ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance



Abstract
1. Introduction
2. Cancer Stem Cells (CSCs)
3. ATP Binding Cassette (ABC) Transporter Structure and Location
4. Relevance of ABC Transporters in Cancer Cell Biology
5. ABC Transporter Regulation by Genes and Signalling Pathways
6. Endogenous Role of ABC Transporters in CSCs
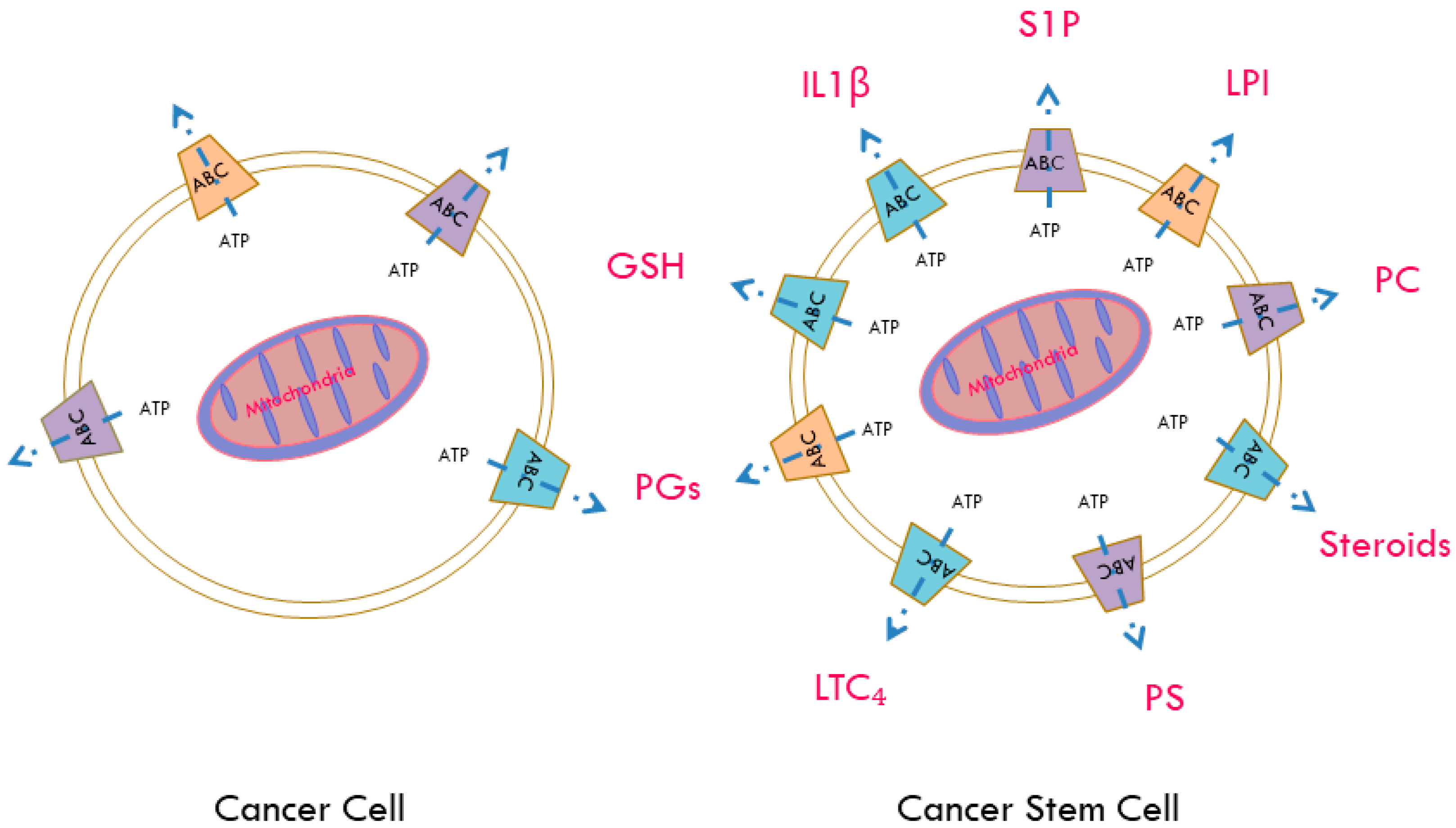
7. ABC Transporters as Regulators of the Release of Active Biomolecules
8. ABC Transporters and Cellular Redox Status
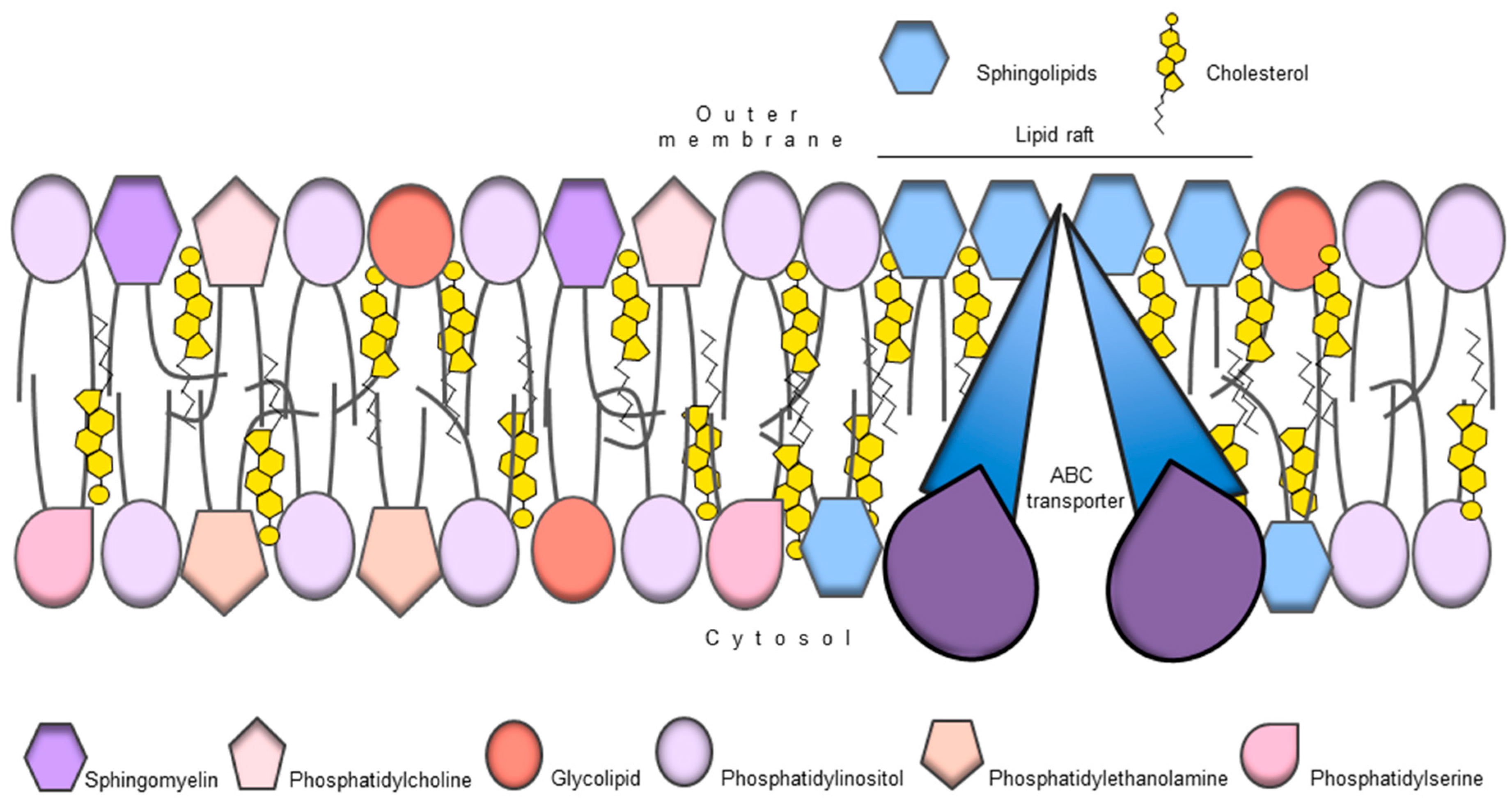
9. ABC Transporters and Membrane Lipid Composition
10. ABC Transporters and Tumour Metabolism
11. ABC Transporters, Tumour Microenvironment and CSC Niche
12. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABC | ATP binding cassettes |
| CSC | Cancer stem cells |
| MDR | Multi drug resistance |
| OCT4 | Octamer-binding transcription factor 4 |
| ALDH | Aldehyde dehydrogenase |
| EpCAM | Epithelial cell adhesion molecule |
| EMT | Epithelial to mesenchymal transition |
| SOX2 | Sex determining region y-box 2 |
| MMIC | Malignant melanoma initiating cells |
| P-gp | Permeability glycoprotein |
| MRP | Multidrug resistance-associated protein |
| BCRP | Breast cancer resistant protein |
| APE | Aminoacyl tRNA-peptidyltRNA-decylated tRNA |
| mTOR | Mammalian target of rapamycin |
| PAK1 | p-21 Activated kinase 1 |
| HMGA | High mobility group A |
| EGFR | Epidermal growth factor receptor |
| NF-κB | Nuclear factor kappa beta |
| PI3K | Phosphoinositide 3-kinase |
| COX2 | Cyclooxygenase 2 |
| ERBB2 | Receptor tyrosine kinase 2 |
| SMO | Smoothened |
| PTEN | Phosphatase and tensin homolog |
| DNAPK | DNA dependent protein kinase |
| LMWH | Low molecular weight heparin |
| HIF | Hypoxia inducible factor |
| IL | Interleukin |
| PG | Prostaglandin |
| PGI2 | Prostacyclin |
| LT | Leukotriene |
| TX | Thromboxane |
| LTC₄ | Leukotriene C 4 |
| CXCR1 | C-X-C chemokine receptor type 1 |
| CoA | Coenzyme A |
| AMPK | AMP-activated protein kinase |
| LPI | Lysophosphatidylinositol |
| PDAC | Pancreatic ductal adenocarcinoma |
| GPCR | G-protein-coupled receptor |
| S1P | Sphingosine-1-phosphate |
| GSH | Glutathione |
| GSX | Glutathione conjugates |
| GSSH | Glutathione disulphide |
| HSC | Hematopoietic stem cells |
| ROS | Reactive oxygen species |
| MAPK | Mitogen activated protein kinase |
| ERK | Extracellular signal-regulated kinase |
| VEGF | Vascular endothelial growth factor |
| PPARγ | Peroxisome proliferator-activated receptor |
| MCT | Monocarbonate transporters |
| ERK | Extracellular signal-regulated kinase |
| ESC | Embryonic stem cells |
| BBB | Blood brain barrier |
| CNS | Central nervous system |
| Nrf2 | Nuclear factor (erythroid-derived 2)-like 2 |
| HDL | High density lipoproteins |
| KRAS | Kirsten rat sarcoma |
| MGMT | Ο6-methylguanine-DNA methyltransferase |
| TCA | Tricarboxylic acid cycle |
| TIS | Therapy induced senescence |
| EC | Endothelial cells |
| ECM | Extracellular matrix |
| CAF | Cancer associated fibroblasts |
| MSC | Mesenchymal stem cells |
| TAM | Tumour associated macrophages |
| GBM | Glioblastoma multiforme |
| HNSCC | Head and neck squamous cell carcinoma |
| EGF | Epidermal growth factor |
| STAT3 | Signal transducer and activator of transcription factor 3 |
| PKM2 | Pyruvate kinase M2 |
| NK | Natural killer |
| MIC | Metastasis initiating cells |
| SETD | SET domain |
| DTC | Disseminated tumour cells |
| CTC | Circulating tumour cells |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Kurth, I.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Discovery of the cancer stem cell related determinants of radioresistance. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2013, 108, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A. Perivascular Nitric Oxide Activates Notch Signaling and Promotes Stem-Like Character in PDGF-Induced Gliomas. Ph.D. Thesis, Weill Medical College of Cornell University, Ann Arbor, MI, USA, 2010. [Google Scholar]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updates 2016, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Copsel, S.; Garcia, C.; Diez, F.; Vermeulem, M.; Baldi, A.; Bianciotti, L.G.; Russel, F.G.M.; Shayo, C.; Davio, C. Multidrug Resistance Protein 4 (MRP4/ABCC4) Regulates cAMP Cellular Levels and Controls Human Leukemia Cell Proliferation and Differentiation. J. Biol. Chem. 2011, 286, 6979–6988. [Google Scholar] [CrossRef] [PubMed]
