Obstacles to Brain Tumor Therapy: Key ABC Transporters

Abstract
1. ATP-Binding Cassette (ABC) Transporters in the Central Nervous System (CNS)
1.1. Overview of Blood–Brain Barrier (BBB)
1.1.1. Development and Regulation of the BBB
1.1.2. Disruption of the BBB in Diseases
1.2. ABC Transporters Provide Critical Barrier Function of BBB
1.2.1. ABCB1 Expression at the BBB
1.2.2. Cellular Localization of ABCB1
1.2.3. ABCB1 Function at the BBB and Clinical Importance
1.2.4. ABCG2 Expression at the BBB
1.2.5. ABCG2 Function at the BBB; Redundancy with ABCB1?
1.3. Regulation of ABC Transporters at the BBB
1.3.1. Ligand-Activated Receptor Signaling
1.3.2. Inflammatory Signaling Pathways
1.3.3. Gender Bias and Age
1.3.4. ABC Transporters and the Blood Brain Barrier in Cancer
2. ABC Transporters in Central Nervous System (CNS) Tumors
2.1. Gliomas
2.1.1. Prognosis, Treatment, Cells of Origin
2.1.2. Glioblastoma
2.1.3. Glioblastoma and ABCB1, ABCG2
2.1.4. ABCB1 and ABCG2: Impact upon Temozolomide (TMZ) Therapy
2.1.5. Improving Glioblastoma Therapy by Altering Blood–Tumor Barrier (BTB) and BBB/ABC Transporter Function
2.2. Medulloblastoma
2.2.1. Medulloblastoma Overview
2.2.2. Etiology of Medulloblastoma
2.2.3. Demographics, Treatments and Clinical Outcome
2.2.4. ABC Transporters and Medulloblastoma
2.2.5. Improving Medulloblastoma Therapy by Concurrently Altering the BTB and BBB by Targeting ABC Transporter Function
3. Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Saunders, N.R.; Dreifuss, J.-J.; Dziegielewska, K.M.; Johansson, P.A.; Habgood, M.D.; Møllgård, K.; Bauer, H.-C. The rights and wrongs of blood-brain barrier permeability studies: A walk through 100 years of history. Front. Neurosci. 2014, 8, 404. [Google Scholar] [CrossRef] [PubMed]
- Motoike, T.; Loughna, S.; Perens, E.; Roman, B.L.; Liao, W.; Chau, T.C.; Richardson, C.D.; Kawate, T.; Kuno, J.; Weinstein, B.M.; et al. Universal GFP reporter for the study of vascular development. Genesis 2000, 28, 75–81. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Agalliu, D.; Cahoy, J.D.; Kaushal, A.; Barres, B.A. The Mouse Blood-Brain Barrier Transcriptome: A New Resource for Understanding the Development and Function of Brain Endothelial Cells. PLoS ONE 2010, 5, e13741. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/β-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/β-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Donahue, S.; Pappas, G.D. The fine structure of capillaries in the cerebral cortex of the rat at various stages of development. Am. J. Anat. 1961, 108, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, H.E.; Witte, J.; Schreibelt, E.M.; de Vries, G. Radical changes in multiple sclerosis pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Olmez, I.; Ozyurt, H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochem. Int. 2012, 60, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Pun, P.B.L.; Lu, J.; Moochhala, S. Involvement of ROS in BBB dysfunction. Free Radic. Res. 2009, 43, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Cardo, C.; O’Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.; Melamed, M.R.; Bertino, J.R. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Immunohistochemical localization in normal tissues of different epitopes in the multidrug transport protein P170: Evidence for localization in brain capillaries and crossreactivity of one antibody with a muscle protein. J. Histochem. Cytochem. 1989, 37, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Sato, T.N. Mouse multidrug resistance 1a/3 gene is the earliest known endothelial cell differentiation marker during blood-brain barrier development. Dev. Dyn. 1995, 202, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Strazielle, N.; Schmitt, C.; Fevre-Montange, M.; Ostrow, J.D.; Tiribelli, C.; Ghersi-Egea, J.-F. Differential expression of the multidrug resistance-related proteins ABCb1 and ABCc1 between blood-brain interfaces. J. Comp. Neurol. 2008, 510, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Berengeno, A.L.; Strazielle, N.; Fazzari, F.; Raseni, A.; Ostrow, J.D.; Wennberg, R.; Ghersi-Egea, J.-F.; Tiribelli, C. Modulation of Mrp1 (ABCc1) and Pgp (ABCb1) by Bilirubin at the Blood-CSF and Blood-Brain Barriers in the Gunn Rat. PLoS ONE 2011, 6, e16165. [Google Scholar] [CrossRef] [PubMed]
- Goralski, K.B.; Acott, P.D.; Fraser, A.D.; Worth, D.; Sinal, C.J. Brain Cyclosporin a Levels Are Determined by Ontogenic Regulation of Mdr1a Expression. Drug Metab. Dispos. 2005, 34, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, É.; Demeule, M.; Ghitescu, L.; Béliveau, R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem. J. 1997, 326, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Smit, J.J.M.; van Tellingen, O.; Beijnen, J.H.; Wagenaar, E.; van Deemter, L.; Mol, C.A.A.M.; van der Valk, M.A.; Robanus-Maandag, E.C.; te Riele, H.P.; et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994, 77, 491–502. [Google Scholar] [CrossRef]
- Lankas, G.R.; Cartwright, M.E.; Umbenhauer, D. P-Glycoprotein Deficiency in a Subpopulation of CF-1 Mice Enhances Avermectin-Induced Neurotoxicity. Toxicol. Appl. Pharmacol. 1997, 143, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Umbenhauer, D.R.; Lankas, G.R.; Pippert, T.R.; Wise, L.D.; Cartwright, M.E.; Hall, S.J.; Beare, C.M. Identification of a P-Glycoprotein-Deficient Subpopulation in the CF-1 Mouse Strain Using a Restriction Fragment Length Polymorphism. Toxicol. Appl. Pharmacol. 1997, 146, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Mealey, K.L.; Bentjen, S.A.; Gay, J.M.; Cantor, G.H. Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics 2001, 11, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Cooray, H.C.; Blackmore, C.G.; Maskell, L.; Barrand, M.A. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport 2002, 13, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Harati, R.; Benech, H.; Villégier, A.S.; Mabondzo, A. P-Glycoprotein, Breast Cancer Resistance Protein, Organic Anion Transporter 3, and Transporting Peptide 1a4 during Blood–Brain Barrier Maturation: Involvement of Wnt/β-Catenin and Endothelin-1 Signaling. Mol. Pharm. 2013, 10, 1566–1580. [Google Scholar] [CrossRef] [PubMed]
- Cisternino, S.; Mercier, C.; Bourasset, F.; Roux, F.; Scherrmann, J.-M. Expression, Up-Regulation, and Transport Activity of the Multidrug-Resistance Protein Abcg2 at the Mouse Blood-Brain Barrier. Cancer Res. 2004, 64, 3296–3301. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Römermann, K.; Karch, R.; Wulkersdorfer, B.; Stanek, J.; Philippe, C.; Maier-Salamon, A.; Haslacher, H.; Jungbauer, C.; Wadsak, W.; et al. Pilot PET Study to Assess the Functional Interplay Between ABCB1 and ABCG2 at the Human Blood-Brain Barrier. Clin. Pharmacol. Ther. 2016, 100, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Orphan nuclear receptors in drug discovery. Drug Discov. Today 2007, 12, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Geick, A.; Eichelbaum, M.; Burk, O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001, 276, 14581–14587. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.O.; Hartz, A.M.S.; Fricker, G.; Miller, D.S. Pregnane X Receptor Up-Regulation of P-Glycoprotein Expression and Transport Function at the Blood-Brain Barrier. Available online: http://molpharm.aspetjournals.org (accessed on 13 August 2017).
