Recent Advances in Comprehending the Signaling Pathways Involved in the Progression of Breast Cancer


Abstract
1. Introduction
2. Alterations of Signaling Pathways in Some Widespread Biological Mechanisms Involved in Breast Cancer Progression
2.1. The Calcium-Sensing Receptor (CaSR) Signaling
2.1.1. Normal Parathyroid and Mammary Glands
2.1.2. Breast Cancer Cells
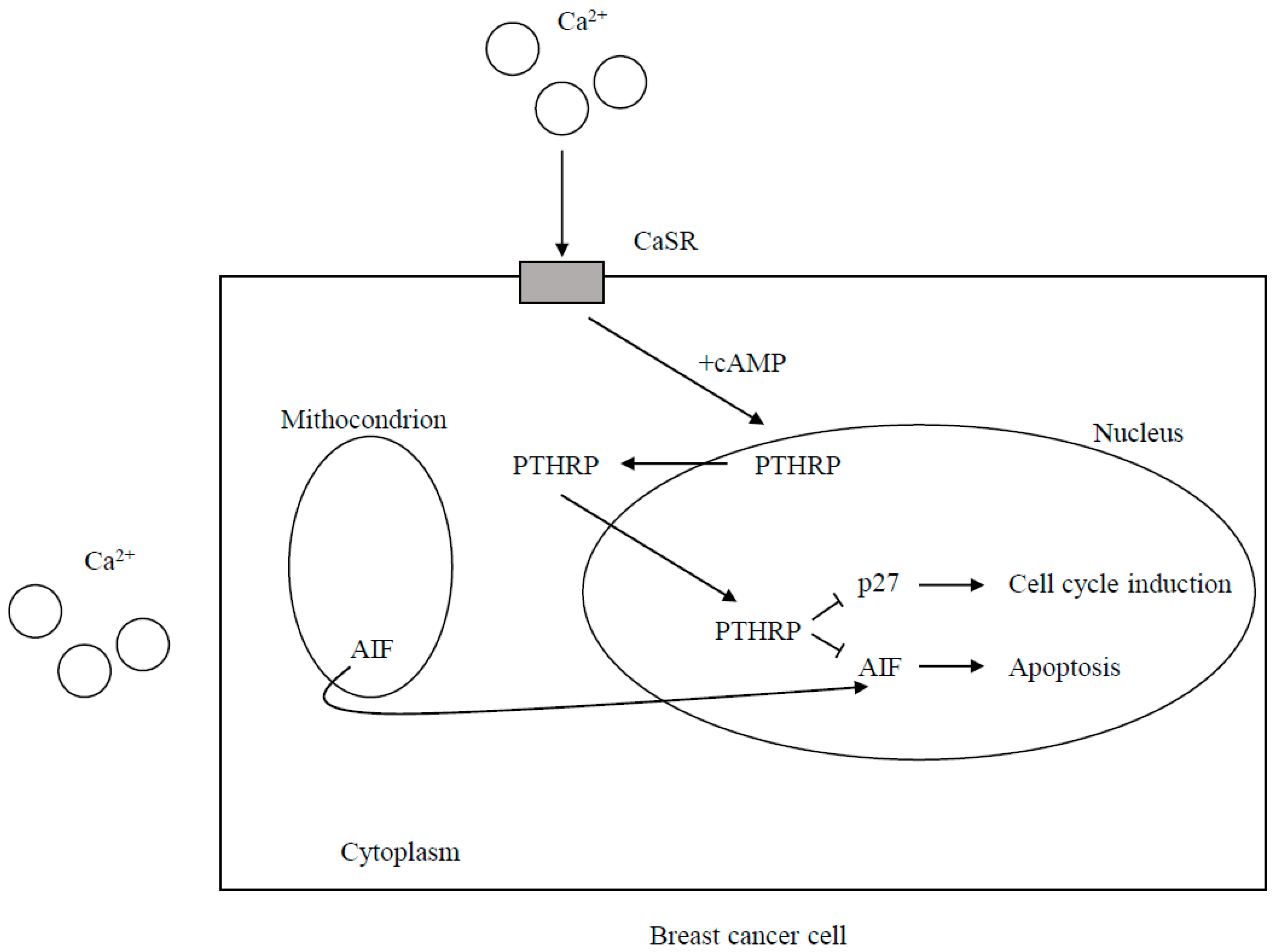
CaSR Expression and the Effects of CaSR Signaling on Cell Proliferation and Death
CaSR-Nuclear PTHRP Pathway
2.2. The Caveolae and Signaling
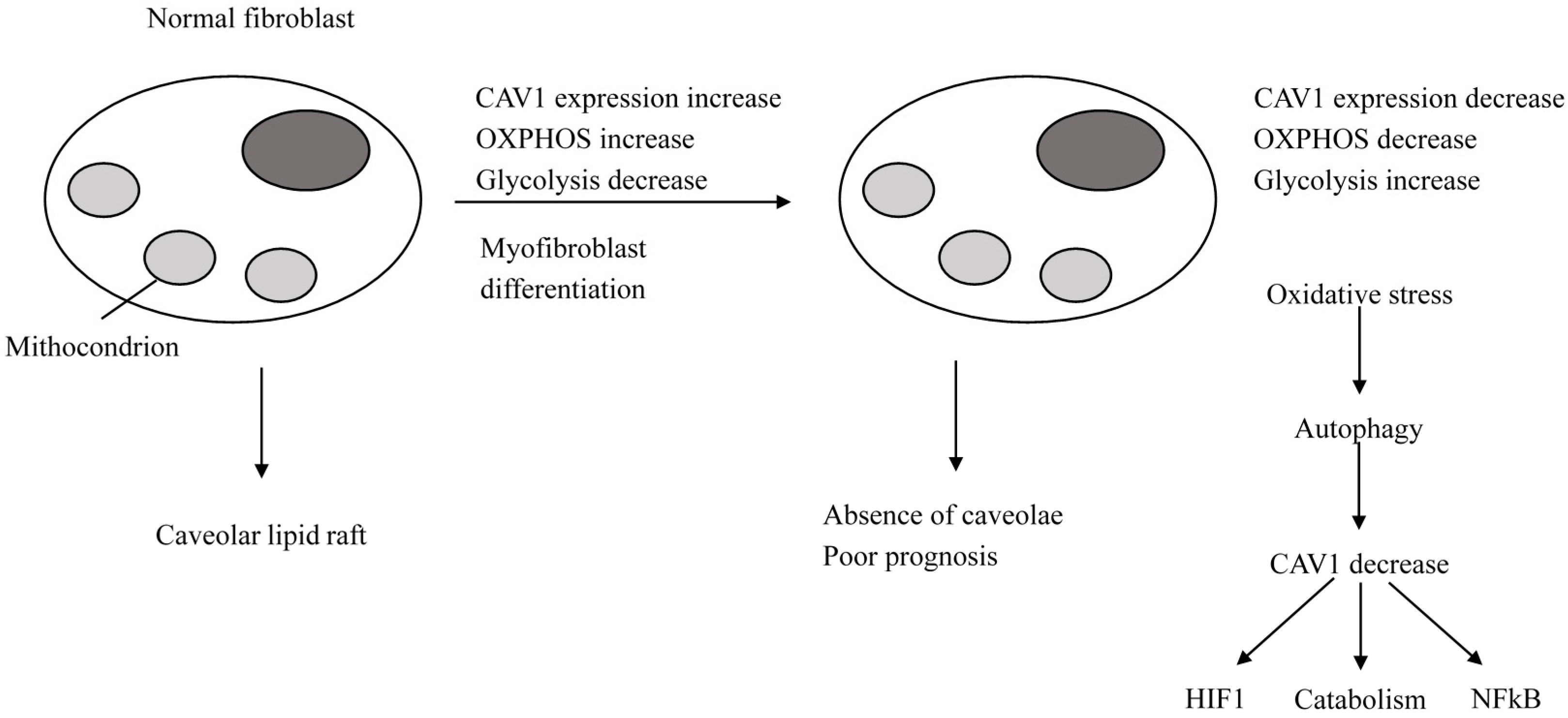
2.2.1. Normal Cells
2.2.2. Breast Cancer Cells
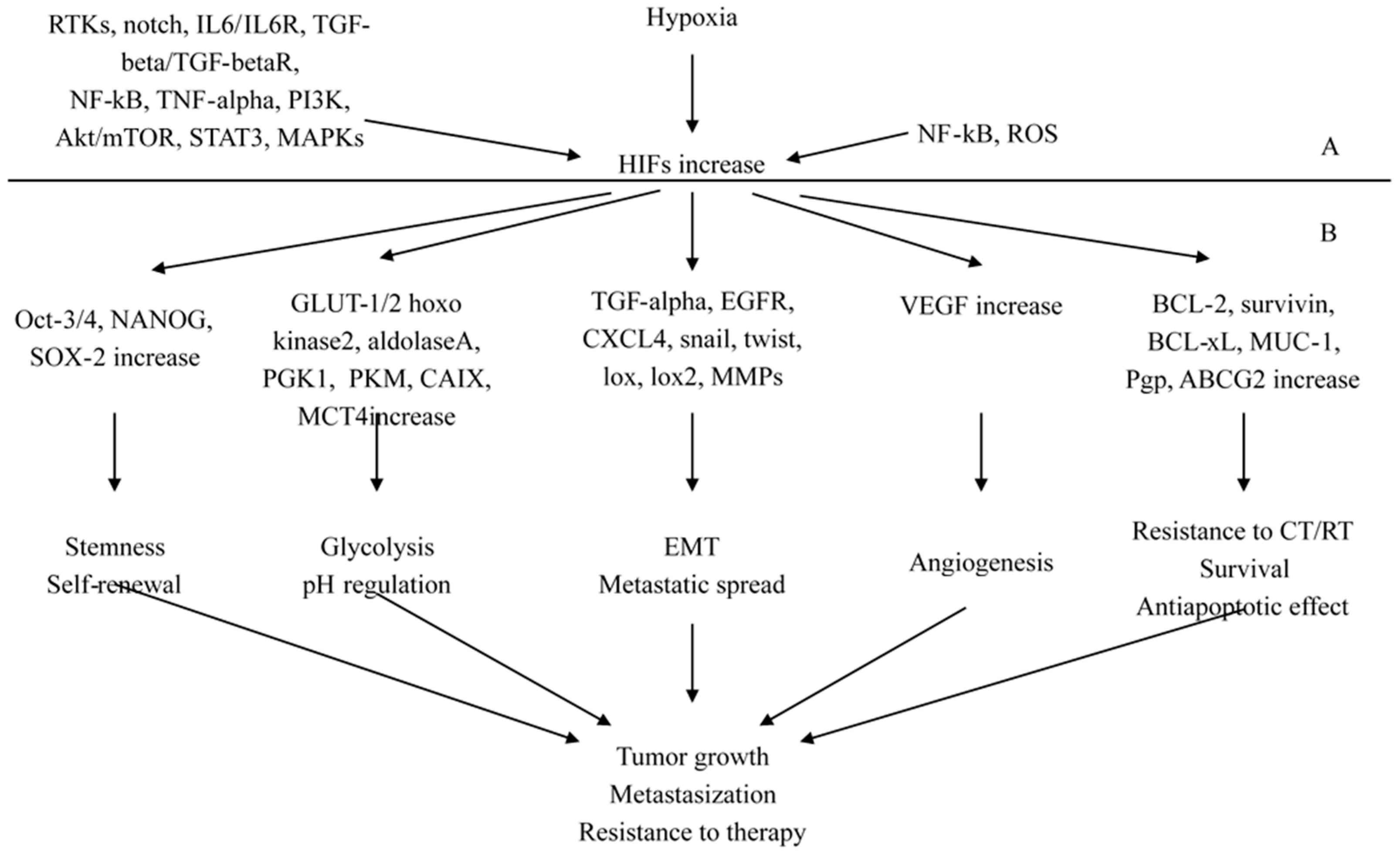
2.3. The Pathological Pathways Promoted by Hypoxia-Inducing Factors (HIFs)
2.3.1. HIFs as Key Regulators of Stemness
2.3.2. HIFs as Regulators of Cancer Progression and Metastasization
2.3.3. Molecular Signaling Mechanisms and Novel Interconnections Involving HIF-1 Pathway
2.3.4. HIFs-NF-κB Crosstalk and Inflammation in Cancer
2.4. Disturbances in the Apoptotic Machinery
2.5. Recently Unraveled Molecular Pathways Related to Epithelial to Mesenchymal Transition (EMT)
2.6. HER-2/Neu Gene Amplification and Protein Expression and the Expression of Other Members of the Epithelial Growth Factor Receptor Family
3. Conclusions
Conflicts of Interest
References
- Verigos, J.; Magklara, A. Revealing the Complexity of Breast Cancer by Next Generation Sequencing. Cancers 2015, 7, 2183–2200. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Zardavas, D.; Irrthum, A.; Swanton, C.; Piccart, M. Clinical management of breast cancer heterogeneity. Nat. Rev. Clin. Oncol. 2015, 12, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. New advances on critical implications of tumor- and metastasis-initiating cells in cancer progression, treatment resistance and disease recurrence. Histol. Histopathol. 2010, 25, 1057–1073. [Google Scholar] [PubMed]
- Mimeault, M.; Batra, S.K. Novel biomarkers and therapeutic targets for optimizing the therapeutic management of melanomas. World J. Clin. Oncol. 2012, 3, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bräuner-Osborne, H.; Wellendorph, P.; Jensen, A.A. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr. Drug Targets 2007, 8, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.M.; MacLeod, R.J. Extracellular calcium sensing and extracellular calcium signalling. Physiol. Rev. 2001, 81, 239–297. [Google Scholar] [PubMed]
- Brennan, S.C.; Thiem, U.; Roth, S.; Aggarwal, A.; Fetahu, I.S.; Tennakoon, S.; Gomes, A.R.; Brandi, M.L.; Bruggeman, F.; Mentaverri, R.; et al. Calcium sensing receptor signalling in physiology and cancer. Biochim. Biophys. Acta 2013, 1833, 1732–1744. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Wysolmerski, J.J. Calcium-Sensing Receptor in Breast Physiology and Cancer. Front. Physiol. 2016, 7, 440. [Google Scholar] [CrossRef] [PubMed]
- Wysolmerski, J.J. Parathyroid hormone-related protein: An update. J. Clin. Endocrinol. Metab. 2012, 97, 2947–2956. [Google Scholar] [CrossRef] [PubMed]
- Cheng, I.; Klingensmith, M.E.; Chattopadhyay, N.; Kifor, O.; Butters, R.R.; Soybel, D.I.; Brown, E.M. Identification and localization of the extracellular calcium-sensing receptor in human breast. J. Clin. Endocrinol. Metab. 1998, 83, 703–707. [Google Scholar] [CrossRef] [PubMed]
- VanHouten, J.; Dann, P.; McGeoch, G.; Brown, E.M.; Krapcho, K.; Neville, M.; Wysolmerski, J.J. The calcium-sensing receptor regulates mammary gland parathyroid hormone-related protein production and calcium transport. J. Clin. Investig. 2004, 113, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Mamillapalli, R.; VanHouten, J.; Zawalich, W.; Wysolmerski, J. Switching of G-protein usage by the calcium-sensing receptor reverses its effect on parathyroid hormone-related protein secretion in normal versus malignant breast cells. J. Biol. Chem. 2008, 283, 24435–24447. [Google Scholar] [CrossRef] [PubMed]
