Ascorbic Acid Attenuates Senescence of Human Osteoarthritic Osteoblasts

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
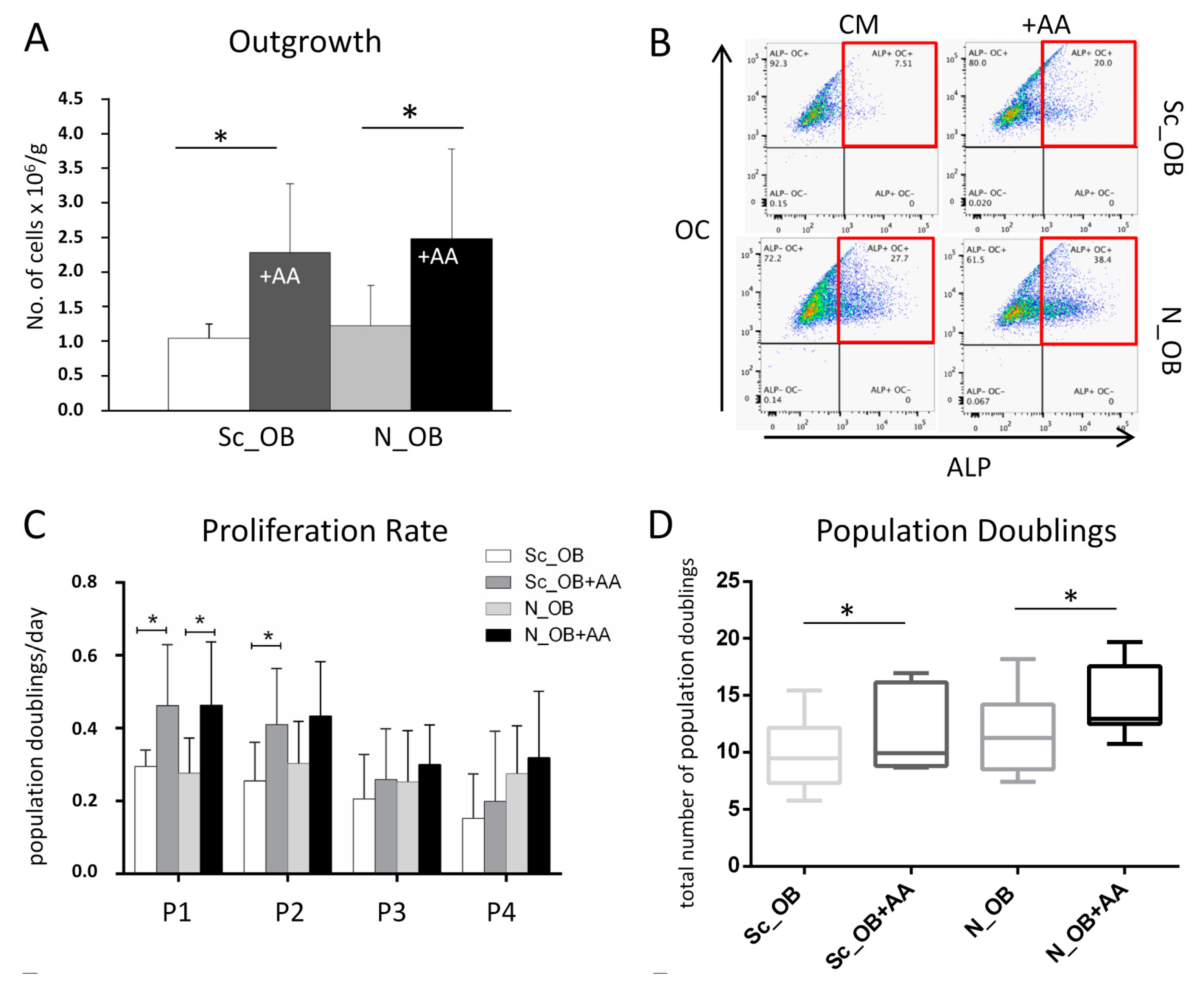
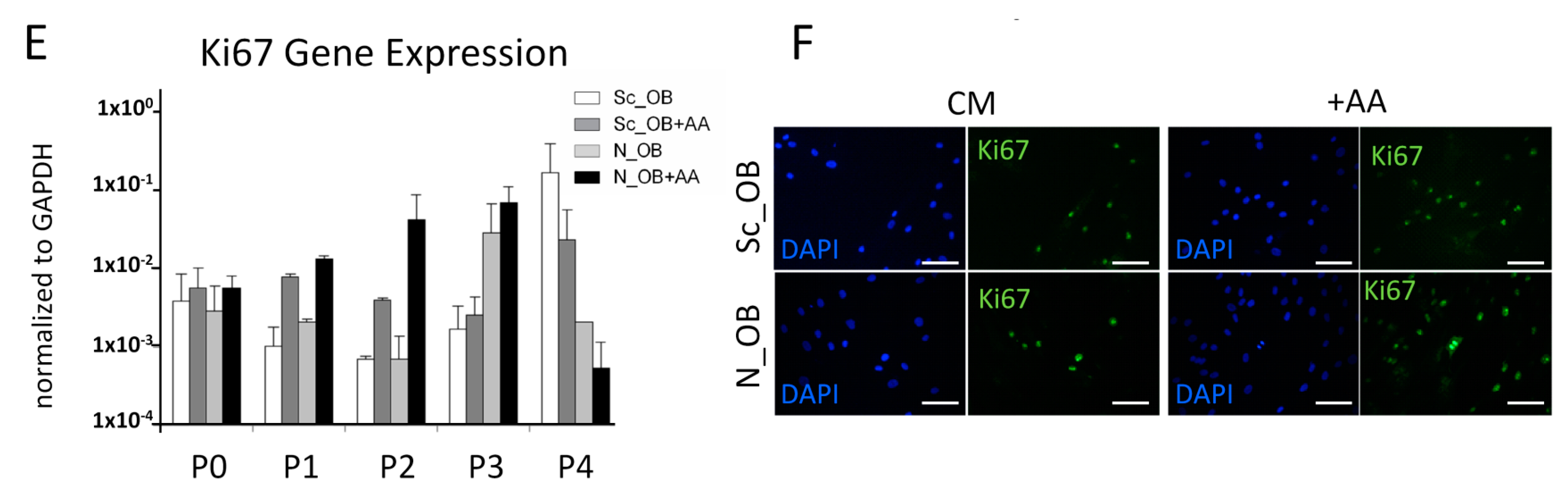
2.1. Effect of Ascorbic Acid on the Outgrowth and Proliferation Rate of Human Osteoarthritic OB
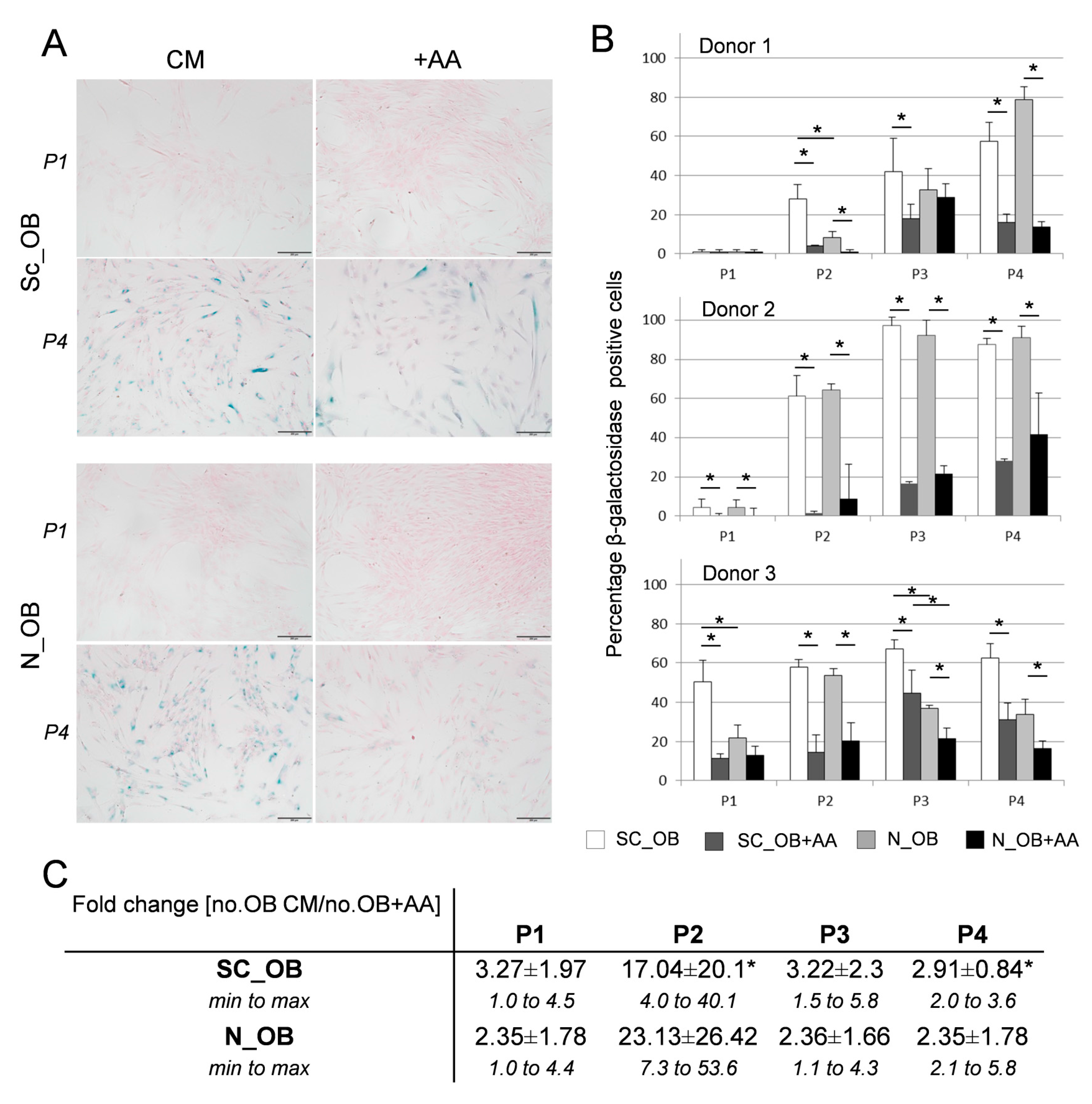
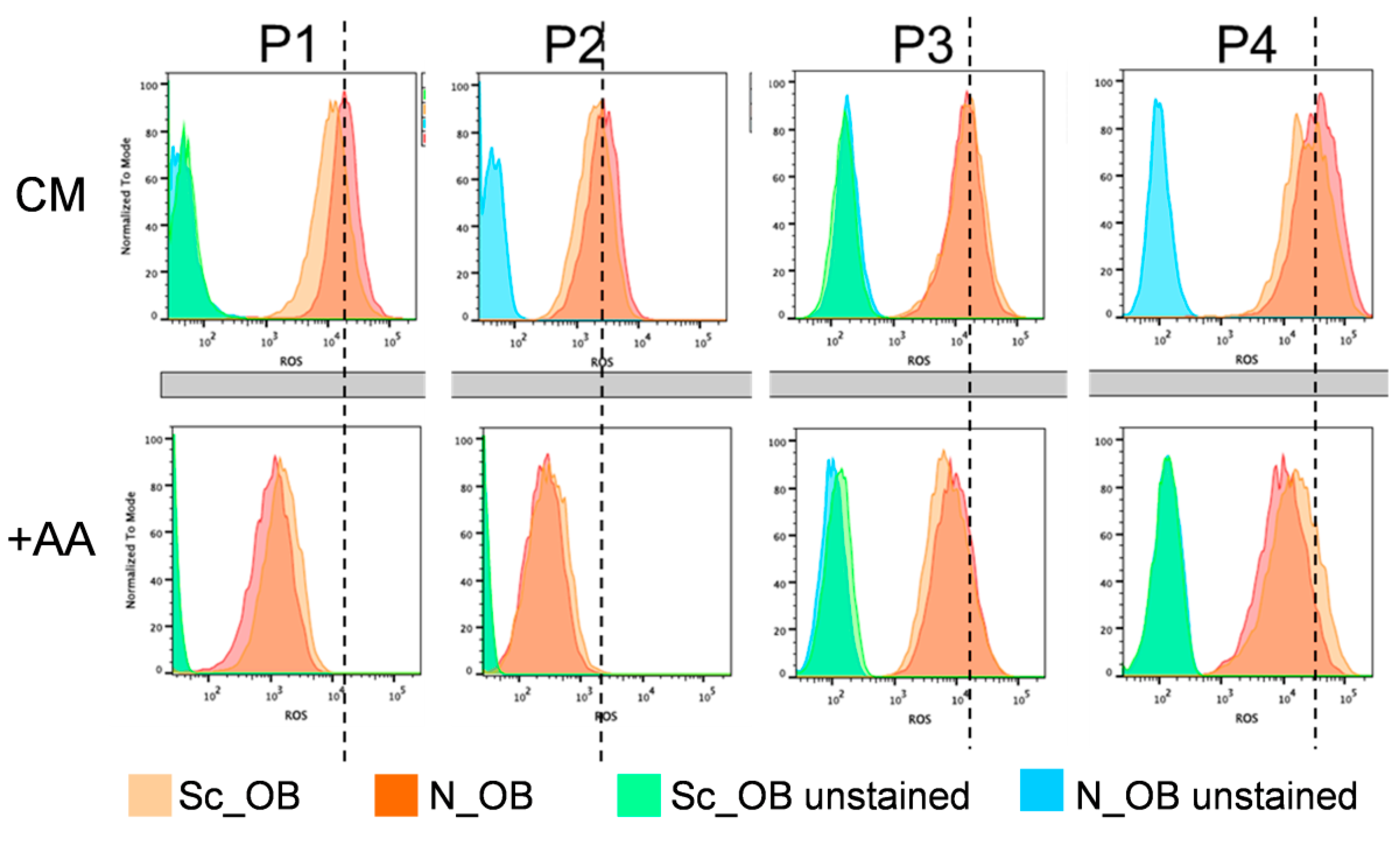
2.2. Attenuation of Human Osteoarthritic OB Senescence in the Presence of Ascorbic Acid
2.3. Osteogenic Differentiation of Human Osteoarthritic OB in the Presence of Ascorbic Acid
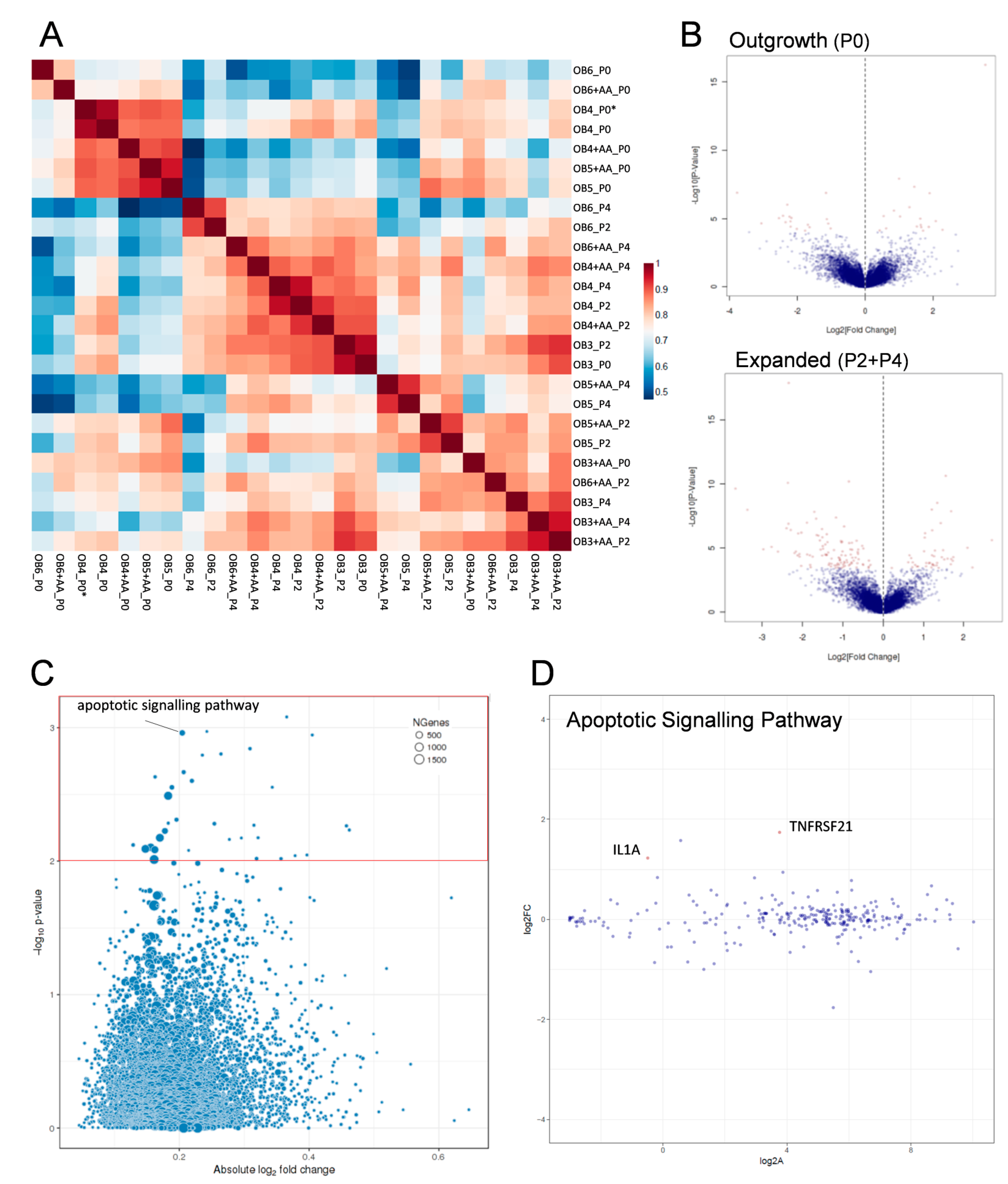
2.4. Transcriptomic Analysis of Human Osteoarthritic OB in the Absence and Presence of Ascorbic Acid
3. Discussion
4. Materials and Methods
4.1. Isolation and Expansion of Human Osteoarthritic Osteoblasts
4.2. Surface Marker Expression
4.3. Immunohistochemistry
4.4. Quantitative Real-Time Reverse Transcriptase PCR
4.5. Identification and Quantification of Senescent Cells
4.6. Quantification of Reactive Oxygen Species (ROS)
4.7. Osteogenic Differentiation
4.8. In Vivo Implantation of Osteogenic Constructs
4.9. Histological Processing
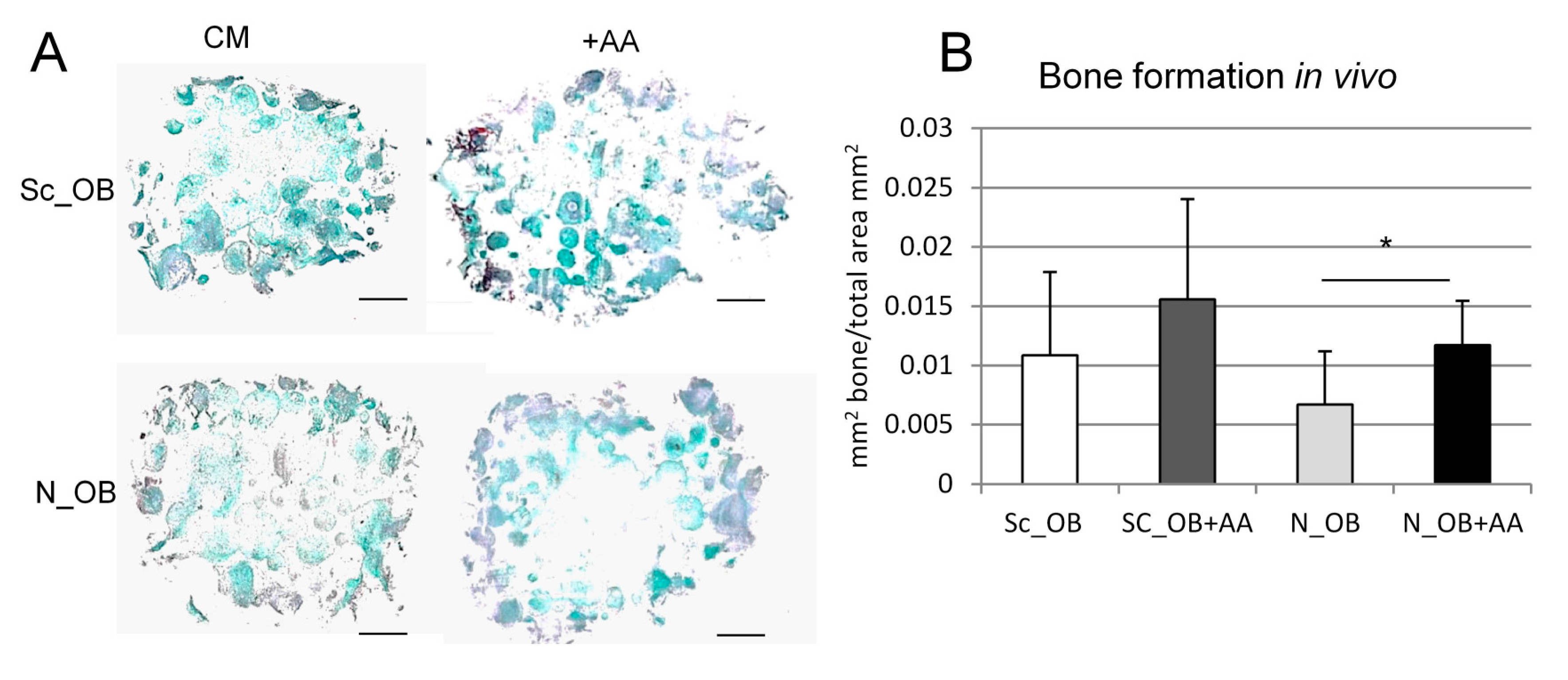
4.10. Analysis of Bone Formation In Vivo
4.11. Statistical Analyses
4.12. Transcriptomic Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Buckwalter, J.A.; Martin, J.A. Osteoarthritis. Adv. Drug Deliv. Rev. 2006, 58, 150–167. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, C.; Nguyen, C.; Lefevre-Colau, M.M.; Rannou, F.; Poiraudeau, S. Risk factors and burden of osteoarthritis. Ann. Phys. Rehabil. Med. 2016, 59, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B. Articular cartilage degradation in osteoarthritis. HSS J. 2012, 8, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A.; McWilliams, D.F.; Turley, M.J.; Dixon, M.R.; Fransès, R.E.; Mapp, P.I.; Wilson, D. Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatology 2010, 49, 1852–1861. [Google Scholar] [CrossRef]
- Yusup, A.; Kaneko, H.; Liu, L.; Ning, L.; Sadatsuki, R.; Hada, S.; Kamagata, K.; Kinoshita, M.; Futami, I.; Shimura, Y.; et al. Bone marrow lesions, subchondral bone cysts and subchondral bone attrition are associated with histological synovitis in patients with end-stage knee osteoarthritis: A cross-sectional study. Osteoarthr. Cartil. 2015, 23, 1858–1864. [Google Scholar] [CrossRef] [PubMed]