- Hedditch, E.L.; Gao, B.; Russell, A.J.; Lu, Y.; Emmanuel, C.; Beesley, J.; Johnatty, S.E.; Chen, X.; Harnett, P.; George, J.; et al. ABCA Transporter Gene Expression and Poor Outcome in Epithelial Ovarian Cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.J.; Haber, M.; Porro, A.; Munoz, M.A.; Iraci, N.; Xue, C.; Murray, J.; Flemming, C.L.; Smith, J.; Fletcher, J.I.; et al. ABCC Multidrug Transporters in Childhood Neuroblastoma: Clinical and Biological Effects Independent of Cytotoxic Drug Efflux. J. Natl. Cancer Inst. 2011, 103, 1236–1251. [Google Scholar] [CrossRef] [PubMed]
- Copsel, S.; Bruzzone, A.; May, M.; Beyrath, J.; Wargon, V.; Cany, J.; Russel, F.G.M.; Shayo, C.; Davio, C. Multidrug resistance protein 4/ATP binding cassette transporter 4: A new potential therapeutic target for acute myeloid leukemia. Oncotarget 2014, 5, 9308–9321. [Google Scholar] [CrossRef] [PubMed]
- Mochida, Y.; Taguchi, K.-I.; Taniguchi, S.; Tsuneyoshi, M.; Kuwano, H.; Tsuzuki, T.; Kuwano, M.; Wada, M. The role of P-glycoprotein in intestinal tumorigenesis: Disruption of mdr1a suppresses polyp formation in ApcMin/+ mice. Carcinogenesis 2003, 24, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Mori, Y.; Hayashi, R.; Takada, M.; Ino, Y.; Naishiro, Y.; Kondo, T.; Hirohashi, S. Suppression of intestinal polyposis in Mdr1-deficient ApcMin/+ mice. Cancer Res. 2003, 63, 895–901. [Google Scholar] [PubMed]
- Huls, M.; Russel, F.G.M.; Masereeuw, R. The role of ATP binding cassette transporters in tissue defense and organ regeneration. J. Pharmacol. Exp. Ther. 2009, 328, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov. 2015, 14, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, R.; Scheffer, G.L.; Scheper, R.J.; de Gruijl, T.D. The ABC of dendritic cell development and function. Trends Immunol. 2009, 30, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Skrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, B.; Fried, J.; Strife, A.; Sakai, Y.; Ota, K.; Okita, T. Studies of cellular proliferation in human leukemia. III. Behavior of leukemic cells in three adults with acute leukemia given continuous infusions of 3H-thymidine for 8 or 10 days. Cancer 1970, 25, 1237–1260. [Google Scholar] [CrossRef]
- Clarkson, B.; Ohkita, T.; Ota, K.; Fried, J. Studies of cellular proliferation in human leukemia. I. Estimation of growth rates of leukemic and normal hematopoietic cells in two adults with acute leukemia given single injections of tritiated thymidine. J. Clin. Investig. 1967, 46, 506–529. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, B.D.; Dowling, M.D.; Gee, T.S.; Cunningham, I.B.; Burchenal, J.H. Treatment of acute leukemia in adults. Cancer 1975, 36, 775–795. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.F.B.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of Bronchioalveolar Stem Cells in Normal Lung and Lung Cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; Ruggero De, M. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.; Cusimano, M.; Dirks, P. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Welte, Y.; Adjaye, J.; Lehrach, H.R.; Regenbrecht, C.R.A. Cancer stem cells in solid tumors: Elusive or illusive? Cell Commun. Signal. 2010, 8. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Dent, S.Y.R. Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.V.; Simeone, D.M. Biology and clinical applications of pancreatic cancer stem cells. Gastroenterology 2013, 144, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Moitra, K. Overcoming Multidrug Resistance in Cancer Stem Cells. BioMed Res. Int. 2015, 2015, 635745. [Google Scholar] [CrossRef] [PubMed]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The Role of ABC Transporters in Drug Resistance, Metabolism and Toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, R.; Maffucci, T.; Falasca, M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 2011, 30, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Linton, K.J. Investigational ABC transporter inhibitors. Expert Opin. Investig. Drugs 2012, 21, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug Resistance in Cancer: Role of ATP-Dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The Human ATP-Binding Cassette (ABC) Transporter Superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007, 67, 4827–4833. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; McArthur, C.; Jaffe, R.B. Ovarian cancer stem-like side-population cells are tumourigenic and chemoresistant. Br. J. Cancer 2010, 102, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, C.-Y.; Liu, T.; Wu, B.; Zhou, F.; Xiong, J.-X.; Wu, H.-S.; Tao, J.; Zhao, G.; Yang, M.; et al. Persistence of side population cells with high drug efflux capacity in pancreatic cancer. World J. Gastroenterol. 2008, 14, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, C.W.; Harkey, M.A.; Torok-Storb, B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 2002, 99, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Chuthapisith, S.; Eremin, J.; El-Sheemey, M.; Eremin, O. Breast cancer chemoresistance: Emerging importance of cancer stem cells. Surg. Oncol. 2010, 19, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Eyre, R.; Harvey, I.; Stemke-hale, K.; Lennard, T.W.; Tyson-capper, A.; Meeson, A.P. Reversing paclitaxel resistance in ovarian cancer cells via inhibition of the ABCB1 expressing side population. Tumor Biol. 2014, 35, 9879–9892. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Schatton, T.; Kim, S.; Zhan, Q.; Wilson, B.J.; Ma, J.; Saab, K.R.; Osherov, V.; Widlund, H.R.; Gasser, M.; et al. VEGFR-1 expressed by malignant melanoma-initiating cells is required for tumor growth. Cancer Res. 2011, 71, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Mohelnikova-Duchonova, B.; Brynychova, V.; Oliverius, M.; Honsova, E.; Kala, Z.; Muckova, K.; Soucek, P. Differences in transcript levels of ABC transporters between pancreatic adenocarcinoma and nonneoplastic tissues. Pancreas 2013, 42, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Chou, I.-L.; Chen, L.-Y.; Su, H.-Y.; Lin, S.-J.; Huang, Y.-W.; Deatherage, D.; Yan, P.; Nephew, K.; Lee, C.-I.; Huang, T.; et al. Hypomethylation of TGF-β target gene, ABCA1 in ovarian cancer and cancer initialing cell and is associated with poor prognosis in cancer patients. In Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research, Orlando, FL, USA, 2–6 April 2011. [Google Scholar]
- Chou, J.-L.; Huang, R.-L.; Shay, J.; Chen, L.-Y.; Lin, S.-J.; Yan, P.S.; Chao, W.-T.; Lai, Y.-H.; Lai, Y.-L.; Chao, T.-K.; et al. Hypermethylation of the TGF-β target, ABCA1 is associated with poor prognosis in ovarian cancer patients. Clin. Epigenet. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Bachmeier, B.E.; Iancu, C.M.; Killian, P.H.; Kronski, E.; Mirisola, V.; Angelini, G.; Jochum, M.; Nerlich, A.G.; Pfeffer, U. Overexpression of the ATP binding cassette gene ABCA1 determines resistance to Curcumin in M14 melanoma cells. Mol. Cancer 2009, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Quazi, F.; Molday, R.S. Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J. Biol. Chem. 2013, 288, 34414–34426. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Abe-Dohmae, S.; Sato, R.; Yokoyama, S. ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J. Lipid Res. 2006, 47, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Karl, T.; Garner, B. Understanding the function of ABCA7 in Alzheimer’s disease. Biochem. Soc. Trans. 2015, 43, 920–923. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.-F.; Yu, J.-T.; Tan, M.-S.; Tan, L. ABCA7 in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, B.C.; Gillet, J.-P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug resistance: Still a daunting challenge to the successful treatment of AML. Drug Resist. Updates 2012, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Bleau, A.-M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Kolenda, J.; Jensen, S.S.; Aaberg-Jessen, C.; Christensen, K.; Andersen, C.; Brünner, N.; Kristensen, B.W. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J. Neuro-Oncol. 2011, 103, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Hayes, E.; Lewis, R.; Ichi, S.; Mania-Farnell, B.; Shim, K.; Takao, T.; Allender, E.; Mayanil, C.S.; Tomita, T. CD133 and DNA-PK regulate MDR1 via the PI3K- or Akt-NF-κB pathway in multidrug-resistant glioblastoma cells in vitro. Oncogene 2016, 35, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.-Y.; Kwon, Y.-S.; Nam, K.-S.; Kim, S. Lapatinib enhances the cytotoxic effects of doxorubicin in MCF-7 tumorspheres by inhibiting the drug efflux function of ABC transporters. Biomed. Pharmacother. 