overexpression induces an epithelial to mesenchymal transition and cancer stem cell-like properties in SCC9 cells. Lab. Investig. 2012, 92, 744–752. [Google Scholar] [CrossRef] [PubMed]
- La Fleur, L.; Johansson, A.C.; Roberg, K. A CD44high/EGFRlow subpopulation within head and neck cancer cell lines shows an epithelial-mesenchymal transition phenotype and resistance to treatment. PLoS ONE 2012, 7, e44071. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427. [Google Scholar] [CrossRef] [PubMed]
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug transporters in drug efficacy and toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef]
- Hawley, T.S.; Riz, I.; Yang, W.; Wakabayashi, Y.; DePalma, L.; Chang, Y.T.; Peng, W.; Zhu, J.; Hawley, R.G. Identification of an ABCB1 (P-glycoprotein)-positive carfilzomib-resistant myeloma subpopulation by the pluriopotent stem cell fluorescent dye Cdy1. Am. J. Hematol. 2013, 88, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Chuthapisith, S.; Eremin, J.; El-Sheemey, M.; Eremin, O. Breast cancer chemoresistance: Emerging importance of cancer stem cells. Surg. Oncol. 2010, 19, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, C.W.; Harkey, M.A.; Torok-Storb, B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 2002, 99, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008, 9, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Tsuchida, R.; Malkin, D.; Koren, G.; Baruchel, S.; Yeger, H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells 2008, 26, 1818–1830. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Li, H.; Hao, X. The self-renewal signaling pathways utilized by gastric cancer stem cells. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Jogi, A.; Ora, I.; Nilsson, H.; Lindeheim, A.; Makino, Y.; Poellinger, L.; Axelson, H.; Pahlman, S. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc. Natl. Acad. Sci. USA 2002, 99, 7021–7026. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology 2004, 19, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Physiological and pathological responses to hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef]
- Liu, L.; Salnikov, A.V.; Bauer, N.; Aleksandrowicz, E.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Schemmer, P.; Werner, J.; et al. Triptolide reverses hypoxia-induced epithelial-mesenchymal transition and stem-like features in pancreatic cancer by NF-κB downregulation. Int. J. Cancer 2014, 134, 2489–2503. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Almog, N. Molecular mechanisms underlying tumor dormancy. Cancer Lett. 2010, 294, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313 Pt 1, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.W.; Palle, K. Aldehyde dehydrogenases in cancer stem cells: Potential as therapeutic targets. Ann. Transl. Med. 2016, 4, 518. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Chen, Y.S.; Chou, S.H.; Han, C.L.; Chen, Y.J.; Yang, C.C.; Huang, C.Y.; Lo, J.F. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 2014, 74, 6291–6305. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, Q.; Fu, X.; Luo, W. ALDH1A1 overexpression is associated with the progression and prognosis in gastric cancer. BMC Cancer 2014, 14, 705. [Google Scholar] [CrossRef] [PubMed]
- Balicki, D. Moving forward in human mammary stem cell biology and breast cancer prognostication using ALDH1. Cell Stem Cell 2007, 1, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Huo, W.; Du, M.; Pan, X.; Zhu, X.; Li, Z. Prognostic value of ALDH1 expression in lung cancer: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 2045–2051. [Google Scholar] [PubMed]