- Bauer, B.; Yang, X.; Hartz, A.M.S.; Olson, E.R.; Zhao, R.; Kalvass, J.C.; Pollack, G.M.; Miller, D.S. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol. Pharmacol. 2006, 70, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sykes, D.B.; Miller, D.S. Constitutive Androstane Receptor-Mediated Up-Regulation of ATP-Driven Xenobiotic Efflux Transporters at the Blood-Brain Barrier. Mol. Pharmacol. 2010, 78, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hawkins, B.T.; Miller, D.S. Aryl hydrocarbon receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. FASEB J. 2011, 25, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Mahringer, A.; Fricker, G. BCRP at the Blood–Brain Barrier: Genomic Regulation by 17β-Estradiol. Mol. Pharm. 2010, 7, 1835–1847. [Google Scholar] [CrossRef] [PubMed]
- Synold, T.W.; Dussault, I.; Forman, B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Othman, R.T.; Kimishi, I.; Bradshaw, T.D.; Storer, L.C.; Korshunov, A.; Pfister, S.M.; Grundy, R.G.; Kerr, I.D.; Coyle, B. Overcoming multiple drug resistance mechanisms in medulloblastoma. Acta Neuropathol. Commun. 2014, 2, 57. [Google Scholar] [CrossRef] [PubMed]
- Goldwirt, L.; Beccaria, K.; Carpentier, A.; Farinotti, R.; Fernandez, C. Irinotecan and temozolomide brain distribution: A focus on ABCB1. Cancer Chemother. Pharmacol. 2014, 74, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Schaich, M.; Kestel, L.; Pfirrmann, M.; Robel, K.; Illmer, T.; Kramer, M.; Dill, C.; Ehninger, G.; Schackert, G.; Krex, D. A MDR1 (ABCB1) gene single nucleotide polymorphism predicts outcome of temozolomide treatment in glioblastoma patients. Ann. Oncol. 2009, 20, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Tivnan, A.; Zakaria, Z.; O’Leary, C.; Kogel, D.; Pokorny, J.L.; Sarkaria, J.N.; Prehn, J.H. Inhibition of multidrug resistance protein 1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Front. Neurosci. 2015, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Martín, V.; Sanchez-Sanchez, A.M.; Herrera, F.; Gomez-Manzano, C.; Fueyo, J.; Alvarez-Vega, M.A.; Antolín, I.; Rodriguez, C. Melatonin-induced methylation of the ABCG2/BCRP promoter as a novel mechanism to overcome multidrug resistance in brain tumour stem cells. Br. J. Cancer 2013, 108, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Durmaz, R.; Deliorman, S.; Uyar, R.; Işiksoy, S.; Erol, K.; Tel, E. The effects of anticancer drugs in combination with nimodipine and verapamil on cultured cells of glioblastoma multiforme. Clin. Neurol. Neurosurg. 1999, 101, 238–244. [Google Scholar] [CrossRef]
- Nakai, E.; Park, K.; Yawata, T.; Chihara, T.; Kumazawa, A.; Nakabayashi, H.; Shimizu, K. Enhanced MDR1 Expression and Chemoresistance of Cancer Stem Cells Derived from Glioblastoma. Cancer Investig. 2009, 27, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Lum, B.L.; Kaubisch, S.; Yahanda, A.M.; Adler, K.M.; Jew, L.; Ehsan, M.N.; Brophy, N.A.; Halsey, J.; Gosland, M.P. Alteration of etoposide pharmacokinetics and pharmacodynamics by cyclosporine in a phase I trial to modulate modulate multidrug resistance. J. Clin. Oncol. 1992, 10, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Morfouace, M.; Cheepala, S.; Jackson, S.; Fukuda, Y.; Patel, Y.T.; Fatima, S.; Kawauchi, D.; Shelat, A.A.; Stewart, C.F.; Sorrentino, B.P.; et al. ABCG2 Transporter Expression Impacts Group 3 Medulloblastoma Response to Chemotherapy. Cancer Res. 2015, 75, 3879–3889. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.F.; Mukthavavam, R.; Chao, Y.; Bharati, S.; Fogal, V.; Pastorino, S.; Cong, X.; Nomura, N.; Gallagher, M.; Vali, S.; et al. Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. J. Transl. Med. 2014, 12. [Google Scholar] [CrossRef]
- Rao, V.K.; Wangsa, D.; Robey, R.W.; Huff, L.; Honjo, Y.; Hung, J.; Knutsen, T.; Ried, T.; Bates, S.E. Characterization of ABCG2 gene amplification manifesting as extrachromosomal DNA in mitoxantrone-selected SF295 human glioblastoma cells. Cancer Genet. Cytogenet. 2005, 160, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Balayssac, D.; Cayre, A.; Authier, N.; Bourdu, S.; Penault-Llorca, F.; Gillet, J.P.; Maublant, J.; Eschalier, A.; Coudore, F. Patterns of P-glycoprotein activity in the nervous system during vincristine-induced neuropathy in rats. J. Peripher. Nerv. Syst. 2005, 10, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Kemper, E.M.; van Zandbergen, A.E.; Cleypool, C.; Mos, H.A.; Boogerd, W.; Beijnen, J.H.; van Tellingen, O. Increased Penetration of Paclitaxel into the Brain by Inhibition of P-Glycoprotein. Clin. Cancer Res. 2003, 9, 2847–2855. [Google Scholar]
- Fellner, S.; Bauer, B.; Miller, D.S.; Schaffrik, M.; Fankhänel, M.; Spruss, T.; Bernhardt, G.; Graeff, C.; Färber, L.; Gschaidmeier, H.; et al. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J. Clin. Investig. 2002, 110, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; de Gooijer, M.C.; Roig, E.M.; Buil, L.C.M.; Christner, S.M.; Beumer, J.H.; Eurdinger, T.W.; Beijnen, J.H.; van Tellingen, O. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin. Cancer Res. 2014, 20, 2703–2713. [Google Scholar] [CrossRef] [PubMed]
