Development of A Chimeric Antigen Receptor Targeting C-Type Lectin-Like Molecule-1 for Human Acute Myeloid Leukemia

{kind=link}
{kind=link}
Abstract
1. Introduction
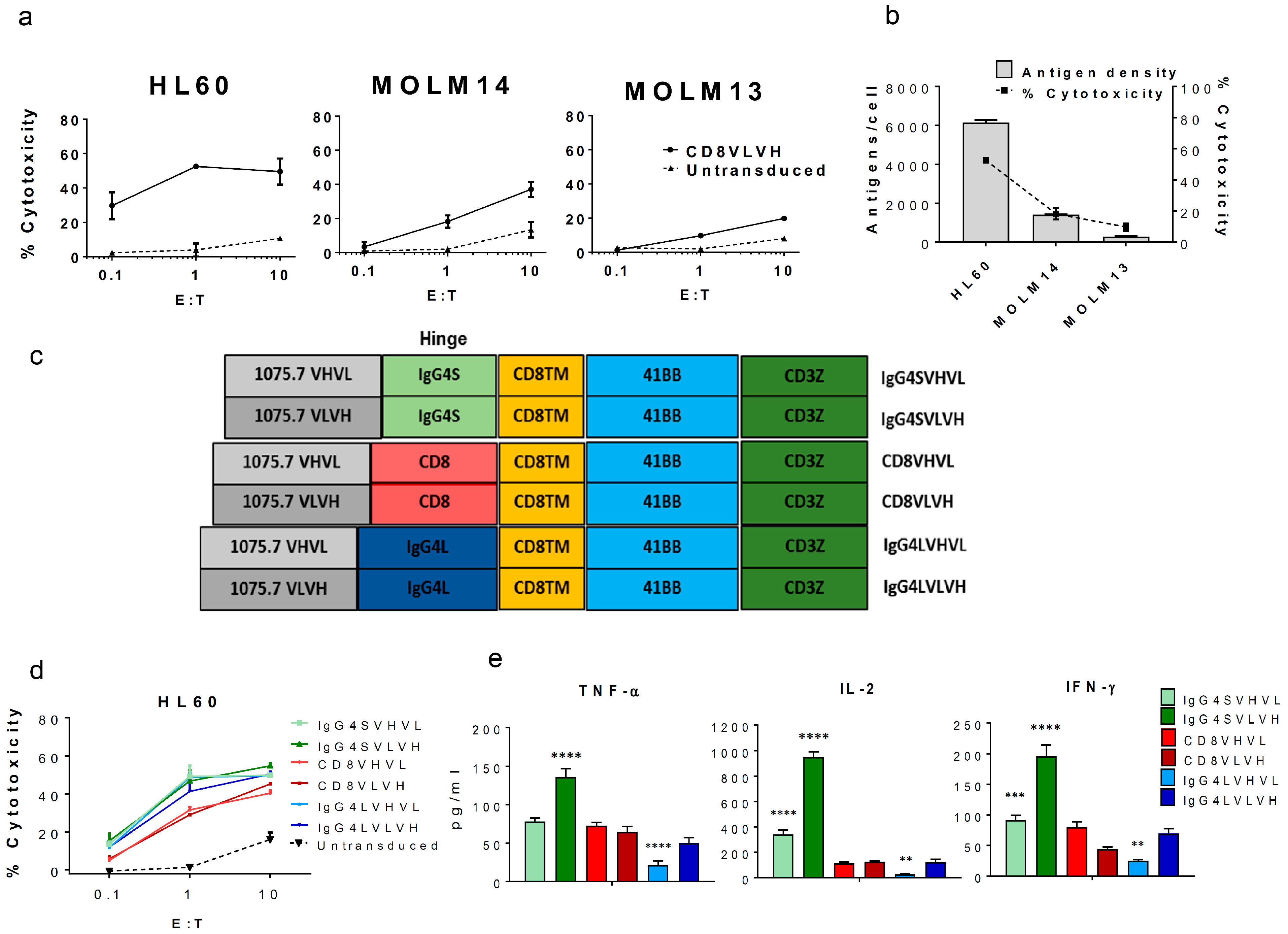
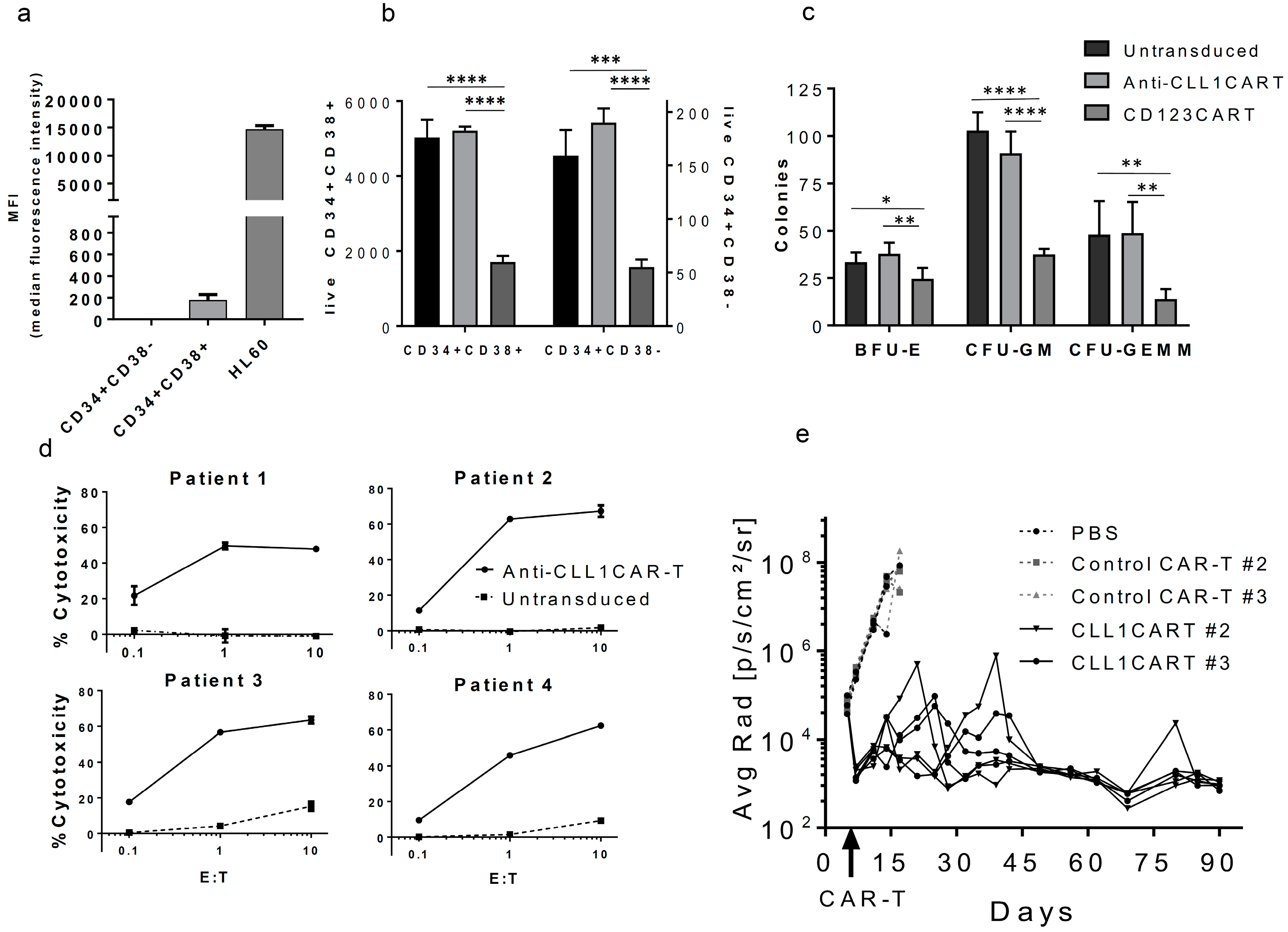
2. Results and Discussion
3. Materials and Methods
3.1. Cells and Human Derived Samples
3.2. In Vitro Assays
3.3. Xenograft Model
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rotiroti, M.C.; Arcangeli, S.; Casucci, M.; Perriello, V.; Bondanza, A.; Biondi, A.; Tettamanti, S.; Biagi, E. Acute myeloid leukemia targeting by chimeric antigen receptor t cells: Bridging the gap from preclinical modeling to human studies. Hum. Gene Ther. 2017, 28, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Buggins, A.G.; Milojkovic, D.; Arno, M.J.; Lea, N.C.; Mufti, G.J.; Thomas, N.S.; Hirst, W.J. Microenvironment produced by acute myeloid leukemia cells prevents t cell activation and proliferation by inhibition of nf-kappab, c-myc, and prb pathways. J. Immunol. 2001, 167, 6021–6030. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed]
- O’Hear, C.; Heiber, J.F.; Schubert, I.; Fey, G.; Geiger, T.L. Anti-cd33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica 2015, 100, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Pizzitola, I.; Anjos-Afonso, F.; Rouault-Pierre, K.; Lassailly, F.; Tettamanti, S.; Spinelli, O.; Biondi, A.; Biagi, E.; Bonnet, D. Chimeric antigen receptors against cd33/cd123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 2014, 28, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, S.; Marin, V.; Pizzitola, I.; Magnani, C.F.; Attianese, G.M.P.G.; Cribioli, E.; Maltese, F.; Galimberti, S.; Lopez, A.F.; Biondi, A.; et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel cd123-specific chimeric antigen receptor. Br. J. Haematol. 