- VanHouten, J.N.; Wysolmerski, J.J. Transcellular calcium transport in mammary epithelial cells. J. Mammary Gland Biol. Neoplasia 2007, 12, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.L.; Chattopadhyay, N.; Kifor, O.; Yamaguchi, T.; Butters, R.R.; Brown, E.M. Extracellular calcium-sensing receptor expression and its potential role in regulating parathyroid hormone-related peptide secretion in human breast cancer cell lines. Endocrinology 2000, 141, 4357–4364. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Takyar, F.M.; Swan, K.; Jeong, J.; VanHouten, J.; Sullivan, C.; Dann, P.; Yu, H.; Fiaschi-Taesch, N.; Chang, W.; et al. Calcium-Sensing Receptor Promotes Breast Cancer by Stimulating Intracrine Actions of Parathyroid Hormone-Related Protein. Cancer Res. 2016, 76, 5348–5360. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kong, X.; Jiang, L.; Ma, T.; Yan, S.; Yuan, C.; Yang, Q. A genetic polymorphism (rs17251221) in the calcium-sensing receptor is associated with breast cancer susceptibility and prognosis. Cell. Physiol. Biochem. 2014, 33, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, L.; Moran, M.S.; Jiang, L.; Kong, X.; Zhang, H.; Zhang, X.; Haffty, B.G.; Yang, Q. Prognostic significance of calcium-sensing receptor in breast cancer. Tumour Biol. 2014, 35, 5709–5715. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Hydo, L.M.; Liu, S.; Miller, R.T. Activation of choline kinase by extracellular Ca2+ is Ca2+-sensing receptor, Gα12 and Rho-dependent in breast cancer cells. Cell Signal. 2009, 21, 1894–1900. [Google Scholar] [CrossRef] [PubMed]
- Vanhouten, J.N.; Wysolmerski, J.J. The calcium-sensing receptor in the breast. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Mihai, R.; Stevens, J.; McKinney, C.; Ibrahim, N.B. Expression of the calcium receptor in human breast cancer—A potential new marker predicting the risk of bone metastases. Eur. J. Surg. Oncol. 2006, 32, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Camp, R.L.; Chung, G.G.; Rimm, D.L. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat. Med. 2002, 8, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Haddad, S.A.; Hu, Q.; Liu, S.; Lunetta, K.L.; Ruiz-Narvaez, E.A.; Hong, C.C.; Zhu, Q.; Sucheston-Campbell, L.; Cheng, T.Y.; et al. Genetic variations in vitamin D-related pathways and breast cancer risk in African American women in the AMBER consortium. Int. J. Cancer 2016, 138, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Promkan, M.; Liu, G.; Patmasiriwat, P.; Chakrabarty, S. BRCA1 suppresses the expression of survivin and promotes sensitivity to paclitaxel through the calcium sensing receptor (CaSR) in human breast cancer cells. Cell Calcium 2011, 49, 79–88. [Google Scholar] [CrossRef] [PubMed]
- El Hiani, Y.; Ahidouch, A.; Lehen’kyi, V.; Hague, F.; Gouilleux, F.; Mentaverri, R.; Kamel, S.; Lassoued, K.; Brûlé, G.; Ouadid-Ahidouch, H. Extracellular signal-regulated kinases 1 and 2 and TRPC1 channels are required for calcium-sensing receptor-stimulated MCF-7 breast cancer cell proliferation. Cell. Physiol. Biochem. 