- Ikeda, J.; Mamat, S.; Tian, T.; Wang, Y.; Luo, W.; Rahadiani, N.; Aozasa, K.; Morii, E. Reactive oxygen species and aldehyde dehydrogenase activity in Hodgkin lymphoma cells. Lab. Investig. 2012, 92, 606–614. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aponte, P.M.; Caicedo, A. Stemness in cancer: Stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Gasparetto, M.; Smith, C.A. ALDHs in normal and malignant hematopoietic cells: Potential new avenues for treatment of AML and other blood cancers. Chem. Biol. Interact. 2017, 276, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.L.; Yu, C.C.; Lo, J.F.; Sung, W.W.; Lee, H.; Chen, S.L.; Chou, M.Y. Enhanced cisplatin resistance in oral-cancer stem-like cells is correlated with upregulation of excision-repair cross-complementation group 1. J. Dent. Sci. 2012, 7, 111–117. [Google Scholar] [CrossRef][Green Version]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Ren, Y.; Zhang, J.; Chen, J.; Zhou, W.; Guo, W.; Wang, X.; Chen, H.; Li, M.; et al. Cisplatin-enriching cancer stem cells confer multidrug resistance in non-small cell lung cancer via enhancing TRIB1/HDAC activity. Cell Death Dis. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M.; Calin, G.A. miRNAs, cancer and stem cell division. Cell 2005, 122, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Hao, X.; Zhang, M.; Tang, W.; Meng, Y.; Li, L.; Xiang, D.; DeSano, J.T.; Bommer, G.T.; Fan, D.; et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Ding, Y.C.; Yu, W.; Wang, Q.; Meng, B.; Huang, T. MicroRNA-200c overexpression plays an inhibitory role in human pancreatic cancer stem cells by regulating epithelial-mesenchymal transition. Minerva Med. 2015, 106, 193–202. [Google Scholar] [PubMed]
- Song, S.J.; Ito, K.; Ala, U.; Kat, L.; Webster, W.; Sun, S.M.; Jongen-Lavrenic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The oncogenic MicroRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 2013, 13, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Polisen, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef] [PubMed]
- De Carolis, S.; Bertoni, S.; Nati, M.; D’Anello, L.; Papi, A.; Tesei, A.; Cricca, M.; Bonafe, M. Carbonic anhydrase 9 mRNA/microRNA34a interplay in hypoxic human mammospheres. J. Cell. Physiol. 2016, 231, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; de Carolis, S.; Bertoni, S.; Stroci, G.; Sceberras, V.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; Orlandi, M.; Bonafe, M. PPARγ and RXR ligands disrupt the inflammatory cross-talk in the hypoxic breast cancer stem cells niche. J. Cell. Physiol. 2014, 229, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Doellgast, G.J. Quantitation of peroxidase-antibody binding to membrane fragments using column chromatography. Anal. Biochem. 1979, 98, 53–59. [Google Scholar] [CrossRef]
- Bao, B.; Ali, S.; Ahmad, A.; Azmi, A.A.; Li, Y.; Banerjee, S.; Kong, D.; Sethi, S.; Sboukameel, A.; Padhye, S.B.; et al. Hypoxia-induced aggressiveness of pancreatic cancer cells is due to increased expression of VEGF, IL-6 and miR-21, which can be attenuated by CDF treatment. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.U.; Miyazaki, H.; Takeshita, F.; Yamamoto, Y.; Minoura, K.; Ono, M.; Kodaira, M.; Tamura, K.; Mori, M.; Ochiya, T. Loss of microRNA-27b contributes to breast cancer stem cell generation by activating ENPP1. Nat. Commun. 2015, 6, 7318. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Martelotto, L.G.; Ng, C.K.; Piscuoglio, S.; Weigelt, B.; Reis-Filho, J.S. Breast cancer intra-tumor heterogeneity. Breast Cancer Res. 2014, 16, 210. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Fuller, M. Stem cells and cancer: Two faces of eve. Cell 2006, 124, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xie, T. Stem cell niche: Structure and function. Annu. Rev. Cell Dev. Biol. 2005, 21, 605–631. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, W.; Jia, Y.; Yu, Q.; Grau, G.E.; Peng, L.; Ran, Y.; Yang, Z.; Deng, H.; Lou, J. Single-cell clones of liver cancer stem cells have the potential of differentiating into different types of tumor cells. Cell Death Dis. 2013, 4, e857. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Sawayama, H.; Sugihara, H.; Baba, H. Interaction between gastric cancer stem cells and the tumor microenvironment. J. Gastroenterol. 2014, 49, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Daverey, A.; Drain, A.P.; Kidambi, S. Physical intimacy of breast cancer cells with mesenchymal stem cells elicits trastuzumab resistance through src activation. Sci. Rep. 2015, 5, 13744. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, C.-H.; Tan, L.-D.; Wang, Q.-S.; Li, X.-Q.; Feng, Y.-M. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Han, M.E.; Kim, H.J.; Shin, D.H.; Hwang, S.H.; Kang, C.D.; Oh, S.O. Overexpression of NRG1 promotes progression of gastric cancer by regulating the self-renewal of cancer stem cells. J. Gastroenterol. 2015, 50, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef] [PubMed]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Victor, C.T.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, M.; Krüger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Investig. 2006, 116, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ginestier, C.; Ou, S.J.; Clouthier, S.G.; Patel, S.H.; Monville, F.; Korkaya, H.; Heath, A.; Dutcher, J.; Kleer, C.G.; et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2012, 71, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Rafii, A.; Mirshahi, P.; Poupot, M.; Faussat, A.M.; Simon, A.; Ducros, E.; Mery, E.; Couderc, B.; Lis, R.; Capdet, J.; et al. Oncologic trogocytosis of an original stromal cells induces chemoresistance of ovarian tumours. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Dong, Z.; Lauxen, I.S.; Filho, M.S.A.; Nör, J.E. Endothelial cell-secreted EGF induces epithelial to mesenchymal transition and endows head and neck cancer cells with stem-like phenotype. Cancer Res. 2014, 74, 2869–2881. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular Nitric Oxide Activates Notch Signaling and Promotes Stem-like Character in PDGF-induced Glioma Cells. Cell Stem Cell 2013, 6, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Jain, R.K. Tumor microenvironment abnormalities: Causes, consequences, and strategies to normalize. J. Cell. Biochem. 2007, 101, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Bonde, A.-K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Kim, G.; Davis, A.; Malik, F.; Henry, N.L.; Quraishi, A.A.; Tawakkol, N.; Angelo, R.D.; Chung, S.; Luther, T.; et al. Activation of an IL6 inflamatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Murai, T. Lipid raft-mediated regulation of hyaluronan-CD44 interactions in inflammation and cancer. Front. Immunol. 2015, 6, 420. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.-F.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A.; Augsten, M. Cancer-associated fibroblasts and tumor growth-bystanders turning into key players. Curr. Opin. Genet. Dev. 2009, 19, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G.; Ryan, G.B.; Majne, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Goldberg, A.; Lin, Z.; Ko, Y.H.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol. Ther. 2011, 12, 924–938. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Dong, Z.; Vodopyanov, D.; Imai, A.; Joseph, I.; Prince, M.E.; Wicha, M.S.; Nör, J.E. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancre stem cells. Cancer Res. 2010, 70, 9969–9978. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.S.; Rustgi, A.K. Matricellular proteins: Priming the tumour microenvironment for cancer development and metastasis. Br. J. Cancer 2013, 108, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.M.; Nguyen, M.; Barsky, S.H. Human breast carcinoma desmoplasia is PDGF initiated. Oncogene 2000, 19, 4337–4345. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.J.; Lebret, S.; Tomaskovic-Crook, E.; Ahmed, N.; Blick, T.; Newgreen, D.F.; Thompson, E.W.; Ackland, M.L. Contribution of fibroblast and mast cell (afferent) and tumor (efferent) IL-6 effects within the tumor microenvironment. Cancer Microenviron. 2012, 5, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef] [PubMed]