2015, 72, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Fu, S.J.; Fan, W.Z.; Wang, Z.H.; Chen, Z.B.; Guo, S.J.; Chen, J.X.; Qiu, S.P. PKCε inhibits isolation and stemness of side population cells via the suppression of ABCB1 transporter and PI3K/Akt, MAPK/ERK signaling in renal cell carcinoma cell line 769P. Cancer Lett. 2016, 376, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.A.; Shepard, E.M.; Scotto, K.W. Differential regulation of MDR1 transcription by the p53 family members. Role of the DNA binding domain. J. Biol. Chem. 2005, 280, 13213–13219. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.R.; Figueiredo, M.A.; Trindade, G.S.; Marins, L.F. OCT4 mutations in human erythroleukemic cells: Implications for multiple drug resistance (MDR) phenotype. Mol. Cell. Biochem. 2015, 400, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, R.-H.; Tseng, K.-F.; Li, K.-P.; Lu, Z.-G.; Liu, Y.; Han, K.; Gan, Z.-H.; Lin, S.-C.; Hu, H.-Y.; et al. Sirolimus induces apoptosis and reverses multidrug resistance in human osteosarcoma cells in vitro via increasing microRNA-34b expression. Acta Pharmacol. Sin. 2016, 37, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, H.; Liu, X.; Evans, B.R.; Medina, D.J.; Liu, C.-G.; Yang, J.-M. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P- glycoprotein expression in human cancer cells. Biochem. Pharmacol. 2008, 76, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, O.; Filkowski, J.; Meservy, J.; Ilnytskyy, Y.; Tryndyak, V.P.; Chekhun, V.F.; Pogribny, I.P. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol. Cancer Ther. 2008, 7, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bieber, M.M.; Teng, N.N.H. Hedgehog signaling regulates drug sensitivity by targeting ABC transporters ABCB1 and ABCG2 in epithelial ovarian cancer. Mol. Carcinog. 2014, 53, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab. Rev. 2002, 34, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.-H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frank, N.Y.; Pendse, S.S.; Lapchak, P.H.; Margaryan, A.; Shlain, D.; Doeing, C.; Sayegh, M.H.; Frank, M.H. Regulation of progenitor cell fusion by ABCB5 P-glycoprotein, a novel human ATP-binding cassette transporter. J. Biol. Chem. 2003, 278, 47156–47165. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, W.; Fan, D.; Yan, Y.; Zhang, X.; Zhang, Y.; Xiong, D. Expression of ABCB5 gene in hematological malignances and its significance. Leuk. Lymphoma 2012, 53, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.J.; Saab, K.R.; Ma, J.; Schatton, T.; Pütz, P.; Zhan, Q.; Murphy, G.F.; Gasser, M.; Waaga-Gasser, A.M.; Frank, N.Y.; et al. ABCB5 maintains melanoma-initiating cells through a proinflammatory cytokine signaling circuit. Cancer Res. 2014, 74, 4196–4207. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Margaryan, A.; Huang, Y.; Schatton, T.; Waaga-Gasser, A.M.; Gasser, M.; Sayegh, M.H.; Sadee, W.; Frank, M.H. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005, 65, 4320–4333. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Anderle, P.; Bussey, K.J.; Barbacioru, C.; Shankavaram, U.; Dai, Z.; Reinhold, W.C.; Papp, A.; Weinstein, J.N.; Sadée, W. Membrane transporters and channels: Role of the transportome in cancer chemosensitivity and chemoresistance. Cancer Res. 2004, 64, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Haber, M.; Diolaiti, D.; Iraci, N.; Henderson, M.; Gherardi, S.; Valli, E.; Munoz, M.A.; Xue, C.; Flemming, C.; et al. Direct and coordinate regulation of ATP-binding cassette transporter genes by Myc factors generates specific transcription signatures that significantly affect the chemoresistance phenotype of cancer cells. J. Biol. Chem. 2010, 285, 19532–19543. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Wu, H.; Xia, J.; Li, Y.; Zhang, Y.; Huang, K.; Wagar, N.; Yoon, Y.; Cho, H.T.; Scala, S.; et al. Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1. Biochem. Pharmacol. 2010, 79, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Deeley, R.G.; Cole, S.P.C. Substrate recognition and transport by multidrug resistance protein 1 (ABCC1). FEBS Lett. 2006, 580, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-F.; Wang, L.-L.; Di, Y.M.; Xue, C.C.; Duan, W.; Li, C.G.; Li, Y. Substrates and Inhibitors of Human Multidrug Resistance Associated Proteins and the Implications in Drug Development. Curr. Med. Chem. 2008, 15, 1981–2039. [Google Scholar] [CrossRef] [PubMed]