2009, 23, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Macleod, R.J.; Chattopadhyay, N.; Tfelt-Hansen, J.; Kifor, O.; Butters, R.R.; Brown, E.M. Calcium-sensing receptor activation stimulates parathyroid hormone-related protein secretion in prostate cancer cells: Role of epidermal growth factor receptor transactivation. Bone 2004, 35, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Bollinger, N.; Creim, J.; Rodland, K.D. Cross-talk between the calcium-sensing receptor and the epidermal growth factor receptor in Rat-1 fibroblasts. Exp. Cell Res. 2005, 308, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.; Wildes, F.; Penet, M.F.; Winnard, P.T., Jr.; Glunde, K.; Artemov, D.; Ackerstaff, E.; Gimi, B.; Kakkad, S.; Raman, V.; et al. Choline kinase overexpression increases invasiveness and drug resistance of human breast cancer cells. NMR Biomed. 2010, 23, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Baio, G.; Rescinito, G.; Rosa, F.; Pace, D.; Boccardo, S.; Basso, L.; Salvi, S.; Calabrese, M.; Truini, M.; Neumaier, C.E. Correlation between Choline Peak at MR Spectroscopy and Calcium-Sensing Receptor Expression Level in Breast Cancer: A Preliminary Clinical Study. Mol. Imaging Biol. 2015, 17, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Hu, X.; Chakrabarty, S. Calcium sensing receptor down-regulates malignant cell behavior and promotes chemosensitivity in human breast cancer cells. Cell Calcium 2009, 45, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Boudot, C.; Hénaut, L.; Thiem, U.; Geraci, S.; Galante, M.; Saldanha, P.; Saidak, Z.; Six, I.; Clézardin, P.; Kamel, S.; et al. Overexpression of a functional calcium-sensing receptor dramatically increases osteolytic potential of MDA-MB-231 cells in a mouse model of bone metastasis through epiregulin-mediated osteoprotegerin downregulation. Oncotarget 2017, 8, 56460–56472. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Jia, X.; Xiao, G.; Kang, Y.; Partridge, N.C.; Qin, L. EGF-like ligands stimulate osteoclastogenesis by regulating expression of osteoclast regulatory factors by osteoblasts: Implications for osteolytic bone metastases. J. Biol. Chem. 2007, 282, 26656–26664. [Google Scholar] [CrossRef] [PubMed]
- Tao, N.; Wagner, S.J.; Lublin, D.M. CD36 is palmitoylated on both N- and C-terminal cytoplasmic tails. J. Biol. Chem. 1996, 271, 22315–22320. [Google Scholar] [CrossRef] [PubMed]
- Dietzen, D.J.; Hastings, W.R.; Lublin, D.M. Caveolin is palmitoylated on multiple cysteine residues. Palmitoylation is not necessary for localization of caveolin to caveolae. J. Biol. Chem. 1995, 270, 6838–6842. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; Way, M.; Zorzi, N.; Stang, E. Caveolin-3 associates with developing T-tubules during muscle differentiation. J. Cell Biol. 1997, 136, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Frank, P.G.; Cheung, M.W.; Pavlides, S.; Llaverias, G.; Park, D.S.; Lisanti, M.P. Caveolin-1 and regulation of cellular cholesterol homeostasis. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.; Marí, M.; Herms, A.; Fernández, A.; Fajardo, A.; Kassan, A.; Giralt, A.; Colell, A.; Balgoma, D.; Barbero, E.; et al. Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr. Biol. 2011, 21, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Trimmer, C.; Sotgia, F.; Whitaker-Menezes, D.; Balliet, R.M.; Eaton, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Iozzo, R.V.; Pestell, R.G.; et al. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment: a new genetically tractable model for human cancer associated fibroblasts. Cancer Biol. Ther. 2011, 11, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Caveolae and signalling in cancer. Nat. Rev. Cancer 2015, 15, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial lactate shuttle in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.; Lambros, M.B.; Robertson, D.; Jones, R.L.; Jones, C.; Mackay, A.; James, M.; Hornick, J.L.; Pereira, E.M.; Milanezi, F.; et al. Caveolin 1 is overexpressed and amplified in a subset of basal-like and metaplastic breast carcinomas: A morphologic, ultrastructural, immunohistochemical, and in situ hybridization analysis. Clin. Cancer Res. 2007, 13, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Mercier, I.; Camacho, J.; Titchen, K.; Gonzales, D.M.; Quann, K.; Bryant, K.G.; Molchansky, A.; Milliman, J.N.; Whitaker-Menezes, D.; Sotgia, F.; et al. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: Chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am. J. Pathol. 2012, 181, 278–293. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.H.; Takeda, K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008, 15, 635–641. [Google Scholar] [CrossRef] [PubMed]
- D’Alterio, C.; Barbieri, A.; Portella, L.; Palma, G.; Polimeno, M.; Riccio, A.; Ieranò, C.; Franco, R.; Scognamiglio, G.; Bryce, J.; et al. Inhibition of stromal CXCR4 impairs development of lung metastases. Cancer Immunol. Immunother. 2012, 61, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The von Hippel-Lindau tumor suppressor gene and kidney cancer. Clin. Cancer Res. 2004, 10, 6290S–6295S. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and Inflammation in Cancer, Focus on HIF and NF-κB. Biomedicines 2017, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Partch, C.L.; Gardner, K.H. Coactivators necessary for transcriptional output of the hypoxia inducible factor, HIF, are directly recruited by ARNT PAS-B. Proc. Natl. Acad. Sci. USA 2011, 108, 7739–7744. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell. Mol. Med. 2013, 17, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Jubb, A.M.; Buffa, F.M.; Harris, A.L. Assessment of tumour hypoxia for prediction of response to therapy and cancer prognosis. J. Cell. Mol. Med. 2010, 14, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Giuntoli, S.; Tanturli, M.; di Gesualdo, F.; Barbetti, V.; Rovida, E.; Dello Sbarba, P. Glucose availability in hypoxia regulates the selection of chronic myeloid leukemia progenitor subsets with different resistance to imatinib-mesylate. Haematologica 2011, 96, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Costa, J.P.; Zanetti, J.S.; Silveira, G.G.; Soave, D.F.; Oliveira, L.R.; Zorgetto, V.A.; Soares, F.A.; Zucoloto, S.; Ribeiro-Silva, A. Differential expression of HIF-1α in CD44+CD24−/low breast ductal carcinomas. Diagn. Pathol. 2011, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Taylor, M.J.; Walsh, L.A.; Dieters-Castator, D.; Das, P.; Jewer, M.; Zhang, G.; Postovit, L.M. Low oxygen levels induce the expression of the embryonic morphogen Nodal. Mol. Biol. Cell. 2011, 22, 4809–4821. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Li, Y.; Zhang, H.; Nan, F. Breast cancer stromal fibroblasts promote the generation of CD44+CD24− cells through SDF-1/CXCR4 interaction. J. Exp. Clin. Cancer Res. 2010, 29, 80. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Gilkes, D.M.; Zhang, H.; Chen, J.; Wei, H.; Chaturvedi, P.; Fraley, S.I.; Wong, C.M.; Khoo, U.S.; Ng, I.O.; et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl. Acad. Sci. USA 2011, 108, 16369–16374. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.J.; Wang, S.; Liu, X.A.; Shen, E.C.; Ding, Q.; Lu, C.; Xu, J.; Cao, Q.H.; Zhu, H.Q.; Wang, F. A novel mouse model of human breast cancer stem-like cells with high CD44+CD24−/lower phenotype metastasis to human bone. Chin. Med. J. 2008, 121, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.K.; Mohammad, K.S.; Fournier, P.G.; McKenna, C.R.; Davis, H.W.; Niewolna, M.; Peng, X.H.; Chirgwin, J.M.; Guise, T.A. Hypoxia and TGF-β drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS ONE 2009, 4, e6896. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, N.; Wottawa, M.; Katschinski, D.M.; Kristiansen, G.; Kietzmann, T. Hypoxia-inducible factor prolyl hydroxylase 2 (PHD2) is a direct regulator of epidermal growth factor receptor (EGFR) signaling in breast cancer. Oncotarget 2017, 8, 9885–9898. [Google Scholar] [PubMed]