- Klose-Jensen, R.; Hartlev, L.B.; Boel, L.W.; Laursen, M.B.; Stengaard-Pedersen, K.; Keller, K.K.; Hauge, E.-M. Subchondral bone turnover, but not bone volume, is increased in early stage osteoarthritic lesions in the human hip joint. Osteoarthr. Cartil. 2015, 23, 2167–2173. [Google Scholar] [CrossRef]
- Anderson-MacKenzie, J.M.; Quasnichka, H.L.; Starr, R.L.; Lewis, E.J.; Billingham, M.E.J.; Bailey, A.J. Fundamental subchondral bone changes in spontaneous knee osteoarthritis. Int. J. Biochem. Cell Biol. 2005, 37, 224–236. [Google Scholar] [CrossRef]
- Hugle, T.; Geurts, J. What drives osteoarthritis?—Synovial versus subchondral bone pathology. Rheumatology 2017, 56, 1461–1471. [Google Scholar] [PubMed]
- Intema, F.; Hazewinkel, H.A.; Gouwens, D.; Bijlsma, J.W.J.; Weinans, H.; Lafeber, F.P.J.G.; Mastbergen, S.C. In early OA, thinning of the subchondral plate is directly related to cartilage damage: Results from a canine ACLT-meniscectomy model. Osteoarthr. Cartil. 2010, 18, 691–698. [Google Scholar] [CrossRef]
- Boyd, S.K.; Muller, R.; Zernicke, R.F. Mechanical and architectural bone adaptation in early stage experimental osteoarthritis. J. Bone Miner. Res. 2002, 17, 687–694. [Google Scholar] [CrossRef]
- Radin, E.L.; Rose, R.M. Role of subchondral bone in the initiation and progression of cartilage damage. Clin. Orthop. Relat. Res. 1986, 213, 34–40. [Google Scholar] [CrossRef]
- Prasadam, I.; Crawford, R.; Xiao, Y. Aggravation of ADAMTS and matrix metalloproteinase production and role of ERK1/2 pathway in the interaction of osteoarthritic subchondral bone osteoblasts and articular cartilage chondrocytes—Possible pathogenic role in osteoarthritis. J. Rheumatol. 2012, 39, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Westacott, C.I.; Webb, G.R.; Warnock, M.G.; Sims, J.V.; Elson, C.J. Alteration of cartilage metabolism by cells from osteoarthritic bone. Arthritis Rheum. 1997, 40, 1282–1291. [Google Scholar] [CrossRef]
- Sanchez, C.; Deberg, M.A.; Piccardi, N.; Msika, P.; Reginster, J.Y.; Henrotin, Y.E. Subchondral bone osteoblasts induce phenotypic changes in human osteoarthritic chondrocytes. Osteoarthr. Cartil. 2005, 13, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Deberg, M.A.; Piccardi, N.; Msika, P.; Reginster, J.Y.; Henrotin, Y.E. Osteoblasts from the sclerotic subchondral bone downregulate aggrecan but upregulate metalloproteinases expression by chondrocytes. This effect is mimicked by interleukin-6, -1β and oncostatin M pre-treated non-sclerotic osteoblasts. Osteoarthr. Cartil. 