Identification and Initial Characterization of the Effectors of an Anther Smut Fungus and Potential Host Target Proteins


 ,
, 
Abstract
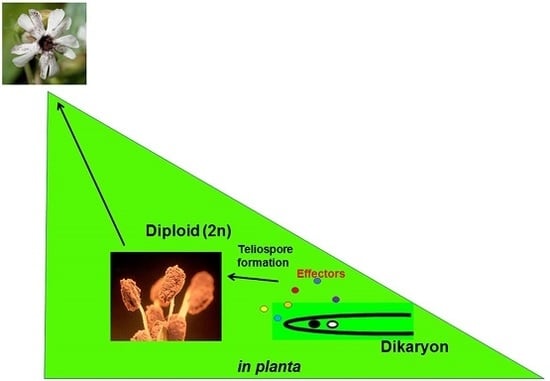
1. Introduction
2. Results
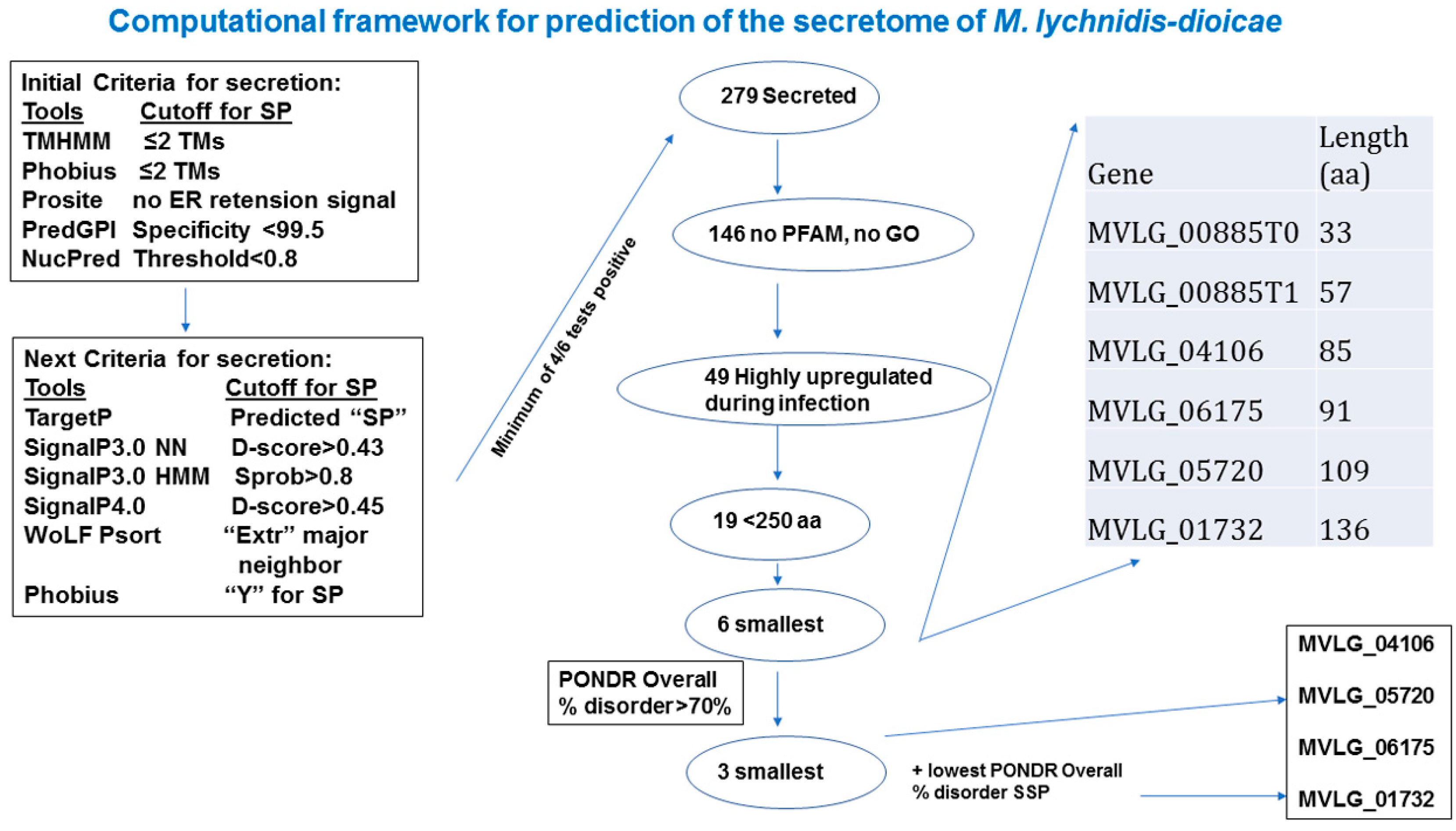
2.1. In Silico Analyses to Identify Potential Effectors
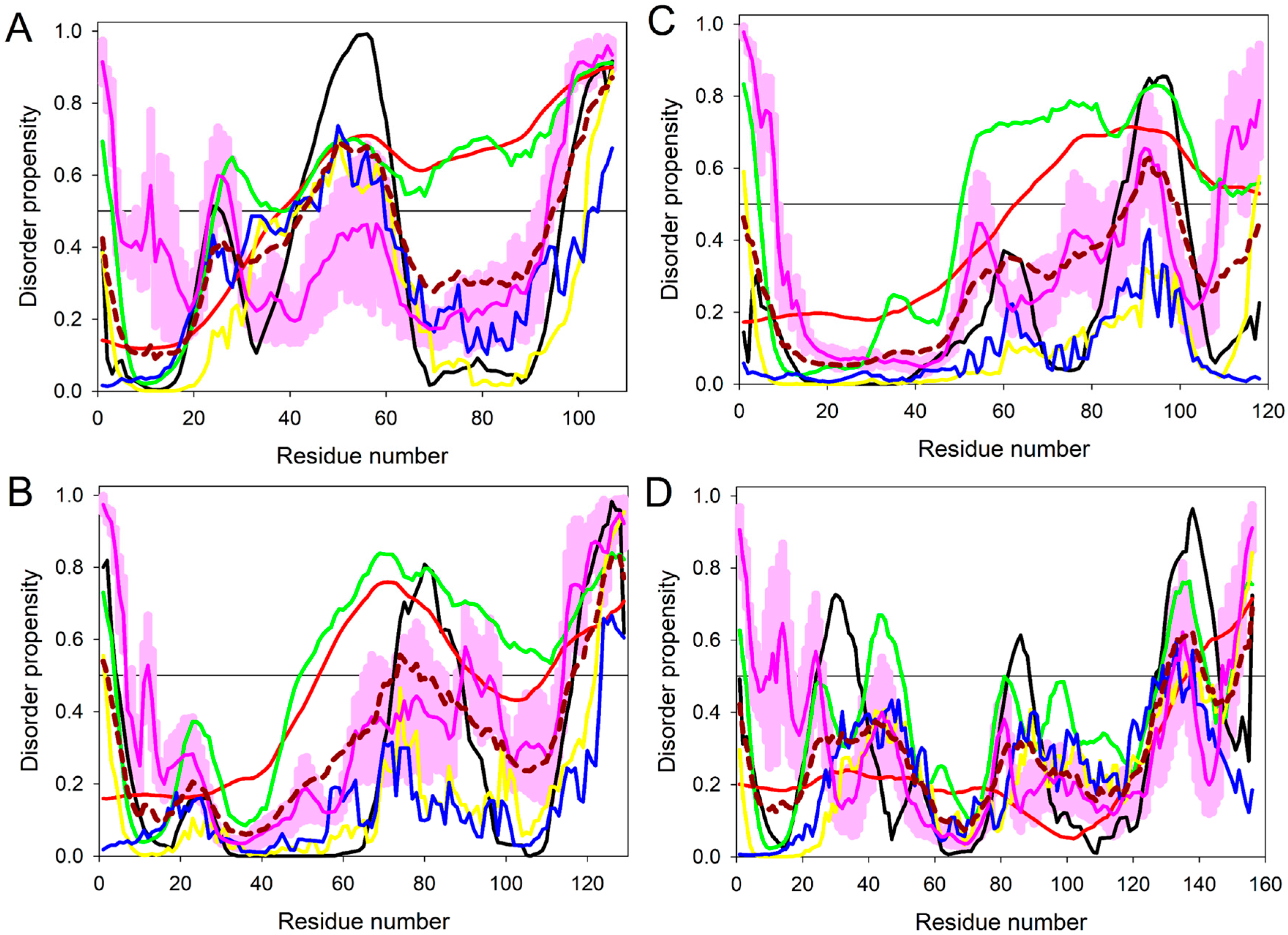
2.2. Intrinsic Disorder in Predicted Small Secreted Proteins (SSPs)
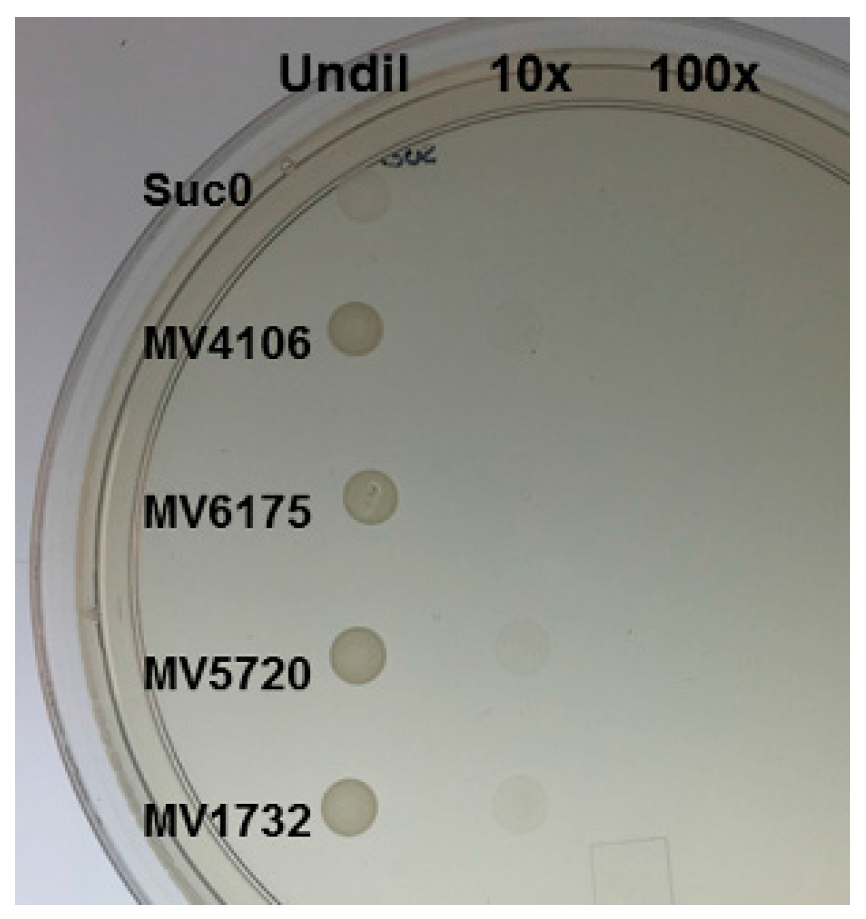
2.3. Yeast Secretion Trap to Verify the Secretory Nature of Predicted Effectors
2.4. Yeast Two-Hybrid Experiment
2.4.1. MVLG_04106 Autoactivates the Reporter Genes in Yeast Two-Hybrid Assay
2.4.2. MVLG_05720 Fungal Protein Interacts with Fungal Proteins
2.4.3. MVLG_06175 Interacts with a Host Protein and a Fungal Protein
2.4.4. MVLG_01732 Interacts with Host Proteins
3. Discussion
3.1. MVLG_04106 Could Serve as a Transcriptional Regulator
3.2. MVLG_05720 Possibly Regulated by Additional Fungal Proteins
3.3. MVLG_06175 Role in Host Entry During the Infection and in Reproduction
3.4. MVLG_01732 Role in Altering the Vesicular Traffic in the Host and Male Sterility
4. Materials and Methods
4.1. Plant and Fungal Growth
4.2. In Silico Analyses
4.2.1. Prediction of Small Secreted Proteins (SSPs)
4.2.2. Prediction of Intrinsic Disorder
4.2.3. Additional Bioinformatic Analyses
4.3. Yeast Secretion Trap (YST) Experiment
4.4. RNA Extraction and cDNA Library Construction
4.5. Yeast Two Hybrid Screen
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ellis, J.; Catanzariti, A.-M.; Dodds, P. The problem of how fungal and oomycete avirulence proteins enter plant cells. Trends Plant Sci. 2006, 11, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Kemen, E.; Kemen, A.; Ehlers, A.; Voegele, R.; Mendgen, K. A novel structural effector from rust fungi is capable of fibril formation. Plant J. Cell Mol. Biol. 2013, 75, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Giraud, T.; Yockteng, R.; Lopez-Villavicencio, M.; Refregier, G.; Hood, M.E. Mating system of the anther smut fungus Microbotryum violaceum: Selfing under heterothallism. Eukaryot. Cell 2008, 7, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Uchida, W.; Matsunaga, S.; Sugiyama, R.; Kazama, Y.; Kawano, S. Morphological development of anthers induced by the dimorphic smut fungus Microbotryum violaceum in female flowers of the dioecious plant Silene latifolia. Planta 2003, 218, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Giraud, T.; Hood, M.E. Maximized