- Nakaoka, H.J.; Tanei, Z.; Hara, T.; Weng, J.S.; Kanamori, A.; Hayashi, T.; Sato, H.; Orimo, A.; Otsuji, K.; Tada, K.; et al. Mint3-mediated L1CAM expression in fibroblasts promotes cancer cell proliferation via integrin α5β1 and tumour growth. Oncogenesis 2017, 6, e334. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; He, C.; Li, Z.; Wang, Z.; Zhang, Q. FBP1 modulates cell metabolism of breast cancer cells by inhibiting the expression of HIF-1α. Neoplasma 2017, 64, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kutomi, G.; Kajiwara, T.; Kukita, K.; Kochin, V.; Kanaseki, T.; Tsukahara, T.; Hirohashi, Y.; Torigoe, T.; Okamoto, Y.; et al. Cancer-associated oxidoreductase ERO1-α promotes immune escape through up-regulation of PD-L1 in human breast cancer. Oncotarget 2017, 8, 24706–24718. [Google Scholar] [CrossRef] [PubMed]
- Padró, M.; Louie, R.J.; Lananna, B.V.; Krieg, A.J.; Timmerman, L.A.; Chan, D.A. Genome-independent hypoxic repression of estrogen receptor α in breast cancer cells. BMC Cancer 2017, 17, 203. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, D.; Yao, Z.; Lin, X.; Liu, J.; Gu, Q.; Dong, X.; Liu, F.; Wang, Y.; Yao, N.; et al. Anti-angiogenic treatment promotes triple-negative breast cancer invasion via vasculogenic mimicry. Cancer Biol. Ther. 2017, 18, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Ikegami, Y.; Nakazawa, H.; Kuriyama, N.; Oki, M.; Hanai, J.; Sukhatme, V.P.; Kaneki, M. Low-Dose Farnesyltransferase Inhibitor Suppresses HIF-1α and Snail Expression in Triple-Negative Breast Cancer MDA-MB-231 Cells In Vitro. J. Cell. Physiol. 2017, 232, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Bandarra, D.; Rocha, S. Tale of two transcription factors: NF-κB and HIF crosstalk. OA Mol. Cell Biol. 2013, 1, 6. [Google Scholar] [CrossRef]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Rodriguez, J.; Pickel, C.; Burr, S.; Fabrizio, J.A.; Nolan, K.A.; Spielmann, P.; Cavadas, M.A.; Crifo, B.; Halligan, D.N.; et al. FIH Regulates Cellular Metabolism through Hydroxylation of the Deubiquitinase OTUB1. PLoS Biol. 2016, 14, e1002347. [Google Scholar] [CrossRef] [PubMed]
- Melvin, A.; Mudie, S.; Rocha, S. Further insights into the mechanism of hypoxia-induced NF-κB. Cell Cycle 2011, 10, 879–882, Erratum in: Cell Cycle 2011, 10, 2041. [Google Scholar] [CrossRef] [PubMed]
- Shmakova, A.; Batie, M.; Druker, J.; Rocha, S. Chromatin and oxygen sensing in the context of JmjC histone demethylases. Biochem. J. 2014, 462, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Sun, H.; Chen, H.; Zavadil, J.; Kluz, T.; Arita, A.; Costa, M. Hypoxia induces trimethylated H3 lysine 4 by inhibition of JARID1A demethylase. Cancer Res. 2010, 70, 4214–4221. [Google Scholar] [CrossRef] [PubMed]
- Prickaerts, P.; Adriaens, M.E.; Beucken, T.V.D.; Koch, E.; Dubois, L.; Dahlmans, V.E.H.; Gits, C.; Evelo, C.T.A.; Chan-Seng-Yue, M.; Wouters, B.G.; et al. Hypoxia increases genome-wide bivalent epigenetic marking by specific gain of H3K27me3. Epigenetics Chromatin 2016, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Yang, M.; Huang, D.B.; Wei, H.; Ozer, G.H.; Ghosh, G.; Stark, G.R. Role of lysine methylation of NF-κB in differential gene regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 13510–13515. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Jackson, M.W.; Wang, B.; Yang, M.; Chance, M.R.; Miyagi, M.; Gudkov, A.V.; Stark, G.R. Regulation of NF-κB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. USA 2010, 107, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Batie, M.; Druker, J.; D’Ignazio, L.; Rocha, S. KDM2 Family Members are Regulated by HIF-1 in Hypoxia. Cells 2017, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Bandarra, D.; Biddlestone, J.; Mudie, S.; Muller, H.A.; Rocha, S. Hypoxia activates IKK-NF-κB and the immune response in Drosophila melanogaster. Biosci. Rep. 2014, 34, e00127. [Google Scholar] [CrossRef] [PubMed]