2005, 13, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Couchourel, D.; Aubry, I.; Delalandre, A.; Lavigne, M.; Martel-Pelletier, J.-P.; Lajeunesse, D. Altered mineralization of human osteoarthritic osteoblasts is attributable to abnormal type I collagen production. Arthritis Rheum. 2009, 60, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Hilal, G.; Martel-Pelletier, J.; Pelletier, J.P.; Ranger, P.; Lajeunesse, D. Osteoblast-like cells from human subchondral osteoarthritic bone demonstrate an altered phenotype in vitro: Possible role in subchondral bone sclerosis. Arthritis Rheum. 1998, 41, 891–899. [Google Scholar] [CrossRef]
- Massicotte, F.; Lajeunesse, D.; Benderdour, M.; Pelletier, J.-P.; Hilal, G.; Duval, N.; Martel-Pelletier, J. Can altered production of interleukin-1β, interleukin-6, transforming growth factor-β and prostaglandin E(2) by isolated human subchondral osteoblasts identify two subgroups of osteoarthritic patients. Osteoarthr. Cartil. 2002, 10, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Mansell, J.P.; Bailey, A.J. Abnormal cancellous bone collagen metabolism in osteoarthritis. J. Clin. Investig. 1998, 101, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di, F.F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Yudoh, K.; Matsuno, H.; Osada, R.; Nakazawa, F.; Katayama, R.; Kimura, T. Decreased cellular activity and replicative capacity of osteoblastic cells isolated from the periarticular bone of rheumatoid arthritis patients compared with osteoarthritis patients. Arthritis Rheum. 2000, 43, 2178–2188. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Cha, B.H.; Lee, J.S.; Kim, S.W.; Cha, H.J.; Lee, S.H. The modulation of the oxidative stress response in chondrocytes by Wip1 and its effect on senescence and dedifferentiation during in vitro expansion. Biomaterials 2013, 34, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Yudoh, K.; Nguyen, V.; Nakamura, H.; Hongo-Masuko, K.; Kato, T.; Nishioka, K. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: Oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res. Ther. 2005, 7, R380–R391. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Buckwalter, J.A. The role of chondrocyte senescence in the pathogenesis of osteoarthritis and in limiting cartilage repair. J. Bone Jt. Surg. Am. 2003, 85 (Suppl. 2), 106–110. [Google Scholar] [CrossRef]