- Tomaszowski, K.H.; Schirrmacher, R.; Kaina, B. Multidrug Efflux Pumps Attenuate the Effect of MGMT Inhibitors. Mol. Pharm. 2015, 12, 3924–3934. [Google Scholar] [CrossRef] [PubMed]
- Lagas, J.S.; van Waterschoot, R.A.B.; van Tilburg, V.A.C.J.; Hillebrand, M.J.; Lankheet, N.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin. Cancer Res. 2009, 15, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Mittapalli, R.K.; Zellmer, D.M.; Gallardo, J.L.; Donelson, R.; Seiler, C.; Decker, S.A.; Santacruz, K.S.; Pokorny, J.L.; Sarkaria, J.N.; et al. Active efflux of Dasatinib from the brain limits efficacy against murine glioblastoma: Broad implications for the clinical use of molecularly targeted agents. Mol. Cancer Ther. 2012, 11, 2183–2192. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Lagas, J.S.; Lankheet, N.A.G.; Poller, B.; Hillebrand, M.J.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int. J. Cancer 2012, 130, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, R.K.; Mittapalli, R.K.; Elmquist, W.F. Pharmacokinetic assessment of efflux transport in sunitinib distribution to the brain. J. Pharmacol. Exp. Ther. 2013, 347, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Lagas, J.S.; van Waterschoot, R.A.B.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol. Cancer Ther. 2010, 9, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takaoka, A.S.; Naishiro, Y.; Hayashi, R.; Maruyama, K.; Maesawa, C.; Ochiai, A.; Hirohashi, S. Transactivation of the Multidrug Resistance 1 Gene by T-Cell Factor 4/β-Catenin Complex in Early Colorectal Carcinogenesis. Cancer Res. 2000, 60, 4761–4766. [Google Scholar] [PubMed]
- Lim, J.C.; Kania, K.D.; Wijesuriya, H.; Chawla, S.; Sethi, J.K.; Pulaski, L.; Romero, I.A.; Couraud, P.O.; Weksler, B.B.; Hladky, S.B.; et al. Activation of β-catenin signalling by GSK-3 inhibition increases P-glycoprotein expression in brain endothelial cells. J. Neurochem. 2008, 106, 1855–1865. [Google Scholar] [CrossRef] [PubMed]
- Pinzón-Daza, M.L.; Salaroglio, I.C.; Kopecka, J.; Garzòn, R.; Couraud, P.-O.; Ghigo, D.; Riganti, C. The Cross-Talk between Canonical and Non-Canonical Wnt-Dependent Pathways Regulates P-Glycoprotein Expression in Human Blood–Brain Barrier Cells. J. Cereb. Blood Flow Metab. 2014, 34, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Sims-Mourtada, J.; Izzo, J.G.; Ajani, J.; Chao, K.S.C. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007, 26, 5674–5679. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Kunkalla, K.; Qu, C.; Schlette, E.; Neelapu, S.S.; Samaniego, F.; Vega, F. ABCG2 is a direct transcriptional target of hedgehog signaling and involved in stroma-induced drug tolerance in diffuse large B-cell lymphoma. Oncogene 2011, 30, 4874–4886. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.J.; van der Sandt, I.C.J.; Voorwinden, L.H.; Vu, D.; Nielsen, J.L.; de Boer, A.G.; Breimer, D.D. Astrocytes Increase the Functional Expression of P-Glycoprotein in an In Vitro Model of The Blood-Brain Barrier. Pharm. Res. 2000, 17, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- De Vries, H.E.; Blom-Roosemalen, M.C.M.; van Oosten, M.; de Boer, A.G.; van Berkel, T.J.C.; Breimer, D.D.; Kuiper, J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J. Neuroimmunol. 1996, 64, 37–43. [Google Scholar] [CrossRef]
- Théron, D.; de Lagerie, S.B.; Tardivel, S.; Pélerin, H.; Demeuse, P.; Mercier, C.; Mabondzo, A.; Farinotti, R.; Lacour, B.; Roux, F.; et al. Influence of tumor necrosis factor-α on the expression and function of P-glycoprotein in an immortalised rat brain capillary endothelial cell line, GPNT. Biochem. Pharmacol. 2003, 66, 579–587. [Google Scholar] [CrossRef]
- Hartz, A.M.S.; Miller, D.S.; Carolina, N. Tumor Necrosis Factor α and Endothelin-1 Increase P-Glycoprotein Expression and Transport Activity at the Blood-brain Barrier. Mol. Pharmacol. 2007, 71, 667–675. [Google Scholar]
- Schuetz, E.G.; Furuya, K.N.; Schuetz, J.D. Interindividual variation in expression of P-glycoprotein in normal human liver and secondary hepatic neoplasms. J. Pharmacol. Exp. Ther. 1995, 275, 1011–1018. [Google Scholar] [PubMed]