2013, 161, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.S.; Wang, Y.; Lv, H.Y.; Han, Q.W.; Fan, H.; Guo, B.; Wang, L.L.; Han, W.D. Treatment of cd33-directed chimeric antigen receptor-modified t cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Mardiros, A.; Dos Santos, C.; McDonald, T.; Brown, C.E.; Wang, X.; Budde, L.E.; Hoffman, L.; Aguilar, B.; Chang, W.C.; Bretzlaff, W.; et al. T cells expressing cd123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood 2013, 122, 3138–3148. [Google Scholar] [CrossRef] [PubMed]
- Cellectis. Cellectis Reports Clinical Hold of ucart123 Studies. Available online: http://www.cellectis.com/en/press/cellectis-reports-clinical-hold-of-ucart123-studies (accessed on 22 September 2017).
- Bakker, A.B.; van den Oudenrijn, S.; Bakker, A.Q.; Feller, N.; van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.; Visser, T.J.; Bijl, N.; Geuijen, C.A.; et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, H.; Sauer, T.; Shum, T.; Parikh, K.; Mamonkin, M.; Omer, B.; Rouce, R.H.; Lulla, P.; Rooney, C.M.; Gottschalk, S.; et al. Treatment of acute myeloid leukemia with t cells expressing chimeric antigen receptors directed to C-type lectin-like molecule 1. Mol. Ther. 2017, 25, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhou, Q.; Deshmukh, V.; Phull, H.; Ma, J.; Tardif, V.; Naik, R.R.; Bouvard, C.; Zhang, Y.; Choi, S.; et al. Targeting human C-type lectin-like molecule-1 (cll1) with a bispecific antibody for immunotherapy of acute myeloid leukemia. Angew. Chem. Int. Ed. Engl. 2014, 53, 9841–9845. [Google Scholar] [CrossRef] [PubMed]
- Moshaver, B.; van Rhenen, A.; Kelder, A.; van der Pol, M.; Terwijn, M.; Bachas, C.; Westra, A.H.; Ossenkoppele, G.J.; Zweegman, S.; Schuurhuis, G.J. Identification of a small subpopulation of candidate leukemia-initiating cells in the side population of patients with acute myeloid leukemia. Stem Cells 2008, 26, 3059–3067. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; van Dongen, G.A.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Stigter-van Walsum, M.; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The novel aml stem cell associated antigen cll-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Singh, S.; Pardoux, C.; Zhao, J.; Hsi, E.D.; Abo, A.; Korver, W. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica 2010, 95, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing cd137 signal transduction domains mediate enhanced survival of t cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of car-t cells for b-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: Evaluation of four different scfvs and antigens. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer. Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ror1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [PubMed]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human t cells containing cd28 and cd137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [PubMed]
- Krzemieniecki, K.; Sevelda, P.; Erdkamp, F.; Smakal, M.; Schwenkglenks, M.; Puertas, J.; Trojan, A.; Szabo, Z.; Bendall, K.; Maenpaa, J. Neutropenia management and granulocyte colony-stimulating factor use in patients with solid tumours receiving myelotoxic chemotherapy--findings from clinical practice. Support. Care Cancer 2014, 22, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Micheletti, A.; Cassatella, M.A. Modulation of human neutrophil survival and antigen expression by activated cd4+ and cd8+ T cells. J. Leukoc. Biol. 2010, 88, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Mishalian, I.; Bayuh, R.; Eruslanov, E.; Michaeli, J.; Levy, L.; Zolotarov, L.; Singhal, S.; Albelda, S.M.; Granot, Z.; Fridlender, Z.G. Neutrophils recruit regulatory T-cells into tumors via secretion of ccl17—A new mechanism of impaired antitumor immunity. Int. J. Cancer 2014, 135, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Eruslanov, E.B.; Bhojnagarwala, P.S.; Quatromoni, J.G.; Stephen, T.L.; Ranganathan, A.; Deshpande, C.; Akimova, T.; Vachani, A.; Litzky, L.; Hancock, W.W.; et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J. Clin. Investig. 2014, 124, 5466–5480. [Google Scholar] [CrossRef] [PubMed]