- Mobasheri, A.; Matta, C.; Zakany, R.; Musumeci, G. Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 2015, 80, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K. The emerging role of senescent cells in tissue homeostasis and pathophysiology. Pathobiol. Aging Age Relat. Dis. 2015, 5, 27743. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Finkel, T. Free radicals and senescence. Exp. Cell Res. 2008, 314, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Kassem, M.; Ankersen, L.; Eriksen, E.F.; Clark, B.F.; Rattan, S.I. Demonstration of cellular aging and senescence in serially passaged long-term cultures of human trabecular osteoblasts. Osteoporos. Int. 1997, 7, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Kveiborg, M.; Rattan, S.I.; Clark, B.F.; Eriksen, E.F.; Kassem, M. Treatment with 1,25-dihydroxyvitamin D3 reduces impairment of human osteoblast functions during cellular aging in culture. J. Cell. Physiol. 2001, 186, 298–306. [Google Scholar] [CrossRef]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M.; et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297. [Google Scholar] [CrossRef] [PubMed]
- Jäger, M.; Fischer, J.; Dohrn, W.; Li, X.; Ayers, D.C.; Czibere, A.; Prall, W.C.; Lensing-Höhn, S.; Krauspe, R. Dexamethasone modulates BMP-2 effects on mesenchymal stem cells in vitro. J. Orthop. Res. 2008, 26, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.J.; Liang, C.R.; Kim, D.J.; Widmer, M.S.; Mikos, A.G. Osteoblastic phenotype of rat marrow stromal cells cultured in the presence of dexamethasone, β-glycerolphosphate, and l-ascorbic acid. J. Cell. Biochem. 1998, 71, 55–62. [Google Scholar] [CrossRef]
- Rovere, P.; Peri, G.; Fazzini, F.; Bottazzi, B.; Doni, A.; Bondanza, A.; Zimmermann, V.S.; Garlanda, C.; Fascio, U.; Sabbadini, M.G.; et al. The long pentraxin PTX3 binds to apoptotic cells and regulates their clearance by antigen-presenting dendritic cells. Blood 2000, 96, 4300–4306. [Google Scholar] [PubMed]