virulence in a sterilizing pathogen: The anther-smut fungus and its co-evolved hosts. J. Evol. Biol. 2008, 21, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Toh, S.S.; Chen, Z.; Schultz, D.J.; Cuomo, C.A.; Perlin, M.H. Transcriptional analysis of mating and pre-infection stages of the anther smut, Microbotryum lychnidis-dioicae. Microbiology 2017. [Google Scholar] [CrossRef] [PubMed]
- Perlin, M.H.; Amselem, J.; Fontanillas, E.; Toh, S.S.; Chen, Z.; Goldberg, J.; Duplessis, S.; Henrissat, B.; Young, S.; Zeng, Q.; et al. Sex and parasites: Genomic and transcriptomic analysis of Microbotryum lychnidis-dioicae, the biotrophic and plant-castrating anther smut fungus. BMC Genom. 2015, 16, 461. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Nakamura, H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006, 580, 2041–2045. [Google Scholar] [CrossRef] [PubMed]
- Ekman, D.; Light, S.; Bjorklund, A.K.; Elofsson, A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol. 2006, 7, R45. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.; Oldfield, C.J.; Fei, J.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic Disorder Is a Common Feature of Hub Proteins from Four Eukaryotic Interactomes. PLoS Comput. Biol. 2006, 2, e100. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and Sequence Repeats in Hub Proteins and Their Implications for Network Evolution. J. Proteome Res. 2006, 5, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.P.; Dash, D. Intrinsic disorder in yeast transcriptional regulatory network. Proteins 2007, 68, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.P.; Ganapathi, M.; Dash, D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins 2007, 66, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol. Biosyst. 2008, 4, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.T.; Roth, A.P.; Xue, B.; Sun, R.; Dunker, A.K.; Uversky, V.N.; LaCount, D.J. Intrinsic disorder mediates hepatitis C virus core-host cell protein interactions. Protein Sci. 2015, 24, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.R.; Saraiva, A.M.; Azzoni, A.R.; Miranda, H.R.; de Toledo, M.A.; Pelloso, A.C.; Souza, A.P. Overexpression and purification of PWL2D, a mutant of the effector protein PWL2 from Magnaporthe grisea. Protein Expr. Purif. 2010, 74, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.H.; Garner, E.C.; Brown, C.J. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Meszaros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Simon, I.; Dosztanyi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Kurgan, L. On the complementarity of the consensus-based disorder prediction. Pac. Symp. Biocomput. 2012, 176–187. [Google Scholar]