- Dolen, Y.; Esendagli, G. Myeloid leukemia cells with a b7-2(+) subpopulation provoke th-cell responses and become immuno-suppressive through the modulation of b7 ligands. Eur. J. Immunol. 2013, 43, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Dudley, M.E.; Wunderlich, J.; El-Gamil, M.; Li, Y.F.; Zhou, J.; Huang, J.; Powell, D.J.; Rosenberg, S.A. Cutting edge: Persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004, 173, 7125–7130. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.D.; Carroll, M.; et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laborda, E.; Mazagova, M.; Shao, S.; Wang, X.; Quirino, H.; Woods, A.K.; Hampton, E.N.; Rodgers, D.T.; Kim, C.H.; Schultz, P.G.; et al. Development of A Chimeric Antigen Receptor Targeting C-Type Lectin-Like Molecule-1 for Human Acute Myeloid Leukemia. Int. J. Mol. Sci. 2017, 18, 2259. https://doi.org/10.3390/ijms18112259
Laborda E, Mazagova M, Shao S, Wang X, Quirino H, Woods AK, Hampton EN, Rodgers DT, Kim CH, Schultz PG, et al. Development of A Chimeric Antigen Receptor Targeting C-Type Lectin-Like Molecule-1 for Human Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2017; 18(11):2259. https://doi.org/10.3390/ijms18112259
Chicago/Turabian StyleLaborda, Eduardo, Magdalena Mazagova, Sida Shao, Xinxin Wang, Herlinda Quirino, Ashley K. Woods, Eric N. Hampton, David T. Rodgers, Chan Hyuk Kim, Peter G. Schultz, and et al. 2017. "Development of A Chimeric Antigen Receptor Targeting C-Type Lectin-Like Molecule-1 for Human Acute Myeloid Leukemia" International Journal of Molecular Sciences 18, no. 11: 2259. https://doi.org/10.3390/ijms18112259
APA StyleLaborda, E., Mazagova, M., Shao, S., Wang, X., Quirino, H., Woods, A. K., Hampton, E. N., Rodgers, D. T., Kim, C. H., Schultz, P. G., & Young, T. S. (2017). Development of A Chimeric Antigen Receptor Targeting C-Type Lectin-Like Molecule-1 for Human Acute Myeloid Leukemia. International Journal of Molecular Sciences, 18(11), 2259. https://doi.org/10.3390/ijms18112259