- Pye, S.R.; Almusalam, B.; Boonen, S.; Vanderschueren, D.; Borghs, H.; Gielen, E.; Adams, J.E.; Ward, K.A.; Bartfai, G.; Casanueva, F.F.; et al. Influence of insulin-like growth factor binding protein (IGFBP)-1 and IGFBP-3 on bone health: Results from the European Male Ageing Study. Calcif. Tissue Int. 2011, 88, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; Verlinden, L.; Meyer, M.B.; Gijsbers, R.; Pike, J.W.; Bouillon, R.; Verstuyf, A. 1,25-Dihydroxyvitamin D3 and the aging-related forkhead box O and sestrin proteins in osteoblasts. J. Steroid Biochem. Mol. Biol. 2013, 136, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Siiskonen, H.; Oikari, S.; Pasonen-Seppanen, S.; Rilla, K. Hyaluronan synthase 1: A mysterious enzyme with unexpected functions. Front. Immunol. 2015, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.D.; Xiao, W.F.; Li, J.; de la Motte, C.A.; Sandy, J.D.; Plaas, A. Deficiency of hyaluronan synthase 1 (Has1) results in chronic joint inflammation and widespread intra-articular fibrosis in a murine model of knee joint cartilage damage. Osteoarthr. Cartil. 2015, 23, 1879–1889. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Tan, W.; Feng, G.; Meng, Y.; Shen, B.; Liu, H.; Cheng, C. Wnt/β-catenin signaling mediates the senescence of bone marrow-mesenchymal stem cells from systemic lupus erythematosus patients through the p53/p21 pathway. Mol. Cell. Biochem. 2014, 387, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Schuijers, J.; Clevers, H. Adult mammalian stem cells: The role of Wnt, Lgr5 and R-spondins. EMBO J. 2012, 31, 2685–2696. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Carames, B.; Ronfani, L.; Ulmer, U.; Komiya, S.; Bianchi, M.E.; Lotz, M. Aging-related loss of the chromatin protein HMGB2 in articular cartilage is linked to reduced cellularity and osteoarthritis. Proc. Natl. Acad. Sci. USA 2009, 106, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Kouhkan, F.; Mobarra, N.; Soufi-Zomorrod, M.; Keramati, F.; Rad, S.M.A.H.; Fathi-Roudsari, M.; Tavakoli, R.; Hajarizadeh, A.; Ziaei, S.; Lahmi, R.; et al. MicroRNA-129-1 acts as tumour suppressor and induces cell cycle arrest of GBM cancer cells through targeting IGF2BP3 and MAPK1. J. Med. Genet. 2016, 53, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, R.; Imamachi, N.; Suzuki, Y.; Yoshida, H.; Tochigi, N.; Oonishi, T.; Suzuki, Y.; Akimitsu, N. Oncofetal protein IGF2BP3 facilitates the activity of proto-oncogene protein eIF4E through the destabilization of EIF4E-BP2 mRNA. Oncogene 2016, 35, 3495–3502. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Roforth, M.M.; Atkinson, E.J.; Peterson, J.M.; Drake, M.T.; McCready, L.K.; Farr, J.N.; Monroe, D.G.; Khosla, S. Isolation and characterization of human osteoblasts from needle biopsies without in vitro culture. Osteoporos. Int. 2014, 25, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.P. Cellular defenses against damage from reactive oxygen species. Physiol. Rev. 1994, 74, 139–162. [Google Scholar] [PubMed]