- Fan, X.; Kurgan, L. Accurate prediction of disorder in protein chains with a comprehensive and empirically designed consensus. J. Biomol. Struct. Dyn. 2014, 32, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Giollo, M.; Di Domenico, T.; Ferrari, C.; Zimmermann, O. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics 2015, 31, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Rose, J.K. A yeast secretion trap assay for identification of secreted proteins from eukaryotic phytopathogens and their plant hosts. Methods Mol. Biol. 2012, 835, 519–530. [Google Scholar] [PubMed]
- Lu, S.; Edwards, M.C. Genome-Wide analysis of small secreted cysteine-rich proteins identifies candidate effector proteins potentially involved in fusarium graminearum-wheat interactions. Phytopathology 2016, 106, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, J.; Storchan, G.B.; MacCarthy, C.M.; Uversky, V.N.; Tcherkasskaya, O. Local flexibility in molecular function paradigm. Mol. Cell. Proteom. 2006, 5, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Minezaki, Y.; Homma, K.; Kinjo, A.R.; Nishikawa, K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J. Mol. Biol. 2006, 359, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Hsu, W.-L.; Xin, F.; Dunker, A.K.; Uversky, V.N.; Radivojac, P. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 2014, 23, 1077–1093. [Google Scholar] [CrossRef] [PubMed]
- Branco, S.; Badouin, H.; Rodríguez de la Vega, R.C.; Gouzy, J.; Carpentier, F.; Aguileta, G.; Siguenza, S.; Brandenburg, J.-T.; Coelho, M.A.; Hood, M.E.; et al. Evolutionary strata on young mating-type chromosomes despite the lack of sexual antagonism. Proc. Natl. Acad. Sci. USA 2017, 114, 7067–7072. [Google Scholar] [CrossRef] [PubMed]
- Branco, S.; Carpentier, F.; de la Vega, R.C.R.; Badouin, H.; Snirc, A.; le Prieur, S.; Coelho, M.A.; Bergerow, D.; Hood, M.E.; Giraud, T. Multiple convergent events of supergene evolution in mating-type chromosomes. Submitt. Nat. Commun. Unpublished work. 2017. [Google Scholar]
- Fontanillas, E.; Hood, M.E.; Badouin, H.; Petit, E.; Barbe, V.; Gouzy, J.; de Vienne, D.M.; Aguileta, G.; Poulain, J.; Wincker, P.; et al. Degeneration of the nonrecombining regions in the mating-type chromosomes of the anther-smut fungi. Mol. Biol. Evol. 2015, 32, 928–943. [Google Scholar] [CrossRef] [PubMed]