- Wijnholds, J.; Evers, R.; Leusden, V.; Mol, C.; Zaman, G.J.R.; Mayer, U.; Beijnen, J.H.; Van der Valk, M.; Krimpenfort, P.; Borst, P. Increased sensitivity to anticancer drugs and decreased inflammatory response in mice lacking the multidrug resistance-associated protein. Nat. Med. 1997, 3, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Mitra, P.; Oskeritzian, C.A.; Payne, S.G.; Beaven, M.A.; Milstien, S.; Spiegel, S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16394–16399. [Google Scholar] [CrossRef] [PubMed]
- Zinzi, L.; Contino, M.; Cantore, M.; Capparelli, E.; Leopoldo, M.; Colabufo, N.A. ABC transporters in CSCs membranes as a novel target for treating tumor relapse. Front. Pharmacol. 2014, 5, 163. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Wielinga, P.; Zelcer, N.; Van der Heijden, I.; Kuil, A.; De Haas, M.; Wijnholds, J.; Borst, P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2003, 100, 9244–9249. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Schriml, L.M.; Hutchinson, A.; Romano-Spica, V.; Dean, M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998, 58, 5337–5339. [Google Scholar] [PubMed]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeck, A.; Vankelecom, H.; Van Delm, W.; Gremeaux, L.; Wouters, J.; Allemeersch, J.; Govaere, O.; Roskams, T.; Topal, B. Human pancreatic cancer contains a side population expressing cancer stem cell-associated and prognostic genes. PLoS ONE 2013, 8, e73968. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Kita, K.; Zheng, Y.-W.; Yokosuka, O.; Saisho, H.; Iwama, A.; Nakauchi, H.; Taniguchi, H. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology 2006, 44, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Seo, E.J.; Choi, E.J.; Lee, S.I.; Kwon, Y.W.; Jang, I.H.; Kim, S.-C.; Kim, K.-H.; Suh, D.-S.; Lee, S.C.; et al. Crucial role of HMGA1 in the self-renewal and drug resistance of ovarian cancer stem cells. Exp. Mol. Med. 2016, 48, e255. [Google Scholar] [CrossRef] [PubMed]
- Turrini, E.; Haenisch, S.; Laechelt, S.; Diewock, T.; Bruhn, O.; Cascorbi, I. MicroRNA profiling in K-562 cells under imatinib treatment: Influence of miR-212 and miR-328 on ABCG2 expression. Pharmacogenet. Genom. 2012, 22, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Hasanabady, M.H.; Kalalinia, F. ABCG2 inhibition as a therapeutic approach for overcoming multidrug resistance in cancer. J. Biosci. 2016, 41, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, N.G.; Miller, A.; Titus, M.A.; Huss, W.J. The Efflux Transporter ABCG2 Maintains Prostate Stem Cells. Mol. Cancer Res. 2017, 15, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Callaghan, D.; Jones, A.; Bai, J.; Rasquinha, I.; Smith, C.; Pei, K.; Walker, D.; Lue, L.-F.; Stanimirovic, D.; et al. ABCG2 is up-regulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Aβ(1–40) peptides. J. Neurosci. 2009, 29, 5463–5475. [Google Scholar] [CrossRef] [PubMed]
- Brechbuhl, H.M.; Gould, N.; Kachadourian, R.; Riekhof, W.R.; Voelker, D.R.; Day, B.J. Glutathione transport is a unique function of the ATP-binding cassette protein ABCG2. J. Biol. Chem. 2010, 285, 16582–16587. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Desano, J.T.; Xu, L. MicroRNA Regulation of Cancer Stem Cells and Therapeutic Implications. AAPS J. 2009, 11, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. Recent progress on tissue-resident adult stem cell biology and their therapeutic implications. Stem Cell Rev. 2008, 4, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Wang, W.; Li, Y.; Ruden, D.M.; Wang, F.; Li, Y.; Wang, F.; Song, J.; Zheng, K. Low Molecular Weight Heparin Ablates Lung Cancer Cisplatin-Resistance by Inducing Proteasome-Mediated ABCG2 Protein Degradation. PLoS ONE 2012, 7, e41035. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.T. The role of arachidonic acid in normal and malignant hematopoiesis. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Models Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Ruban, E.L.; Ferro, R.; Arifin, S.A.; Falasca, M. Lysophosphatidylinositol: A novel link between ABC transporters and G-protein-coupled receptors. Biochem. Soc. Trans. 2014, 42, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Suzuoki, M.; Miyamoto, M.; Kato, K.; Hiraoka, K.; Oshikiri, T.; Nakakubo, Y.; Fukunaga, A.; Shichinohe, T.; Shinohara, T.; Itoh, T.; et al. Impact of caveolin-1 expression on prognosis of pancreatic ductal adenocarcinoma. Br. J. Cancer 2002, 87, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Staubach, S.; Hanisch, F.-G. Lipid rafts: Signaling and sorting platforms of cells and their roles in cancer. Expert Rev. Proteom. 2011, 8, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Serafimidis, I.; Rodriguez-Aznar, E.; Lesche, M.; Yoshioka, K.; Takuwa, Y.; Dahl, A.; Pan, D.; Gavalas, A. Pancreas lineage allocation and specification are regulated by sphingosine-1-phosphate signalling. PLoS Biol. 2017, 15, e2000949. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, G.; Langmann, T.; Heimerl, S. Role of ABCG1 and other ABCG family members in lipid metabolism. J. Lipid Res. 2001, 42, 1513–1520. [Google Scholar] [PubMed]