- Amatore, C.; Arbault, S. Oxidative Stress at the Single Cell Level. In Electrochemical Methods for Neuroscience; Michael, A.C., Borland, L.M., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2007; Chapter 13. [Google Scholar]
- Guaiquil, V.H.; Vera, J.C.; Golde, D.W. Mechanism of vitamin C inhibition of cell death induced by oxidative stress in glutathione-depleted HL-60 cells. J. Biol. Chem. 2001, 276, 40955–40961. [Google Scholar] [CrossRef] [PubMed]
- Imhof, H.; Sulzbacher, I.; Grampp, S.; Czerny, C.; Youssefzadeh, S.; Kainberger, F. Subchondral bone and cartilage disease: A rediscovered functional unit. Investig. Radiol. 2000, 35, 581–588. [Google Scholar] [CrossRef]
- Levick, J.R. Microvascular architecture and exchange in synovial joints. Microcirculation 1995, 2, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.; Kurz, B. Antioxidant to treat osteoarthritis: Dream or reality? Curr. Drug Targets 2007, 8, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Abramson, S.B. Nitric oxide in inflammation and pain associated with osteoarthritis. Arthritis Res. Ther. 2008, 10 (Suppl. 2), S2. [Google Scholar] [CrossRef] [PubMed]
- Studer, R.; Jaffurs, D.; Stefanovic-Racic, M.; Robbins, P.D.; Evans, C.H. Nitric oxide in osteoarthritis. Osteoarthr. Cartil. 1999, 7, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Kashino, G.; Kodama, S.; Nakayama, Y.; Suzuki, K.; Fukase, K.; Goto, M.; Watanabe, M. Relief of oxidative stress by ascorbic acid delays cellular senescence of normal human and Werner syndrome fibroblast cells. Free Radic. Biol. Med. 2003, 35, 438–443. [Google Scholar] [CrossRef]
- Pearce, V.P.; Sherrell, J.; Lou, Z.; Kopelovich, L.; Wright, W.E.; Shay, J.W. Immortalization of epithelial progenitor cells mediated by resveratrol. Oncogene 2008, 27, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, A.A.; Steding, C.E.; Herbert, B.S. Pharmaceutical regulation of telomerase and its clinical potential. J. Cell. Mol. Med. 2012, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Gronthos, S.; Chen, S.; Reddi, A.; Counter, C.M.; Robey, P.G.; Wang, C.