- Valenti, G.; Quinn, H.M.; Heynen, G.J.J.E.; Lan, L.; Holland, J.D.; Vogel, R.; Wulf-Goldenberg, A.; Birchmeier, W. Cancer stem cells regulate cancer-associated fibroblasts via activation of hedgehog signaling in mammary gland tumors. Cancer Res. 2017, 77, 2134–2147. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 4, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Dong, J.; Huang, Q.; Lou, M.; Wang, A.; Lan, Q. Endothelial cell transdifferentiation of human glioma stem progenitor cells in vitro. Brain Res. Bull. 2010, 82, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Grange, C.; Sapino, A.; Camussi, G. Endothelial cell differentiation of human breast tumour stem/progenitor cells. J. Cell. Mol. Med. 2009, 13, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Ckauser, K.R.; Tam, W.L.; Frose, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammtatory monocytes to facilitate breast tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.C.; Scott, K.A.; Wilson, J.L.; Thompson, R.G.; Proudfoot, A.E.; Balkwill, F.R. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003, 63, 8360–8365. [Google Scholar] [PubMed]
- Botelho, M.; Alves, H. Significance of Cancer Stem Cells in Anti-Cancer Therapies. Int. J. Immunother. Cancer Res. 2016, 2, 14–16. [Google Scholar] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: Final update (30-month) of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Schulze, B.; Meissner, M.; Ghanaati, S.; Burck, I.; Rödel, C.; Balermpas, P. Hedgehog pathway inhibitor in combination with radiation therapy for basal cell carcinomas of the head and neck. Strahlenther. Onkol. 2016, 192, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J. The cancer stem cell gamble. Science 2015, 347, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.T.; Hamai, A.; Hienzsch, A.; Cañeque, T.; Muller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Tallman, M.S.; Altman, J.K. How I treat acute promyelocytic leukemia. Leukemia 2009, 114, 5126–5135. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Li, Z.; Xu, X.; Chen, C.; Wei, W.; Fan, M.; Chen, X.; Li, J.J.; Wang, Y.; Huang, J. All-trans retinoic acids induce differentiation and sensitize a radioresistant breast cancer cells to chemotherapy. BMC Complement. Altern. Med. 2016, 16, 113. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Del Barco, S.; Vazquez-Martin, A.; Cufí, S.; Oliveras-Ferraros, C.; Bosch-Barrera, J.; Joven, J.; Martin-Castillo, B.; Menendez, J.A. Metformin: Multi-faceted protection against cancer. Oncotarget 2011, 2, 896–917. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Agbor-Tarh, D.; Bradbury, I.; di Cosimo, S.; Azim, H.A., Jr.; Fumagalli, D.; Sarp, S.; Wolff, A.C.; Andersson, M.; Kroep, J.; et al. Impact of diabetes, insulin, and metformin use on the outcome of patients with human epidermal growth factor receptor 2-positive primary breast cancer: Analysis from the ALTTO phase III randomized trial. J. Clin. Oncol. 2017, 35, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Son, K.; Fujioka, S.; Iida, T.; Furukawa, K.; Fujita, T.; Yamada, H.; Chiao, P.J.; Yanaga, K. Doxycycline induces apoptosis in PANC-1 pancreatic cancer cells. Anticancer Res. 2009, 29, 3995–4003. [Google Scholar] [PubMed]
- Duivenvoorden, W.C.; Popovic, S.V.; Lhotak, S.; Seidlitz, E.; Hirte, H.W.; Tozer, R.G.; Singh, G. Doxyxycle decreases tumor burden in bone metastasis model of human breast cancer. Cancer Res. 2002, 62, 1588–1591. [Google Scholar] [PubMed]