- Chandra, J.; Samali, A.; Orrenius, S. Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 2000, 29, 323–333. [Google Scholar] [CrossRef]
- Inoue, M.; Sato, E.F.; Nishikawa, M.; Ah-Mee, P.; Yukimi, K.; Imada, I.; Utsumi, K. Mitochondrial Generation of Reactive Oxygen Species and its Role in Aerobic Life. Curr. Med. Chem. 2003, 10, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S. Novel aspects of oxidative stress-associated carcinogenesis. Antioxid. Redox Signal. 2006, 8, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Qiu, W.; Shirihai, O.S. Mitochondrial ABC transporters function: The role of ABCB10 (ABC-me) as a novel player in cellular handling of reactive oxygen species. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1945–1957. [Google Scholar] [CrossRef] [PubMed]
- Demicco, E.G.; Kavanagh, K.T.; Romieu-Mourez, R.; Wang, X.; Shin, S.R.; Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. RelB/p52 NF-κB complexes rescue an early delay in mammary gland development in transgenic mice with targeted superrepressor IκB-α expression and promote carcinogenesis of the mammary gland. Mol. Cell. Biol. 2005, 25, 10136–10147. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Campos, C.R.; Peart, J.C.; Smith, L.K.; Boni, J.L.; Cannon, R.E.; Miller, D.S. Nrf2 Upregulates ATP Binding Cassette Transporter Expression and Activity at the Blood-Brain and Blood-Spinal Cord Barriers. J. Neurosci. 2014, 34, 8585–8593. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Pagler, T.A.; Seimon, T.A.; Thorp, E.; Welch, C.L.; Witztum, J.L.; Tabas, I.; Tall, A.R. ABCA1 and ABCG1 Protect Against Oxidative Stress-Induced Macrophage Apoptosis During Efferocytosis. Circ. Res. 2010, 106, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, Y.; Wong, J.; Conklin, D.S. PPARγ maintains ERBB2-positive breast cancer stem cells. Oncogene 2013, 32, 5512–5521. [Google Scholar] [CrossRef] [PubMed]
- Tirinato, L.; Liberale, C.; Di Franco, S.; Candeloro, P.; Benfante, A.; La Rocca, R.; Potze, L.; Marotta, R.; Ruffilli, R.; Rajamanickam, V.P.; et al. Lipid droplets: A new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells 2015, 33, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.-X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Rose-Sperling, D.; Hellmich, U.A. Diverse relations between ABC transporters and lipids: An overview. Biochim. Biophys. Acta 2017, 1859, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Pomorski, T.; Hrafnsdóttir, S.; Devaux, P.F.; van Meer, G. Lipid distribution and transport across cellular membranes. Semin. Cell Dev. Biol. 2001, 12, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Tarling, E.J.; Vallim, T.Q.D.A.; Edwards, P.A. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol. Metab. 2013, 24, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Clay, A.T.; Sharom, F.J. Lipid Bilayer Properties Control Membrane Partitioning, Binding, and Transport of P-Glycoprotein Substrates. Biochemistry 2013, 52, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Romsicki, Y.; Sharom, F.J. The membrane lipid environment modulates drug interactions with the P-glycoprotein multidrug transporter. Biochemistry 1999, 38, 6887–6896. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, R.; Stafford, A.; Epand, R.M. Increased accumulation of drugs in a multidrug resistant cell line by alteration of membrane biophysical properties. Biochim. Biophys. Acta 1993, 1175, 277–282. [Google Scholar] [CrossRef]