-Y. Bone formation by human postnatal bone marrow stromal stem cells is enhanced by telomerase expression. Nat. Biotechnol. 2002, 20, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, J.L.; Rosada, C.; Serakinci, N.; Justesen, J.; Stenderup, K.; Rattan, S.I.S.; Jensen, T.G.; Kassem, M. Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat. Biotechnol. 2002, 20, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.E.; Shay, J.W. Historical claims and current interpretations of replicative aging. Nat. Biotechnol. 2002, 20, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Szychlinska, M.A.; Stoddart, M.J.; D’Amora, U.; Ambrosio, L.; Alini, M.; Musumeci, G. Mesenchymal stem cell-based cartilage regeneration approach and cell senescence: Can we manipulate cell aging and function? Tissue Eng. Part B Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Deberg, M.A.; Bellahcene, A.; Castronovo, V.; Msika, P.; Delcour, J.P.; Crielaard, J.M.; Henrotin, Y.E. Phenotypic characterization of osteoblasts from the sclerotic zones of osteoarthritic subchondral bone. Arthritis Rheum. 2008, 58, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Suda, R.K.; Billings, P.C.; Egan, K.P.; Kim, J.-H.; McCarrick-Walmsley, R.; Glaser, D.L.; Porter, D.L.; Shore, E.M.; Pignolo, R.J. Circulating osteogenic precursor cells in heterotopic bone formation. Stem Cells 2009, 27, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; de Kreutzenberg, S.V.; Agostini, C.; Cabrelle, A.; Binotto, G.; Rattazzi, M.; Bertacco, E.; et al. Widespread increase in myeloid calcifying cells contributes to ectopic vascular calcification in type 2 diabetes. Circ. Res. 2011, 108, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Pippenger, B.E.; Duhr, R.; Muraro, M.G.; Pagenstert, G.I.; Hugle, T.; Geurts, J. Multicolor flow cytometry-based cellular phenotyping identifies osteoprogenitors and inflammatory cells in the osteoarthritic subchondral bone marrow compartment. Osteoarthr. Cartil. 2015, 23, 1865–1969. [Google Scholar] [CrossRef] [PubMed]
- Barbero, A.; Grogan, S.; Schafer, D.; Heberer, M.; Mainil-Varlet, P.; Martin, I. Age related changes in human articular chondrocyte yield, proliferation and post-expansion chondrogenic capacity. Osteoarthr. Cartil. 2004, 12, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Barbero, A.; Ploegert, S.; Heberer, M.; Martin, I. Plasticity of clonal populations of dedifferentiated adult human articular chondrocytes. Arthritis Rheum. 