- Booy, E.P.; Henson, E.S.; Gibson, S.B. Epidermal growth factor regulates Mcl-1 expression through the MAPK-Elk-1 signalling pathway contributing to cell survival in breast cancer. Oncogene 2011, 30, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.R.; Vaillant, F.; Lim, E.; Lee, L.; Breslin, K.; Feleppa, F.; Deb, S.; Ritchie, M.E.; Takano, E.; Ward, T.; et al. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc. Natl. Acad. Sci. USA 2012, 109, 2766–2771. [Google Scholar] [CrossRef] [PubMed]
- Vitagliano, O.; Addeo, R.; D’Angelo, V.; Indolfi, C.; Indolfi, P.; Casale, F. The Bcl-2/Bax and Ras/Raf/MEK/ERK signaling pathways: Implications in pediatric leukemia pathogenesis and new prospects for therapeutic approaches. Expert Rev. Hematol. 2013, 6, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Mortenson, M.M.; Galante, J.G.; Gilad, O.; Schlieman, M.G.; Virudachalam, S.; Kung, H.J.; Bold, R.J. BCL-2 functions as an activator of the AKT signaling pathway in pancreatic cancer. J. Cell. Biochem. 2007, 102, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Ning, Y.; Polverini, P.J. Endothelial cells expressing Bcl-2 promotes tumor metastasis by enhancing tumor angiogenesis, blood vessel leakiness and tumor invasion. Lab. Investig. 2008, 88, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Tucker, C.A.; Kapanen, A.I.; Chikh, G.; Hoffman, B.G.; Kyle, A.H.; Wilson, I.M.; Masin, D.; Gascoyne, R.D.; Bally, M.; Klasa, R.J. Silencing Bcl-2 in models of mantle cell lymphoma is associated with decreases in cyclin D1, nuclear factor-κB, p53, bax, and p27 levels. Mol. Cancer Ther. 2008, 7, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef] [PubMed]
- Elfadl, D.; Hodgkinson, V.C.; Long, E.D.; Scaife, L.; Drew, P.J.; Lind, M.J.; Cawkwell, L. A pilot study to investigate the role of the 26S proteasome in radiotherapy resistance and loco-regional recurrence following breast conserving therapy for early breast cancer. Breast 2011, 20, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Scully, M.; Dawson, G.; Goodwin, C.; Xia, M.; Lu, X.; Kakkar, A. Perturbation of the heparin/heparin-sulfate interactome of human breast cancer cells modulates pro-tumourigenic effects associated with PI3K/Akt and MAPK/ERK signalling. Thromb. Haemost. 2013, 109, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Winsel, S.; Mäki-Jouppila, J.; Tambe, M.; Aure, M.R.; Pruikkonen, S.; Salmela, A.L.; Halonen, T.; Leivonen, S.K.; Kallio, L.; Børresen-Dale, A.L.; et al. Excess of miRNA-378a-5p perturbs mitotic fidelity and correlates with breast cancer tumourigenesis in vivo. Br. J. Cancer 2014, 111, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Reavie, L.; Couto, J.P.; De Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O.; et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Yadav, V.; Kumar, S.; Saini, N. MicroRNA-195 inhibits proliferation, invasion and metastasis in breast cancer cells by targeting FASN, HMGCR, ACACA and CYP27B1. Sci. Rep. 2015, 5, 17454. [Google Scholar] [CrossRef] [PubMed]
- Al Saleh, S.; Al Mulla, F.; Luqmani, Y.A. Estrogen receptor silencing induces epithelial to mesenchymal transition in human breast cancer cells. PLoS ONE 2011, 6, e20610. [Google Scholar] [CrossRef] [PubMed]