- Cohen, R.; Neuzillet, C.; Tijeras-Raballand, A.; Faivre, S.; de Gramont, A.; Raymond, E. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget 2015, 6, 16832–16847. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.A. Is Reliance on Mitochondrial Respiration a “Chink in the Armor” of Therapy-Resistant Cancer? Cancer Cell 2014, 26, 788–795. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. The Warburg effect: Why and how do cancer cells activate glycolysis in the presence of oxygen? Anti-Cancer Agents Med. Chem. 2008, 8, 305–312. [Google Scholar] [CrossRef]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 15063. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Scandurra, F.M.; Gnaiger, E. Cell Respiration Under Hypoxia: Facts and Artefacts in Mitochondrial Oxygen Kinetics. In Oxygen Transport to Tissue XXXI; Takahashi, E., Bruley, D.F., Eds.; Springer: Boston, MA, USA, 2010; pp. 7–25. [Google Scholar]
- Rumsey, W.L.; Schlosser, C.; Nuutinen, E.M.; Robiolio, M.; Wilson, D.F. Cellular energetics and the oxygen dependence of respiration in cardiac myocytes isolated from adult rat. J. Biol. Chem. 1990, 265, 15392–15402. [Google Scholar] [PubMed]
- Ezashi, T.; Das, P.; Roberts, R.M. Low O2 tensions and the prevention of differentiation of hES cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4783–4788. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-J.; Chen, W.-W.; Zhang, X. Glioblastoma multiforme: Effect of hypoxia and hypoxia inducible factors on therapeutic approaches. Oncol. Lett. 2016, 12, 2283–2288. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Persano, L.; Pistollato, F.; Moro, E.; Frasson, C.; Porazzi, P.; Della Puppa, A.; Bresolin, S.; Battilana, G.; Indraccolo, S.; et al. Wnt activation promotes neuronal differentiation of Glioblastoma. Cell Death Dis. 2013, 4, e500. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhou, C.; Xu, L.; Xiao, H. Hypoxia enhances stemness of cancer stem cells in glioblastoma: An in vitro study. Int. J. Med. Sci. 2013, 10, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Garrido, W.; Rocha, J.D.; Jaramillo, C.; Fernandez, K.; Oyarzun, C.; San Martin, R.; Quezada, C. Chemoresistance in high-grade gliomas: Relevance of adenosine signalling in stem-like cells of glioblastoma multiforme. Curr. Drug Targets 2014, 15, 931–942. [Google Scholar] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Soga, T.; Pollard, P.J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Investig. 2013, 123, 3652–3658. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.E.; Nör, J.E. Perivascular stem cell niche in head and neck cancer. Cancer Lett. 2013, 338, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Filatova, A.; Acker, T.; Garvalov, B.K. The cancer stem cell niche(s): The crosstalk between glioma stem cells and their microenvironment. Biochim. Biophys. Acta 2013, 1830, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Infanger, D.W.; Cho, Y.; Lopez, B.S.; Mohanan, S.; Liu, S.C.; Gursel, D.; Boockvar, J.A.; Fischbach, C. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. 2013, 73, 7079–7089. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Perrin, R.; Hill, R.P.; Dubrovska, A.; Kurth, I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int. J. Radiat. Biol. 2014, 90, 636–652. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Liu, Y. Hypoxia-Inducible Factors in Cancer Stem Cells and Inflammation. Trends Pharmacol. Sci. 2015, 36, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Uribe, D.; Torres, Á.; Rocha, J.D.; Niechi, I.; Oyarzún, C.; Sobrevia, L.; San Martín, R.; Quezada, C. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol. Asp. Med. 2017, 55, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Celià-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable β catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef] [PubMed]
- Francí, C.; Takkunen, M.; Dave, N.; Alameda, F.; Gómez, S.; Rodríguez, R.; Escrivà, M.; Montserrat-Sentís, B.; Baró, T.; Garrido, M.; et al. Expression of Snail protein in tumor-stroma interface. Oncogene 2006, 25, 5134–5144. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.F.; Norton, L.; Massagué, J. Tumor self-seeding by circulating cancer cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, F.; Schlimok, G.; Dirschedl, P.; Witte, J.; Riethmüller, G. Prognostic Significance of Micrometastatic Tumour Cells in Bone Marrow of Colorectal Cancer Patients. Lancet 1992, 340, 685–689. [Google Scholar] [CrossRef]