- Bebawy, M.; Chetty, M. Gender Differences in P-Glycoprotein Expression and Function: Effects on Drug Disposition and Outcome. Curr. Drug Metab. 2009, 10, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Zhao, Y.L.; Nadai, M.; Naruhashi, K.; Shimizu, A.; Takagi, K.; Takagi, K.; Hasegawa, T. Gender-related differences in expression and function of hepatic P-glycoprotein and multidrug resistance-associated protein (Mrp2) in rats. Life Sci. 2006, 79, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Merino, G.; van Herwaarden, A.E.; Wagenaar, E.; Jonker, J.W.; Schinkel, A.H. Sex-Dependent Expression and Activity of the ATP-Binding Cassette Transporter Breast Cancer Resistance Protein (BCRP/ABCG2) in Liver. Mol. Pharmacol. 2005, 67, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Van Assema, D.M.E.; Lubberink, M.; Boellaard, R.; Schuit, R.C.; Windhorst, A.D.; Scheltens, P.; Lammertsma, A.A.; van Berckel, B.N.M. P-Glycoprotein Function at the Blood–Brain Barrier: Effects of Age and Gender. Mol. Imaging Biol. 2012, 14, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.-N.; Hou, W.-Y.; Xu, S.-F.; Lu, Y.-F.; Liu, J. Ontogeny, aging, and gender-related changes in hepatic multidrug resistant protein genes in rats. Life Sci. 2017, 170, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The role of ABC transporters in clinical practice. Oncologist 2003, 8, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, A.; Ierano, C.; Szakacs, G.; Robey, R.W.; Bates, S.E. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011, 50, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Linton, K.J. Investigational ABC transporter inhibitors. Expert Opin. Investig. Drugs 2012, 21, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Wolburg-Buchholz, K.; Kraus, J.; Rascher-Eggstein, G.; Liebner, S.; Hamm, S.; Duffner, F.; Grote, E.-H.; Risau, W.; Engelhardt, B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003, 105, 586–592. [Google Scholar] [PubMed]
- Bhowmik, A.; Khan, R.; Ghosh, M.K. Blood brain barrier: A challenge for effectual therapy of brain tumors. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K.; Vaughan, M.M.; Peress, N.S.; Slocum, H.K.; Rustum, Y.M. MDR1 P-glycoprotein is expressed by endothelial cells of newly formed capillaries in human gliomas but is not expressed in the neovasculature of other primary tumors. Am. J. Pathol. 1996, 149, 853–858. [Google Scholar] [PubMed]
- Fattori, S.; Becherini, F.; Cianfriglia, M.; Parenti, G.; Romanini, A.; Castagna, M. Human brain tumors: Multidrug-Resistance P-glycoprotein expression in tumor cells and intratumoral capillary endothelial cells. Virchows Arch. 2007, 451, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Shedid, D.; Beaulieu, E.; del Maestro, R.F.; Moghrabi, A.; Ghosn, P.B.; Moumdjian, R.; Berthelet, F.; Béliveau, R. Expression of multidrug-resistance P-glycoprotein (MDR1) in human brain tumors. Int. J. Cancer 2001, 93, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, E.R.; Fine, R.L. Increased permeability of the blood-brain barrier to chemotherapy in metastatic brain tumors: Establishing a treatment paradigm. J. Clin. Oncol. 2007, 25, 2306–3212. [Google Scholar] [CrossRef] [PubMed]
- Lockman, P.R.; Mittapalli, R.K.; Taskar, K.S.; Rudraraju, V.; Gril, B.; Bohn, K.A.; Adkins, C.E.; Roberts, A.; Thorsheim, H.R.; Gaasch, J.A.; et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res. 2010, 16, 5664–5678. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Kirkwood, J.M. Temozolomide, a Novel Alkylating Agent with Activity in the Central Nervous System, May Improve the Treatment of Advanced Metastatic Melanoma. Oncologist 2000, 5, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Yung, W.K.A.; Albright, R.E.; Olson, J.; Fredericks, R.; Fink, K.; Prados, M.D.; Brada, M.; Spence, A.; Hohl, R.J.; Shapiro, W.; et al. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br. J. Cancer 2000, 83, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Bleau, A.M.; Huse, J.T.; Holland, E.C. The ABCG2 resistance network of glioblastoma. Cell Cycle 2009, 8, 2936–2944. [Google Scholar] [CrossRef] [PubMed]
- Shai, R.; Shi, T.; Kremen, T.J.; Horvath, S.; Liau, L.M.; Cloughesy, T.F.; Mischel, P.S.; Nelson, S.F. Gene expression profiling identifies molecular subtypes of gliomas. Oncogene 2003, 22, 4918–4923. [Google Scholar] [CrossRef] [PubMed]
- French, P.J.; Swagemakers, S.M.A.; Nagel, J.H.A.; Kouwenhoven, M.C.M.; Brouwer, E.; van der Spek, P.; Luider, T.M.; Kros, J.M.; van den Bent, M.J.; Sillevis Smitt, P.A. Gene Expression Profiles Associated with Treatment Response in Oligodendrogliomas. Cancer Res. 2005, 65, 11335–11344. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Kotliarov, Y.; Steed, M.E.; Christopher, N.; Walling, J.; Su, Q.; Center, A.; Heiss, J.; Rosenblum, M.; Mikkelsen, T.; Zenklusen, J.C.; et al. High-resolution global genomic survey of 178 gliomas reveals novel regions of copy number alteration and allelic imbalances. Cancer Res. 2006, 66, 9428–9436. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Gorter, J.A.; Redeker, S.; van Vliet, E.A.; Ramkema, M.; Scheffer, G.L.; Scheper, R.J.; van der Valk, P.; Leenstra, S.; Baayen, J.C.; et al. Localization of Breast Cancer Resistance Protein (BCRP) in Microvessel Endothelium of Human Control and Epileptic Brain. Epilepsia 2005, 46, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.; Zaiden, N.; Chong, K.-H.; See, S.-J.; Wong, M.-C.; Ang, B.