- Dennison, S.R.; Mura, M.; Harris, F.; Morton, L.H.; Zvelindovsky, A.; Phoenix, D.A. The role of C-terminal amidation in the membrane interactions of the anionic antimicrobial peptide, maximin H5. Biochim. Biophys. Acta 2015, 1848, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Djamei, A.; Schipper, K.; Rabe, F.; Ghosh, A.; Vincon, V.; Kahnt, J.; Osorio, S.; Tohge, T.; Fernie, A.R.; Feussner, I.; et al. Metabolic priming by a secreted fungal effector. Nature 2011, 478, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Poon, H.; Day, A.W. ‘Fimbriae’ in the fungus Ustilago violacea. Nature 1974, 250, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Poon, N.H.N.; Day, A.W. Fungal fimbriae. I. Structure, origin, and synthesis. Can. J. Microbiol. 1975, 21, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Celerin, M.; Ray, J.M.; Schisler, N.J.; Day, A.W.; Stetler-Stevenson, W.G.; Laudenbach, D.E. Fungal fimbriae are composed of collagen. EMBO J. 1996, 15, 4445–4453. [Google Scholar] [PubMed]
- Hubmacher, D.; Bergeron, E.; Fagotto-Kaufmann, C.; Sakai, L.Y.; Reinhardt, D.P. Early Fibrillin-1 assembly monitored through a modifiable recombinant cell approach. Biomacromolecules 2014, 15, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, D.P.; Gambee, J.E.; Ono, R.N.; Bächinger, H.P.; Sakai, L.Y. Initial Steps in Assembly of Microfibrils: Formation of disulfide-cross-linked multimers containing fibrillin-1. J. Biol. Chem. 2000, 275, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Roppolo, D.; Boeckmann, B.; Pfister, A.; Boutet, E.; Rubio, M.C.; Dénervaud-Tendon, V.; Vermeer, J.E.M.; Gheyselinck, J.; Xenarios, I.; Geldner, N. Functional and evolutionary analysis of the casparian strip membrane domain protein family. Plant Physiol. 2014, 165, 1709–1722. [Google Scholar] [CrossRef] [PubMed]
- De Silva, K.; Laska, B.; Brown, C.; Sederoff, H.W.; Khodakovskaya, M. Arabidopsis thaliana calcium-dependent lipid-binding protein (AtCLB): A novel repressor of abiotic stress response. J. Exp. Bot. 2011, 62, 2679–2689. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Hu, G.; Fries, B.; Caza, M.; Wang, J. A defect in ATP-citrate lyase links acetyl-CoA production, virulence factor elaboration and virulence in Cryptococcus neoformans. Mol. Microbiol. 2012, 86, 1404–1423. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Kozik, A.; Griego, A.; Kuang, H.; Michelmore, R.W. Genome-wide analysis of NBS-LRR-encoding genes in arabidopsis. Plant Cell 2003, 15, 809–834. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.-Z.; Song, L.-F.; Zou, J.-J.; Su, Z.; Wu, W.-H. Transcriptome analyses show changes in gene expression to accompany pollen germination and tube growth in arabidopsis. Plant Physiol. 2008, 148, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sato, R.; Brown, M.S.; Hua, X.; Goldstein, J.L. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 1994, 77, 53–62. [Google Scholar] [CrossRef]
- Saheki, Y.; De Camilli, P. The Extended-Synaptotagmins. Biochim. Biophys. Acta 2017, 1864, 1490–1493. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.; Parker, P.J.; Rhee, L.; Yang-Feng, T.L.; Chen, E.; Waterfield, M.D.; Francke, U.; Ullrich, A. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science 1986, 233, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Gao, H.B.; Yuan, M.; Xue, H.W. The Arabidopsis ARCP protein, CSI1, which is required for microtubule stability, is necessary for root and anther development. Plant Cell 2012, 24, 1066–1080. [Google Scholar] [CrossRef] [PubMed]
- Lalanne, E.; Michaelidis, C.; Moore, J.M.; Gagliano, W.; Johnson, A.; Patel, R.; Howden, R.; Vielle-Calzada, J.P.; Grossniklaus, U.; Twell, D. Analysis of transposon insertion mutants highlights the diversity of mechanisms underlying male progamic development in Arabidopsis. Genetics 2004, 167, 1975–1986. [Google Scholar] [CrossRef] [PubMed]
- Malhis, N.; Jacobson, M.; Gsponer, J. MoRFchibi SYSTEM: Software tools for the identification of MoRFs in protein sequences. Nucleic Acids Res. 2016, 44, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. Signalp 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at prosite. Nucleic Acids Res. 2013, 41, D344–D347. [Google Scholar] [CrossRef] [PubMed]
- De Castro, E.; Sigrist, C.J.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. Scanprosite: Detection of prosite signature matches and prorule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef] [PubMed]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.R.D. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Robinson, J.S.; Klionsky, D.J.; Banta, L.M.; Emr, S.D. Protein sorting in Saccharomyces cerevisiae: Isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell. Biol. 1988, 8, 4936–4948. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- McAlister-Henn, L.; Gibson, N.; Panisko, E. Applications of the Yeast Two-Hybrid System. Methods 1999, 19, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Greener, M.; Perkel, J.M. The yeast two-hybrid assay. Scientist 2005, 19, 32. [Google Scholar]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.V.; Dubchak, I. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014, 42, D26–D31. [Google Scholar] [CrossRef] [PubMed]
- Wade Harper, J.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- James, P.; Halladay, J.; Craig, E.A. Genomic Libraries and a Host Strain Designed for Highly Efficient Two-Hybrid Selection in Yeast. Genetics 1996, 144, 1425–1436. [Google Scholar] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted Protein | Expression a | Size (Amino Acids) | No. of Cys | Function |
|---|---|---|---|---|
| MVLG_01732 | 144 rsem vs. 0 | 156 | 1 | Candidate effector |
| MVLG_04106 | 86 rsem vs. 0 | 107 | 6 | Candidate effector |
| MVLG_05720 | 1164 rsem vs. 0 | 129 | 12 | Candidate effector |
| MVLG_06175 | 127 rsem vs. 0 | 118 | 10 | Candidate effector |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuppireddy, V.S.; Uversky, V.N.; Toh, S.S.; Tsai, M.-C.; Beckerson, W.C.; Cahill, C.; Carman, B.; Perlin, M.H. Identification and Initial Characterization of the Effectors of an Anther Smut Fungus and Potential Host Target Proteins. Int. J. Mol. Sci. 2017, 18, 2489. https://doi.org/10.3390/ijms18112489
Kuppireddy VS, Uversky VN, Toh SS, Tsai M-C, Beckerson WC, Cahill C, Carman B, Perlin MH. Identification and Initial Characterization of the Effectors of an Anther Smut Fungus and Potential Host Target Proteins. International Journal of Molecular Sciences. 2017; 18(11):2489. https://doi.org/10.3390/ijms18112489
Chicago/Turabian StyleKuppireddy, Venkata S., Vladimir N. Uversky, Su San Toh, Ming-Chang Tsai, William C. Beckerson, Catarina Cahill, Brittany Carman, and Michael H. Perlin. 2017. "Identification and Initial Characterization of the Effectors of an Anther Smut Fungus and Potential Host Target Proteins" International Journal of Molecular Sciences 18, no. 11: 2489. https://doi.org/10.3390/ijms18112489
APA StyleKuppireddy, V. S., Uversky, V. N., Toh, S. S., Tsai, M.-C., Beckerson, W. C., Cahill, C., Carman, B., & Perlin, M. H. (2017). Identification and Initial Characterization of the Effectors of an Anther Smut Fungus and Potential Host Target Proteins. International Journal of Molecular Sciences, 18(11), 2489. https://doi.org/10.3390/ijms18112489