- Sceneay, J.; Smyth, M.J.; Möller, A. The pre-metastatic niche: Finding common ground. Cancer Metastasis Rev. 2013, 32, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.-Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Krumpochova, P.; Sapthu, S.; Brouwers, J.F.; de Haas, M.; de Vos, R.; Borst, P.; van de Wetering, K. Transportomics: Screening for substrates of ABC transporters in body fluids using vesicular transport assays. FASEB J. 2012, 26, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, K.; Feddema, W.; Helms, J.B.; Brouwers, J.F.; Borst, P. Targeted metabolomics identifies glucuronides of dietary phytoestrogens as a major class of MRP3 substrates in vivo. Gastroenterology 2009, 137, 1725–1735. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ABC Transporters | Tissue Localization | Expression in Cancer | Expression in Cancer Stem Cells (CSCs) | Regulation by Genes & Signaling Pathways | Exogenous Substrates | Endogenous Substrates |
|---|---|---|---|---|---|---|
| ABCA1 | Nervous and hematopoietic system as well as kidney, liver and the blood brain barrier [56] | Pancreas [57], serous ovarian cancer [56] | Serous ovarian cancer initiating cells [58] | Transforming growth factor-β (TGF-β) [59] NF-κB, P65 [60] | Cisplatin [58] | Phosphatidylcholine, phosphatidylserine and sphingomyelin [61] |
| ABCA7 | Pancreas [57] | SREBP2 [62] | Phosphatidylserine [61], amyloid–β peptides [63,64] | |||
| ABCB1/MDR1/P-gp | Small intestine, liver, kidney placenta, BBB [56] | Colorectal, liver, renal cancer [56] | Acute myeloid leukemia (AML) [65] glioblastoma [66,67,68] ovaries [54] breast [69] renal cell carcinoma [70] | P63, P73 [71], OCT4 [72], Mir43b [73], miR-27a [74] hsamiR-451 [75], receptor tyrosine kinase 2 (ERBB2) [69], SMO [76], CD133 and DNA-PK through the PI3K/Akt-NF-κB pathway [68], PKCγ [70], IL6, IL8, hypoxia [67,77] | Anthracyclines actinomycin D, colchicine, etoposide, teniposide, methotrexate, mitomycin C, mitoxantrone, paclitaxel, docetaxel, vincristine, vinblastine [78,79] | Steroids, lipids, bilirubin, bile acids, platelet activating factor [79] |
| ABCB5 | CD133+ progenitor expressed in basal limbal epithelium among epidermal melanocytes [80] | Liver, lung, ovarian, thyroid [56] leukemia cells [81] | Malignant melanoma initiating cells (MMIC) [55,80,82] | Doxorubicin [83], 5-fluorouracil [84], camptothecin [84], irinotecan [84], mitozantrone [84], topotecan [84] | Interlukin 1 beta (IL1β) [82] | |
| ABCC1/MRP1 | Lung, testes, peripheral blood monocellular cells [56] | Endometrial, glioma, head and neck, lymphoma, melanoma, renal, thyroid cancer [56] | Glioblastoma [67] | MYCN [85], OCT4 [72], miR-326 [86], hypoxia [67] | Methotrexate, edatrexate, ZD1694, doxorubicin, daunorubicin, epirubicin, idarubicin, etoposide, vincristine, vinblastine, paclitaxel, irinotecan, SN-38, flutamide, hydroxyflutamide [87,88] | Leukotriene C4 (LTC₄) [89], lysophosphatidylinositol (LPI) [44], sphingosine-1-phosphate (S1P) [90], glutathione (GSH), glutathione disulphide (GSSH) [88] |
| ABCC3/MRP3 | Liver, intestine, colon, prostate, testes, brain, kidney [56] | Colorectal, cervical, lung, liver, thyroid, ovarian, pancreatic cancer [56] | OCT4 [72] | Cisplatin, doxorubicin, etoposide, methotrexate, teniopside, vincristine [88] | GSH [79] | |
| ABCC4/MRP4 | Widely-expressed | Prostate, renal, head and neck, endometrial cancer [56] | Osteocarcinoma [91] | MYCN [85], OCT4 [72], PI3K [91] | Topotecan, PMEA, methotrexate, 6-mercaptopurin [88] | Prostaglandins (PGs), cyclic nucleotides, steroid, GSH conjugates and folate [92] |
| ABCG2/BCRP | Placenta [93], intestine, liver, colon, breast [94] | Cervical, liver, lung, melanoma, testes, breast cancer [56] | Lung [49], pancreas [51,95], liver [96], breast [53,69], ovaries [50,97] | OCT4 [72], miR-212 [98], HMGA1 [97], ERBB2 [69], Hedgehog [99], SMO [76], PI3K/Akt [66] | Mitoxantrone, imatinib, anthracyclins, topotecan, flavopiridol, methotrexate [100] | Androgens [101], amyloid–β peptides [102], GSH [103] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. https://doi.org/10.3390/ijms18112362
Begicevic R-R, Falasca M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. International Journal of Molecular Sciences. 2017; 18(11):2362. https://doi.org/10.3390/ijms18112362
Chicago/Turabian StyleBegicevic, Romana-Rea, and Marco Falasca. 2017. "ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance" International Journal of Molecular Sciences 18, no. 11: 2362. https://doi.org/10.3390/ijms18112362
APA StyleBegicevic, R.-R., & Falasca, M. (2017). ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. International Journal of Molecular Sciences, 18(11), 2362. https://doi.org/10.3390/ijms18112362