-T.; Tang, C. Characterization of a side population of astrocytoma cells in response to temozolomide. J. Neurosurg. 2008, 109, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Emery, I.F.; Gopalan, A.; Wood, S.; Chow, K.; Battelli, C.; George, J.; Blaszyk, H.; Florman, J.; Yun, K. Expression and function of ABCG2 and XIAP in glioblastomas. J. Neurooncol. 2017, 133, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt Pathway Regulates the Side Population Phenotype and ABCG2 Activity in Glioma Tumor Stem-like Cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Rodriguez-Cruz, V.; Ramkissoon, S.H.; Ligon, K.L.; Greco, S.J.; Rameshwar, P.; Munoz, J.L.; Rodriguez-Cruz, V.; Ramkissoon, S.H.; Ligon, K.L.; et al. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget 2015, 6, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Salphati, L.; Heffron, T.P.; Alicke, B.; Nishimura, M.; Barck, K.; Carano, R.A.; Cheong, J.; Edgar, K.A.; Greve, J.; Kharbanda, S.; et al. Targeting the PI3K pathway in the brain—Efficacy of a PI3K inhibitor optimized to cross the blood-brain barrier. Clin. Cancer Res. 2012, 18, 6239–6248. [Google Scholar] [CrossRef] [PubMed]
- Salphati, L.; Lee, L.B.; Pang, J.; Plise, E.G.; Zhang, X. Role of P-glycoprotein and breast cancer resistance protein-1 in the brain penetration and brain pharmacodynamic activity of the novel phosphatidylinositol 3-kinase inhibitor GDC-0941. Drug Metab. Dispos. 2010, 38, 1422–1426. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Blaes, J.; Liao, Y.; Solecki, G.; Gömmel, M.; Berghoff, A.S.; Salphati, L.; Wallin, J.J.; Phillips, H.S.; Wick, W.; et al. Impact of Blood-Brain Barrier Integrity on Tumor Growth and Therapy Response in Brain Metastases. Clin. Cancer Res. 2016, 22, 6078–6087. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. From blood–brain barrier to blood–brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.J.; Robinson, G.W. Medulloblastoma-translating discoveries from the bench to the bedside. Nat. Rev. Clin. Oncol. 2014, 11, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.W.; Jäger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.-J.; Pugh, T.J.; Hovestadt, V.; Stütz, A.M.T.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Remke, M.; Hielscher, T.; Northcott, P.A.; Witt, H.; Ryzhova, M.; Wittmann, A.; Benner, A.; von Deimling, A.; Scheurlen, W.; Perry, A.; et al. Adult medulloblastoma comprises three major molecular variants. J. Clin. Oncol. 2011, 29, 2717–2723. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; Northcott, P.A.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013, 14, 1200–1207. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- King, A.A.; Seidel, K.; Di, C.; Leisenring, W.M.; Perkins, S.M.; Krull, K.R.; Sklar, C.A.; Green, D.M.; Armstrong, G.T.; Zeltzer, L.K.; et al. Long-term neurologic health and psychosocial function of adult survivors of childhood medulloblastoma/PNET: A report from the Childhood Cancer Survivor Study. Neuro Oncol. 2016, 39, 242. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.W.; Onilude, O.E.; Lindsey, J.C.; Lusher, M.E.; Weston, C.L.; Taylor, R.E.; Pearson, A.D.; Clifford, S.C. United Kingdom Children’s Cancer Study Group Brain Tumour Committee, β-Catenin status predicts a favorable outcome in childhood medulloblastoma: The United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J. Clin. Oncol. 2005, 23, 7951–7957. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- Moxon-Emre, I.; Bouffet, E.; Taylor, M.D.; Laperriere, N.; Scantlebury, N.; Law, N.; Spiegler, B.J.; Malkin, D.; Janzen, L.; Mabbott, D. Impact of craniospinal dose, boost volume, and neurologic complications on intellectual outcome in patients with medulloblastoma. J. Clin. Oncol. 2014, 32, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Miller, H.L.; Jensen, P.; Hernan, R.; Connelly, M.; Wetmore, C.; Zindy, F.; Roussel, M.F.; Curran, T.; Gilbertson, R.J.; et al. A molecular fingerprint for medulloblastoma. Cancer Res. 2003, 63, 5428–5437. [Google Scholar] [PubMed]
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Buylla, A.; Ihrie, R.A. Sonic hedgehog signaling in the postnatal brain. Semin. Cell Dev. Biol. 2014, 33, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Wechsler-Reya, R.J.; Scott, M.P. Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron 1999, 22, 103–114. [Google Scholar] [CrossRef]
- Hatten, M.E.; Alder, J.; Zimmerman, K.; Heintz, N. Genes involved in cerebellar cell specification and differentiation. Curr. Opin. Neurobiol. 1997, 7, 40–47. [Google Scholar] [CrossRef]
- Muzio, L.L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet J. Rare Dis. 2008, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Milenković, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; van Sluis, P.; Troost, D.; Meeteren, N.S.; Caron, H.N.; Cloos, J.; et al. Integrated Genomics Identifies Five Medulloblastoma Subtypes with Distinct Genetic Profiles, Pathway Signatures and Clinicopathological Features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef] [PubMed]