- De Francisco, E.M.; Maggiolini, M.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Targeting hypoxic cancer stem cells (CSCs) with Doxyxyxline: Implications for optimizing anti-angiogwnic therapy. Oncotarget 2017, 8, 56126–56142. [Google Scholar] [CrossRef]
- Raha, D.; Wilson, T.R.; Peng, J.; Peterson, D.; Yue, P.; Evangelista, M.; Wilson, C.; Merchant, M.; Settleman, J. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. 2014, 74, 3579–3590. [Google Scholar] [CrossRef] [PubMed]
- Moreb, J.S.; Muhoczy, D.; Ostmark, B.; Zucali, J.R. RNAi-mediated knockdown of aldehyde dehydrogenase class-1A1 and class-3A1 is specific and reveals that each contributes equally to the resistance against 4-hydroperoxycyclophosphamide. Cancer Chemother. Pharmacol. 2007, 59, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Kinders, R.; Rubinstein, L.; Parchment, R.E.; Murgo, A.J.; Collins, J.; Pickeral, O.; Low, J.; Steinberg, S.M.; Gutierrez, M.; et al. Compressing drug development timelines in oncology using phase “0” trials. Nat. Rev. Cancer 2007, 7, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Smalley, W.E.; DuBois, R.N. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv. Pharmacol. 1997, 39, 1–20. [Google Scholar] [PubMed]
- Valverde, A.; Peñarando, J.; Cañas, A.; López-Sánchez, L.M.; Conde, F.; Hernández, V.; Peralbo, E.; López-Pedrera, C.; de la Haba-Rodríguez, J.; Aranda, E.; et al. Simultaneous inhibition of EGFR/VEGFR and cyclooxygenase-2 targets stemness-related pathways in colorectal cancer cells. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; VonHoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Molecule | Effect | Cancer | Ref. |
|---|---|---|---|---|
| CAF | TGFβ secretion | EMT stimulator | Gastric, prostate | [109] |
| NRG1 secretion | Activation of NF-κB signaling pathway | Gastric, breast, prostate, glioma | [110,111,112] | |
| CAF exosomes | Activation of Wnt signaling pathway | Colon | [113,114] | |
| Collagen type I secretion | Decrease of drug uptake | - | [115] | |
| MSC | CXCL12, CXCL7, IL6/IL8 secretion | Activation of NF-κB signaling | Breast | [17,116] |
| Physical interaction | Activation of SCR and its downstream PI3K/Akt | Breast | [117] | |
| Physical interaction | Increase of MDR protein expression | Ovarian | [118] | |
| EC | TNFα secretion | Recruitment of myeloid cells that induce the loop CXCL1/2-S100A8/9 | Breast | [112] |
| EGF secretion | EMT stimulator | Glioblastoma, colorectal, HNSCC | [119,120] | |
| Nitric oxygen, Jagged-1 secretion | Activation of Notch signaling pathway | Glioma, colorectal | [121,122] | |
| - | Direct blockage of drug administration due to irregular vessel shape | - | [123] | |
| TAM | TGFβ and TNFα secretion | EMT stimulator | NSCLC | [124,125] |
| IL6 secretion | Activation of STAT3 | Breast, pancreas | [126] | |
| ECM | ECM stiffness | Physical barrier that physically separates the cells | - | [125] |
| Hyaluronic acid secretion | Activation of CD44 | Breast | [127] | |
| Tenascin C secretion | Activation of Wnt and Notch signaling pathways | Breast | [128] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prieto-Vila, M.; Takahashi, R.-u.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. https://doi.org/10.3390/ijms18122574
Prieto-Vila M, Takahashi R-u, Usuba W, Kohama I, Ochiya T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. International Journal of Molecular Sciences. 2017; 18(12):2574. https://doi.org/10.3390/ijms18122574
Chicago/Turabian StylePrieto-Vila, Marta, Ryou-u Takahashi, Wataru Usuba, Isaku Kohama, and Takahiro Ochiya. 2017. "Drug Resistance Driven by Cancer Stem Cells and Their Niche" International Journal of Molecular Sciences 18, no. 12: 2574. https://doi.org/10.3390/ijms18122574
APA StylePrieto-Vila, M., Takahashi, R.-u., Usuba, W., Kohama, I., & Ochiya, T. (2017). Drug Resistance Driven by Cancer Stem Cells and Their Niche. International Journal of Molecular Sciences, 18(12), 2574. https://doi.org/10.3390/ijms18122574