- Satram-Maharaj, T.; Nyarko, J.N.; Kuski, K.; Fehr, K.; Pennington, P.R.; Truitt, L.; Freywald, A.; Lukong, K.E.; Anderson, D.H.; Mousseau, D.D. The monoamine oxidase-A inhibitor clorgyline promotes a mesenchymal-to-epithelial transition in the MDA-MB-231 breast cancer cell line. Cell Signal. 2014, 26, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Ding, K.; Chong, Q.Y.; Zhao, J.; Liu, Y.; Shao, Y.; Zhang, Y.; Yu, Q.; Xiong, Z.; Zhang, W.; et al. Post-transcriptional regulation of ERBB2 by miR26a/b and HuR confers resistance to tamoxifen in estrogen receptor-positive breast cancer cells. J. Biol. Chem. 2017, 292, 13551–13564. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Ferrari, P.; Biava, P.M.; Carpi, A. Changing the endocrine dependence of breast cancer: Data and hypotheses. Curr. Med. Chem. 2014, 21, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.; Pippen, J., Jr.; Pivot, X.; Lichinitser, M.; Sadeghi, S.; Dieras, V.; Gomez, H.L.; Romieu, G.; Manikhas, A.; Kennedy, M.J.; et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 2009, 27, 5538–5546. [Google Scholar] [CrossRef] [PubMed]
- De Melo Gagliato, D.; Jardim, D.L.; Marchesi, M.S.; Hortobagyi, G.N. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget 2016, 7, 64431–64446. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zi, X.; Zhao, Y.; Mascarenhas, D.; Pollak, M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J. Natl. Cancer Inst. 2001, 93, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Browne, B.C.; Crown, J.; Venkatesan, N.; Duffy, M.J.; Clynes, M.; Slamon, D.; O’Donovan, N. Inhibition of IGF1R activity enhances response to trastuzumab in HER-2-positive breast cancer cells. Ann. Oncol. 2011, 22, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Blumenschein, G.R., Jr.; Mills, G.B.; Gonzalez-Angulo, A.M. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J. Clin. Oncol. 2012, 30, 3287–3296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, W.C.; Li, P.; Guo, H.; Poh, S.B.; Brady, S.W.; Xiong, Y.; Tseng, L.M.; Li, S.H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; Kellokumpu-Lehtinen, P.L.; Bono, P.; Kataja, V.; Desmedt, C.; et al. Tumor-infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann. Oncol. 2014, 25, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Wimana, Z.; Gebhart, G.; Guiot, T.; Vanderlinden, B.; Larsimont, D.; Doumont, G.; van Simaeys, G.; Goldman, S.; Flamen, P.; Ghanem, G. N-Acetylcysteine breaks resistance to trastuzumab caused by MUC4 overexpression in human HER2 positive BC-bearing nude mice monitored by 89Zr-Trastuzumab and 18F-FDG PET imaging. Oncotarget 2017, 8, 56185–56198. [Google Scholar] [PubMed]
- Xu, X.; de Angelis, C.; Burke, K.A.; Nardone, A.; Hu, H.; Qin, L.; Veeraraghavan, J.; Sethunath, V.; Heiser, L.M.; Wang, N.; et al. HER2 Reactivation through Acquisition of the HER2 L755S Mutation as a Mechanism of Acquired Resistance to HER2-targeted Therapy in HER2+ Breast Cancer. Clin. Cancer Res. 2017, 23, 5123–5134. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh, S.; Yazdanparast, R. Activation of STAT3/HIF-1α/Hes-1 axis promotes trastuzumab resistance in HER2-overexpressing breast cancer cells via down-regulation of PTEN. Biochim. Biophys. Acta 2017, 1861, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, J.; de Angelis, C.; Reis-Filho, J.S.; Pascual, T.; Prat, A.; Rimawi, M.F.; Osborne, C.K.; Schiff, R. De-escalation of treatment in HER2-positive breast cancer: Determinants of response and mechanisms of resistance. Breast 2017, S19–S26. [Google Scholar] [CrossRef] [PubMed]