- Schüller, U.; Heine, V.M.; Mao, J.; Kho, A.T.; Dillon, A.K.; Han, Y.-G.; Huillard, E.; Sun, T.; Ligon, A.H.; Qian, Y.; et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 2008, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Grammel, D.; Warmuth-Metz, M.; von Bueren, A.O.; Kool, M.; Pietsch, T.; Kretzschmar, H.A.; Rowitch, D.H.; Rutkowski, S.; Pfister, S.M.; Schüller, U. Sonic hedgehog-associated medulloblastoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol. 2012, 123, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Uziel, T.; Ayrault, O.; Calabrese, C.; Valentine, M.; Rehg, J.E.; Gilbertson, R.J.; Sherr, C.J.; Roussel, M.F. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 2007, 67, 2676–2684. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, D.; Robinson, G.; Uziel, T.; Gibson, P.; Rehg, J.; Gao, C.; Finkelstein, D.; Qu, C.; Pounds, S.; Ellison, D.W.; et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 2012, 21, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Moore, C.E.; Wang, J.; Tewari, A.K.; Eroshkin, A.; Cho, Y.J.; Witt, H.; Korshunov, A.; Read, T.A.; Sun, J.L.; et al. An Animal Model of MYC-Driven Medulloblastoma. Cancer Cell 2012, 21, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Vo, B.T.; Wolf, E.; Kawauchi, D.; Gebhardt, A.; Rehg, J.E.; Finkelstein, D.; Walz, S.; Murphy, B.L.; Youn, Y.H.; Han, Y.-G.; et al. The Interaction of Myc with Miz1 Defines Medulloblastoma Subgroup Identity. Cancer Cell 2016, 29, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Swartling, F.J.; Grimmer, M.R.; Hackett, C.S.; Northcott, P.A.; Fan, Q.-W.; Goldenberg, D.D.; Lau, J.; Masic, S.; Nguyen, K.; Yakovenko, S.; et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010, 24, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Swartling, F.J.; Savov, V.; Persson, A.I.; Chen, J.; Hackett, C.S.; Northcott, P.A.; Grimmer, M.R.; Lau, J.; Chesler, L.; Perry, A.; et al. Distinct Neural Stem Cell Populations Give Rise to Disparate Brain Tumors in Response to N-MYC. Cancer Cell 2012, 21, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Jones, D.T.W.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.-J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.W.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Meeteren, A.S.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Dubuc, A.M.; Pfister, S.; Taylor, M.D. Molecular subgroups of medulloblastoma. Expert Rev. Neurother. 2012, 12, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.H.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Hielscher, T.; Dubuc, A.; Mack, S.; Shih, D.; Remke, M.; Al-Halabi, H.; Albrecht, S.; Jabado, N.; Eberhart, C.G.; et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011, 122, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Pierre, D.; Si, R.; Deng, W.; Ferrin, P.; Nilekar, A.U.; Peng, G.; Herron, J.A.; Bell, D.C.; Saltsburg, H. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2010, 337, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Liu, K.-W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Morfouace, M.; Shelat, A.; Jacus, M.; Freeman, B.B.; Turner, D.; Robinson, S.; Zindy, F.; Wang, Y.-D.; Finkelstein, D.; Ayrault, O.; et al. Pemetrexed and Gemcitabine as Combination Therapy for the Treatment of Group3 Medulloblastoma. Cancer Cell 2014, 25, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Remke, M.; Adamski, J.; Bartels, U.; Tabori, U.; Wang, X.; Huang, A.; Hawkins, C.; Mabbott, D.; Laperriere, N.; et al. Medulloblastoma subgroup-specific outcomes in irradiated children: Who are the true high-risk patients? Neuro Oncol. 2016, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef]
- Rood, B.R.; MacDonald, T.J.; Packer, R.J. Current treatment of medulloblastoma: Recent advances and future challenges. Semin. Oncol. 2004, 31, 666–675. [Google Scholar] [CrossRef] [PubMed]
- De Braganca, K.C.; Packer, R.J. Treatment options for medulloblastoma and CNS primitive neuroectodermal tumor (PNET). Curr. Treat. Options Neurol. 2013, 15, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Hatton, B.A.; Villavicencio, E.H.; Khanna, P.C.; Friedman, S.D.; Ditzler, S.; Pullar, B.; Robison, K.; White, K.F.; Tunkey, C.; et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc. Natl. Acad. Sci. USA 2012, 109, 7859–7864. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Iraci, N.; Soverini, S.; Diolaiti, D.; Gherardi, S.; Terragna, C.; Durante, S.; Valli, E.; Kalebic, T.; Bernardoni, R.; et al. c-MYC Oncoprotein Dictates Transcriptional Profiles of ATP-Binding Cassette Transporter Genes in Chronic Myelogenous Leukemia CD34+ Hematopoietic Progenitor Cells. Mol. Cancer Res. 2011, 9, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Haber, M.; Diolaiti, D.; Iraci, N.; Henderson, M.; Gherardi, S.; Valli, E.; Munoz, M.A.; Xue, C.; Flemming, C.; et al. Direct and coordinate regulation of ATP-binding cassette transporter genes by Myc factors generates specific transcription signatures that significantly affect the chemoresistance phenotype of cancer cells. J. Biol. Chem. 2010, 285, 19532–19543. [Google Scholar] [CrossRef] [PubMed]