2003, 48, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Scherberich, A.; Galli, R.; Jaquiery, C.; Farhadi, J.; Martin, I. Three-dimensional perfusion culture of human adipose tissue-derived endothelial and osteoblastic progenitors generates osteogenic constructs with intrinsic vascularization capacity. Stem Cells 2007, 25, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Kusser, K.L.; Randall, T.D. Simultaneous detection of EGFP and cell surface markers by fluorescence microscopy in lymphoid tissues. J. Histochem. Cytochem. 2003, 51, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Mastrogiacomo, M.; De Leo, G.; Muraglia, A.; Beltrame, F.; Cancedda, R.; Quarto, R. Fluorescence microscopy imaging of bone for automated histomorphometry. Tissue Eng. 2002, 8, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Abramoff, M.D.; Magalhaes, P.J.; Ram, S.J. Image processing with ImageJ. Biophotonics Int. 2004, 7, 36–42. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Gaidatzis, D.; Lerch, A.; Hahne, F.; Stadler, M.B. QuasR: Quantification and annotation of short reads in R. Bioinformatics 2015, 31, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burger, M.G.; Steinitz, A.; Geurts, J.; Pippenger, B.E.; Schaefer, D.J.; Martin, I.; Barbero, A.; Pelttari, K. Ascorbic Acid Attenuates Senescence of Human Osteoarthritic Osteoblasts. Int. J. Mol. Sci. 2017, 18, 2517. https://doi.org/10.3390/ijms18122517
Burger MG, Steinitz A, Geurts J, Pippenger BE, Schaefer DJ, Martin I, Barbero A, Pelttari K. Ascorbic Acid Attenuates Senescence of Human Osteoarthritic Osteoblasts. International Journal of Molecular Sciences. 2017; 18(12):2517. https://doi.org/10.3390/ijms18122517
Chicago/Turabian StyleBurger, Maximilian G., Amir Steinitz, Jeroen Geurts, Benjamin E. Pippenger, Dirk J. Schaefer, Ivan Martin, Andrea Barbero, and Karoliina Pelttari. 2017. "Ascorbic Acid Attenuates Senescence of Human Osteoarthritic Osteoblasts" International Journal of Molecular Sciences 18, no. 12: 2517. https://doi.org/10.3390/ijms18122517
APA StyleBurger, M. G., Steinitz, A., Geurts, J., Pippenger, B. E., Schaefer, D. J., Martin, I., Barbero, A., & Pelttari, K. (2017). Ascorbic Acid Attenuates Senescence of Human Osteoarthritic Osteoblasts. International Journal of Molecular Sciences, 18(12), 2517. https://doi.org/10.3390/ijms18122517