- Ocana, A.; Gil-Martin, M.; Martín, M.; Rojo, F.; Antolín, S.; Guerrero, Á.; Trigo, J.M.; Muñoz, M.; Pandiella, A.; Diego, N.G.; et al. A phase I study of the SRC kinase inhibitor dasatinib with trastuzumab and paclitaxel as first line therapy for patients with HER2-overexpressing advanced breast cancer. GEICAM/2010-04 study. Oncotarget 2017, 8, 73144–73153. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.J.; Zhao, F.; Wang, M.; Swaby, R.F.; Sparano, J.A.; Meropol, N.J.; Bhalla, K.N.; Pellegrino, C.M.; Katherine Alpaugh, R.; Falkson, C.I.; et al. A Phase I/II study of suberoylanilide hydroxamic acid (SAHA) in combination with trastuzumab (Herceptin) in patients with advanced metastatic and/or local chest wall recurrent HER2-amplified breast cancer: A trial of the ECOG-ACRIN Cancer Research Group (E1104). Breast Cancer Res. Treat. 2017, 165, 375–382. [Google Scholar] [PubMed]
- Van Keymeulen, A.; Lee, M.Y.; Ousset, M.; Brohée, S.; Rorive, S.; Giraddi, R.R.; Wuidart, A.; Bouvencourt, G.; Dubois, C.; Salmon, I.; et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015, 525, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, F.L.; Vitorino, R.; Wang, J.; Cardoso, H.; Laranjeira, H.; Simões, J.; Caldas, M.; Henrique, R.; Amado, F.; Williams, C.; et al. The histone H2A isoform Hist2h2ac is a novel regulator of proliferation and epithelial-mesenchymal transition in mammary epithelial and in breast cancer cells. Cancer Lett. 2017, 396, 42–52. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Component(s) | Mechanism | Outcome | References |
|---|---|---|---|
| Heparin/heparin sulfate interactome | Increased PI3K/Akt, MAPK/ERK signaling and TGF-β activity | Tumorigenic phenotype, cell adhesive, invasive properties | [93] |
| miR-37pa-5p overexpression | Inactivation of SAC through induced receptor tyrosine kinase-MAPK pathway and suppression of Aurora kinase | More aggressive and poorly differentiated molecular subtypes | [94] |
| Lineage restriction | Genetic mutation of hyperactivating to PI3K pathway in luminal or basal epithelial cells | Induction of stemness and tumor heterogeneity | [95] |
| has-miR-195 and miR-195 | Decreased cholesterol and triglycerides with mythocondrial dysfunction and involvement of xenobiotic metabolism signaling | Decreased proliferation, invasion and migration | [96] |
| ER-α | ER-α loss | EMT induction | [97,98] |
| MAO-A | MAO inhibitor (clorgyline) through non-canonical pathway | ||
| Hist2h2ac histone isoform | MEK1/2 or PI3K activation | ||
| ER α HER2 amplification and expression of other members of EGFR family | ER-α/HER2 cross-talk, PI3K/Akt/mTOR pathway escape (PI3KCA mutation/PTEN loss, HER2/IGF1R cross-talk), c-met overexpression, src activation, low TILs level | Breast cancer progression | [99,100,101,102,103,104,105,106,107,108,109,110,111,112,113] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolini, A.; Ferrari, P.; Diodati, L.; Carpi, A. Recent Advances in Comprehending the Signaling Pathways Involved in the Progression of Breast Cancer. Int. J. Mol. Sci. 2017, 18, 2321. https://doi.org/10.3390/ijms18112321
Nicolini A, Ferrari P, Diodati L, Carpi A. Recent Advances in Comprehending the Signaling Pathways Involved in the Progression of Breast Cancer. International Journal of Molecular Sciences. 2017; 18(11):2321. https://doi.org/10.3390/ijms18112321
Chicago/Turabian StyleNicolini, Andrea, Paola Ferrari, Lucrezia Diodati, and Angelo Carpi. 2017. "Recent Advances in Comprehending the Signaling Pathways Involved in the Progression of Breast Cancer" International Journal of Molecular Sciences 18, no. 11: 2321. https://doi.org/10.3390/ijms18112321
APA StyleNicolini, A., Ferrari, P., Diodati, L., & Carpi, A. (2017). Recent Advances in Comprehending the Signaling Pathways Involved in the Progression of Breast Cancer. International Journal of Molecular Sciences, 18(11), 2321. https://doi.org/10.3390/ijms18112321