- Ingram, W.J.; Crowther, L.M.; Little, E.B.; Freeman, R.; Harliwong, I.; Veleva, D.; Hassall, T.E.; Remke, M.; Taylor, M.D.; Hallahan, A.R.; et al. ABC transporter activity linked to radiation resistance and molecular subtype in pediatric medulloblastoma. Exp. Hematol. Oncol. 2013, 2, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.F.; Iacono, L.C.; Chintagumpala, M.; Kellie, S.J.; Ashley, D.; Zamboni, W.C.; Kirstein, M.N.; Fouladi, M.; Seele, L.G.; Wallace, D.; et al. Results of a Phase II Upfront Window of Pharmacokinetically Guided Topotecan in High-Risk Medulloblastoma and Supratentorial Primitive Neuroectodermal Tumor. J. Clin. Oncol. 2004, 22, 3357–3365. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
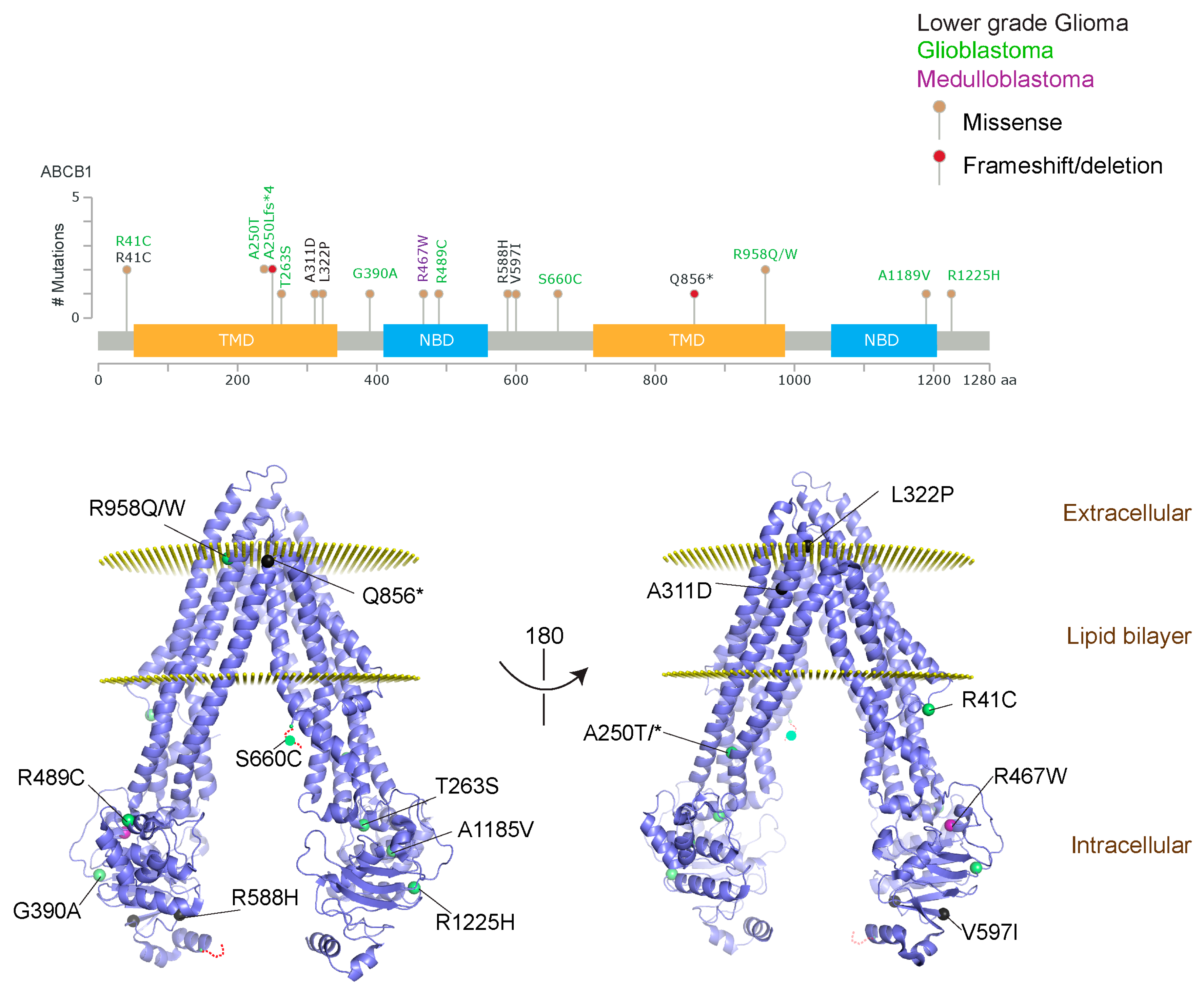{kind=link}
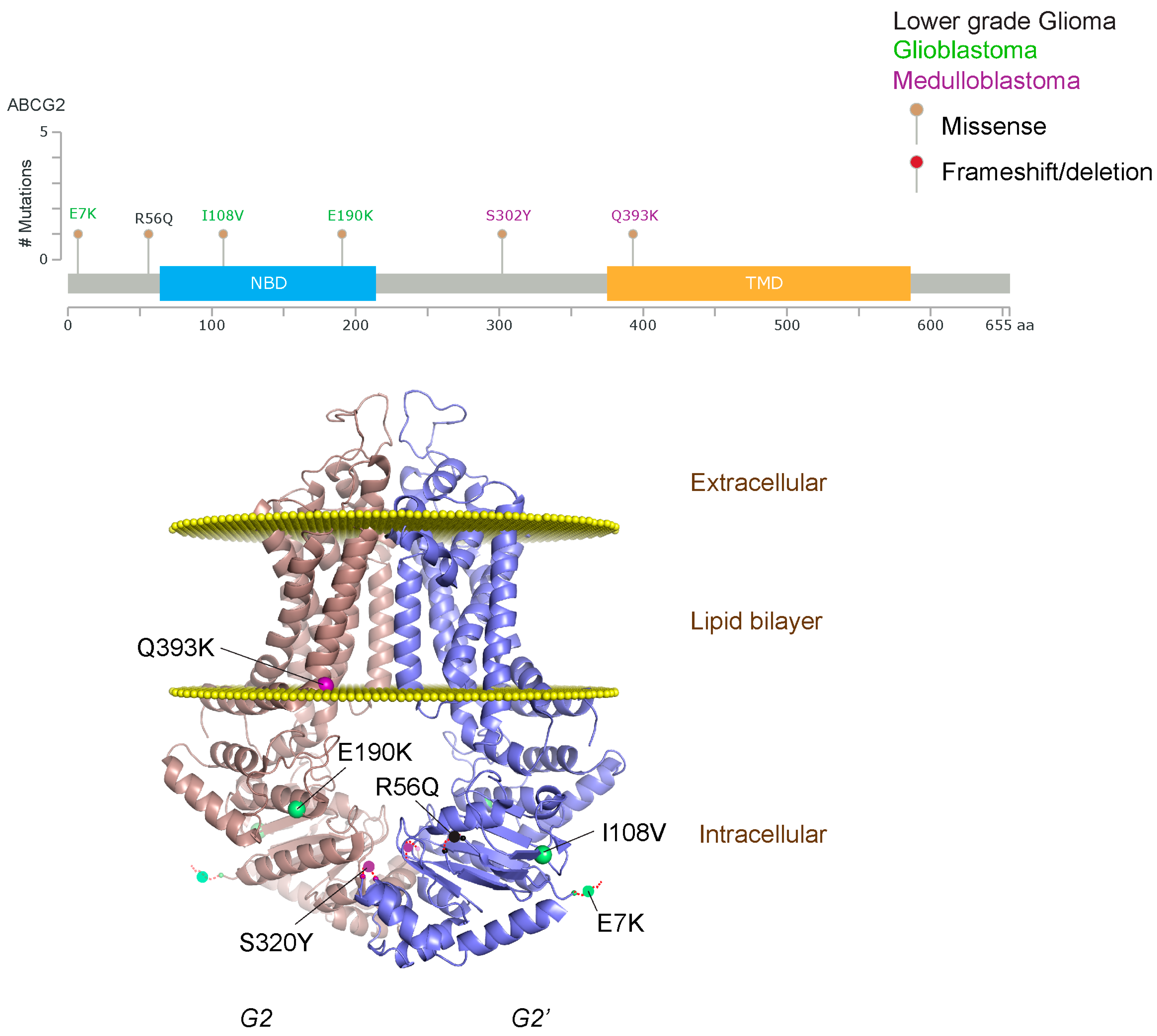{kind=link}
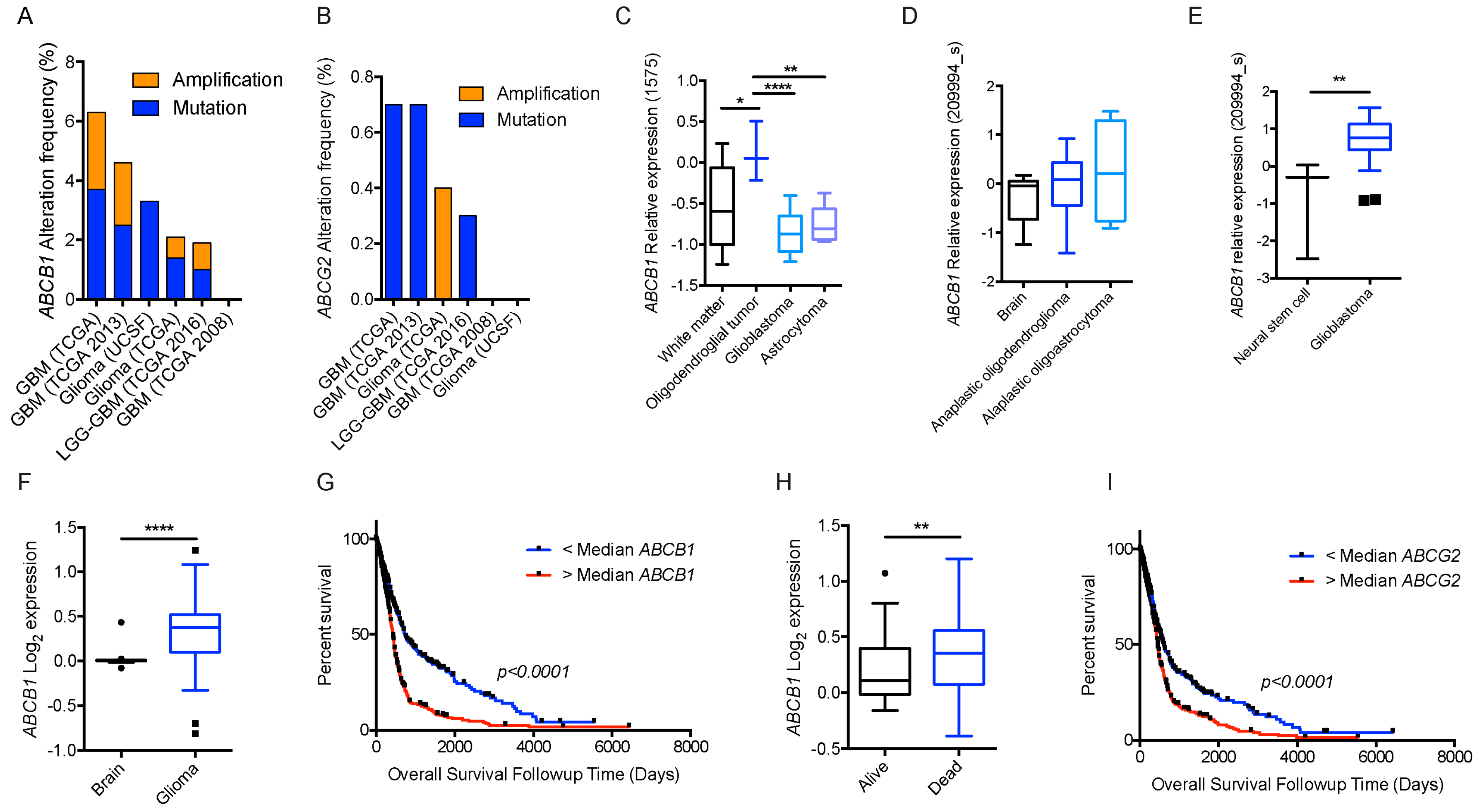{kind=link}
{kind=link}
{kind=link}
| Drug | Type of Drug/Cancer | Transporter Implicated in Drug Resistance | Model System |
|---|---|---|---|
Temozolomide (TMZ)  | Alkylating agent GBM, MB [38] | ABCB1 | Mdr1a−/− mice accumulated higher TMZ in the brain compared to Mdr1a+/+ mice [39] (Mdrla = Abcb1a). |
| ABCB1 | GBM patients with C/C variant at amino acid 1236 of ABCB1 respond better to TMZ than patients with C/T and T/T variants, which results in higher ABCB1 expression [40]. | ||
| ABCG2 | GBM cell lines (U251, A271), cell lines derived from primary tumors (MZ-327, MZ-18), and cell lines from recurrent grade IV tumors (MZ-256, MZ-304) were treated with TMZ. Cell survival was measured by MTT and trypan blue assays. Cells treated with reversan, which inhibits both ABCB1 and ABCG2 showed increased sensitivity to TMZ [41]. | ||
| ABCG2 | GBM cells line (A172, U87, and U373) and neurospheres derived from primary GBM showed increased sensitivity to TMZ in the presence of melatonin. Melatonin treatment affects ABCG2 level via promoter methylation [42]. | ||
Procarbazine  | Alkylating agent GBM and MB | ABCB1 | Primary tumor GBM cells treated with nimodipine, which blocks ABCB1, showed increased sensitivity to procarbazine in an MTT assay [43]. |
Lomustin/CCNU  | Alkylating agent GBM and MB | None reported in GBM/MB context | - |
Carmustin/BCNU  | Alkylating agent GBM | ABCB1 | GBM cancer stem cell line (U87CS), established by growing U87 GBM cell line in neuronal stem cell condition, showed over 8-fold increase in ABCB1 expression and exhibited greater resistance to BCNU [44]. |
Cyloposphamide  | Alkylating agent MB | None reported in GBM/MB context | - |
Carboplatin  | Platinum-based drugs. GBM, MB | ABCB1 | GBM cancer stem cell line (U87CS), established by growing U87 GBM cell line in neuronal stem cell condition, showed over 8-fold increase in ABCB1 expression and exhibited greater resistance tow carboplatin [44]. |
Cisplatin  | Platinum-based drugs. GBM, MB | None reported in GBM/MB context | - |
Etoposide  | Topoisomerase inhibitor | ABCB1 ABCG2 | Patients treated with cyclosporine A, which can inhibit both ABCB1 and ABCG2, had increased systemic exposure to etoposide [45] GBM cancer stem cell line (U87CS), established by growing U87 GBM cell line in neuronal stem cell condition, showed over 8-fold increase in ABCB1 expression and exhibited greater resistance to etoposide [44]. |
| ABCB1 | ABCB1 expression is associated with high-risk MB in large patient cohorts. | ||
| ABCB1 | MB cell lines (DAOY, MED1, MED4, MED4R, MED5R, MED6) express high level of ABCB1. Treatment of these cells with etoposide in combination with ABCB1 inhibitors vardenafil or verapamil sensitizes cells to etoposide measured by clonogenic assay [38]. | ||
Topotecan  | Topoisomerase inhibitor MB | ABCG2 | Inhibition of ABCG2 in Group 3 MB tumorspheres by FTC increased sensitivity of topotecan measured by cell titer glow assay and annexin V staining for apoptotic cells [46]; ABCG2 inhibition with Ko143 increased cytotoxicity of topotecan in Group 3 MB preclinical animal model [46]. |
Irinotecan  | Topoisomerase inhibitor MB and GBM | ABCB1 | CPT-11/SN-38 brain accumulation was found to be higher in Mdr1a+/− and Mdr1−/− mice compared to Mdr1a+/+ mice [39]. |
| ABCB1 | Sensitivity towards irinotecan increased in U118, U87, and SK72 GBM cell lines when ABCB1 was inhibited with pitavastatin as measured by alamar blue assay [47]. ABCB1 mRNA level increased with irinotecan concentration [47]. | ||
Mitoxanthrone  | Topoisomerase inhibitor MB and GBM | ABCG2 | SF395 human GBM cell line treated with mitoxanthrone resulted in ABCG2 being duplicated in mitoxanthrone-resistant clone [48]. |
| ABCG2 | Group 3 tumorspheres treated with ABCG2 inhibitor, FTC, showed about 3-fold increase in sensitivity to mitoxanthrone [46]. | ||
Vinblastine  | Vinca alkaloid/Anti-tubulin MB and GBM | ABCB1 | Mdr1a−/− mice showed 20-fold higher vinblastine level compared to Mdr1a+/+. The most striking difference in drug level was found in the brain [22]. |
Vincristine  | Vinca alkaloid/Anti-tubulin MB and GBM | ABCB1 | Rats injected with vincristine showed elevated level of ABCB1 in the brain. Transport activity, monitored by radioactive tracer, 99 mTc-sestamibi, was subsequently found to increase 4-fold 24 h post vincristine treatment [49]. |
Paclitaxel (Taxol)  | Anti-microtubule Glioma | ABCB1 | Oral administration of ABCB1 inhibitor, zosuquidar, in mice, increased penetration of paclitaxel in the brain [50]. |
| ABCB1 | Valspodar, an ABCB1 inhibitor, increased accumulation of paclitaxel in the brain of nude mice. Paclitaxel, in combination with valspodar, decreased tumor volume by 90% in nude mice bearing U118 MG glioblastoma [51]. | ||
Veliparib (ABT-888)  | PARP1/2 inhibitor GBM | ABCB1 ABCG2 | Tumors from spontaneous high grade glioma model Pten; p16Ink4a/p19Arf; K-Rasv12 were isolated and implanted into nude mice which then received single dose of TMZ, ABT-888, or both with or without elacridar, a dual inhibitor of ABCB1 and ABCG2. Elacridar enhanced brain penetration of ABT-888. Co administration of elacridar enhances efficacy of TMZ and ABT-888 reduced glioblastoma tumor burden [52]. |
Lomeguatrib (O6Benzylguanine/O6BG)  | MGMT inhibitor GBM | ABCB1 | O6BG can compete with Rhodamine 123 and pheophorbide A, substrates of ABCB1 and ABCG2 respectively, in uptake assays, indicating O6BG is a substrate of ABCB1 and ABCG2. |
| ABCG2 | Human glioblastoma GBP61 cells treated with verapamil and Ko143, which inhibit both ABCB1 and ABCG2 increased toxicity of O6BG and TMZ treatment [53]. | ||
Dasatinib  | BCR-ABL/SRC kinase/Tyrosine kinase inhibitor (TKI) GBM | ABCB1 ABCG2 | Abcb1a/b−/−, Abcg2−/−, Abcb1a/b−/−; Abcg2−/− mice received dasatinib by I.P and orally. Mice lacking both ABCB1 and ABCG2 showed higher accumulation in the brain than WT. Comparable level of dasatinib was observed in WT mice treated with ABCB1/ABCG2 dual inhibitor, elacridar and Abcb1a/b−/−; Abcg2−/− mice [54] |
| ABCB1 ABCG2 | mPDGFβ-induced de novo model of murine GBM induced in WT and Abcb1a/b−/−; Abcg2−/− (KO) mice were given 15 mg/kg dasatinib by oral gavage. Dasatinib level was double in both brain and tumor of KO mice compared to WT. KO mice treated with dasatinib also survived longer than WT [55]; In glioma cell lines from humans and murine models, treatment with elacridar (dual ABCB1 and ABCG2) inhibitor sensitizes cells to dasatinib [55] | ||
Sunitinib  | VEGFR, Flk1, PDGFR-α/β inhibitor Tyrosine kinase inhibitor (TKI) GBM | ABCB1 ABCG2 | MDCK-II cells overexpressing human ABCB1 and ABCG2 showed transport of sunitinib. Addition of elacridar inhibited sunitinib transport. Brain accumulation of sunitinib was found to be significantly high in Abcb1a/b−/−; Abcg2−/− mice compared to WT. Elacridar oral administration in WT cells yields similar level of brain sunitinib concentration to Abcb1a/b−/−; Abcg2−/− mice [56] |
| ABCB1 ABCG2 | Sunitinib was administered to WT, Abcb1a/b−/−, Abcg2−/−, Abcb1a/b−/−; Abcg2−/− mice by IP infusion. Brain/plasma ratio was found highest in Abcb1a/b−/−; Abcg2−/− mice. Treatment with elacridar (dual ABCB1/ABCG2) inhibitor gave a brain/plasma ratio similar to Abcb1a/b−/−; Abcg2−/− mice [57]. Zosuquidar (ABCB1 specific inhibitor) only moderately increased sunitinib brain concentration while and Ko143 (ABCG2 inhibitor) had no effect [57]. | ||
Sorafenib  | Tyrosine kinase inhibitor (TKI) GBM | ABCB1 ABCG2 | WT, Abcb1a/b−/−, Abcg2−/−, Abcb1a/b−/−; Abcg2−/− mice were given sorafenib. Sorafenib accumulation did not increase in the brain of ABCB1-lacking mice, but increased moderately in Abcg2−/−, and most significantly in Abcb1a/b−/−; Abcg2−/− mice. Elacridar (dual ABCB1 and ABCG2 inhibitor) increased brain exposure to sorafenib similar to double knockout mice [58]. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wijaya, J.; Fukuda, Y.; Schuetz, J.D. Obstacles to Brain Tumor Therapy: Key ABC Transporters. Int. J. Mol. Sci. 2017, 18, 2544. https://doi.org/10.3390/ijms18122544
Wijaya J, Fukuda Y, Schuetz JD. Obstacles to Brain Tumor Therapy: Key ABC Transporters. International Journal of Molecular Sciences. 2017; 18(12):2544. https://doi.org/10.3390/ijms18122544
Chicago/Turabian StyleWijaya, Juwina, Yu Fukuda, and John D. Schuetz. 2017. "Obstacles to Brain Tumor Therapy: Key ABC Transporters" International Journal of Molecular Sciences 18, no. 12: 2544. https://doi.org/10.3390/ijms18122544
APA StyleWijaya, J., Fukuda, Y., & Schuetz, J. D. (2017). Obstacles to Brain Tumor Therapy: Key ABC Transporters. International Journal of Molecular Sciences, 18(12), 2544. https://doi.org/10.3390/ijms18122544