Zinc Signals and Immunity



Abstract

1. Introduction
2. Zinc Homeostasis and the Immune System: An Overview
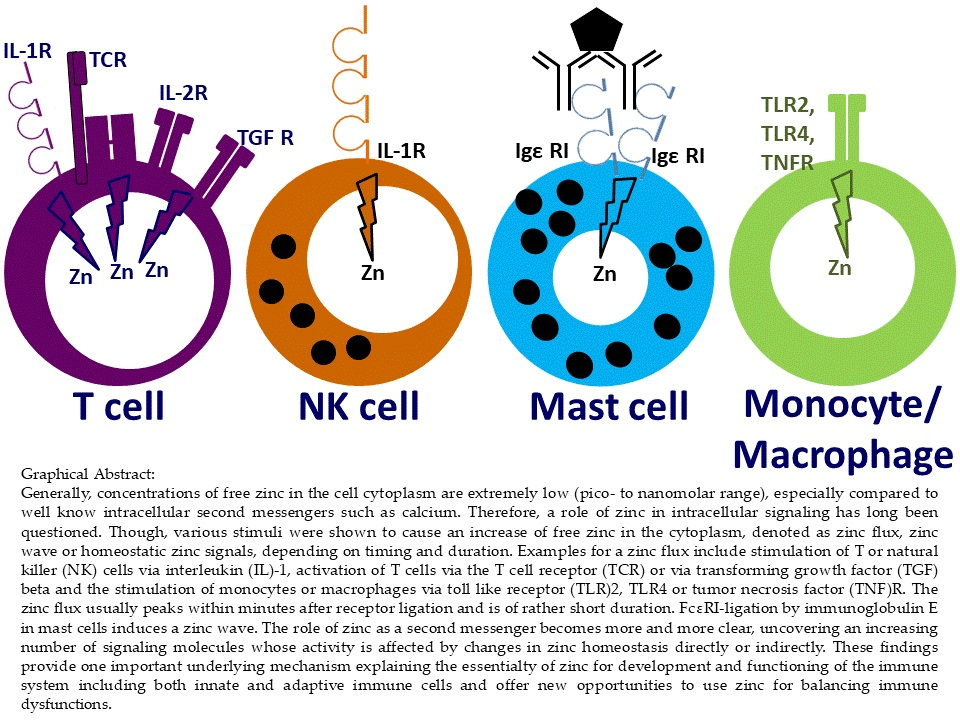
3. Classification of Zinc Signals
4. Zinc, Signaling and Immunity
4.1. Innate Immune Cell Functions
4.2. Zinc and (De-)Phosphorylation
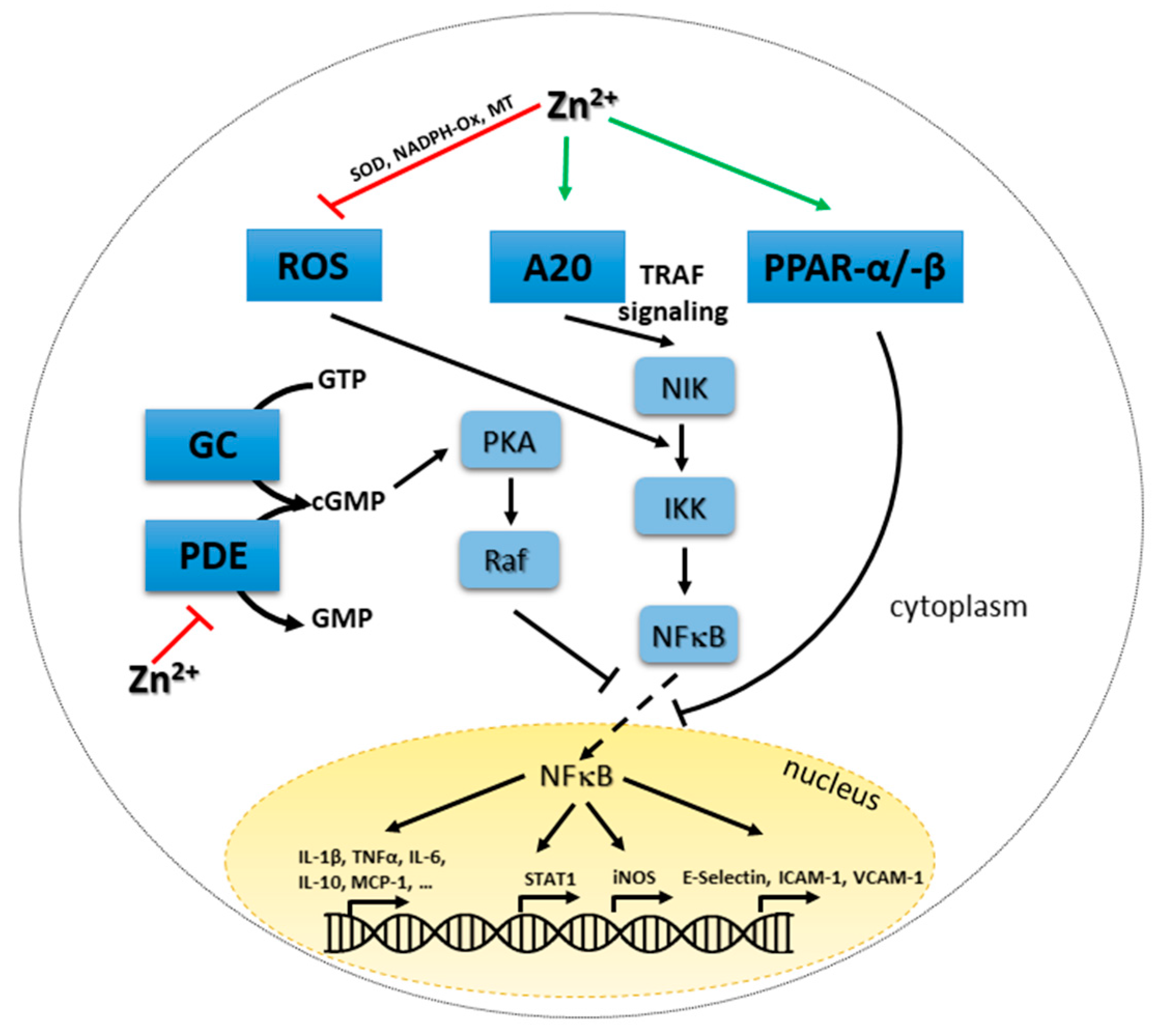
4.3. Zinc and Redox Metabolism: Two Strongly Intertwined Second Messengers
4.4. Zinc, Cyclic Nucleotides and Proteinases A and G
4.5. Zinc-Mediated Regulation of NFκB Signaling and A20 Expression
4.6. Zinc and Signaling Proteome
4.7. Zinc and Transcription Factors
5. From Altered Zinc Homeostasis via Signaling to Innate Immune Cell Functions
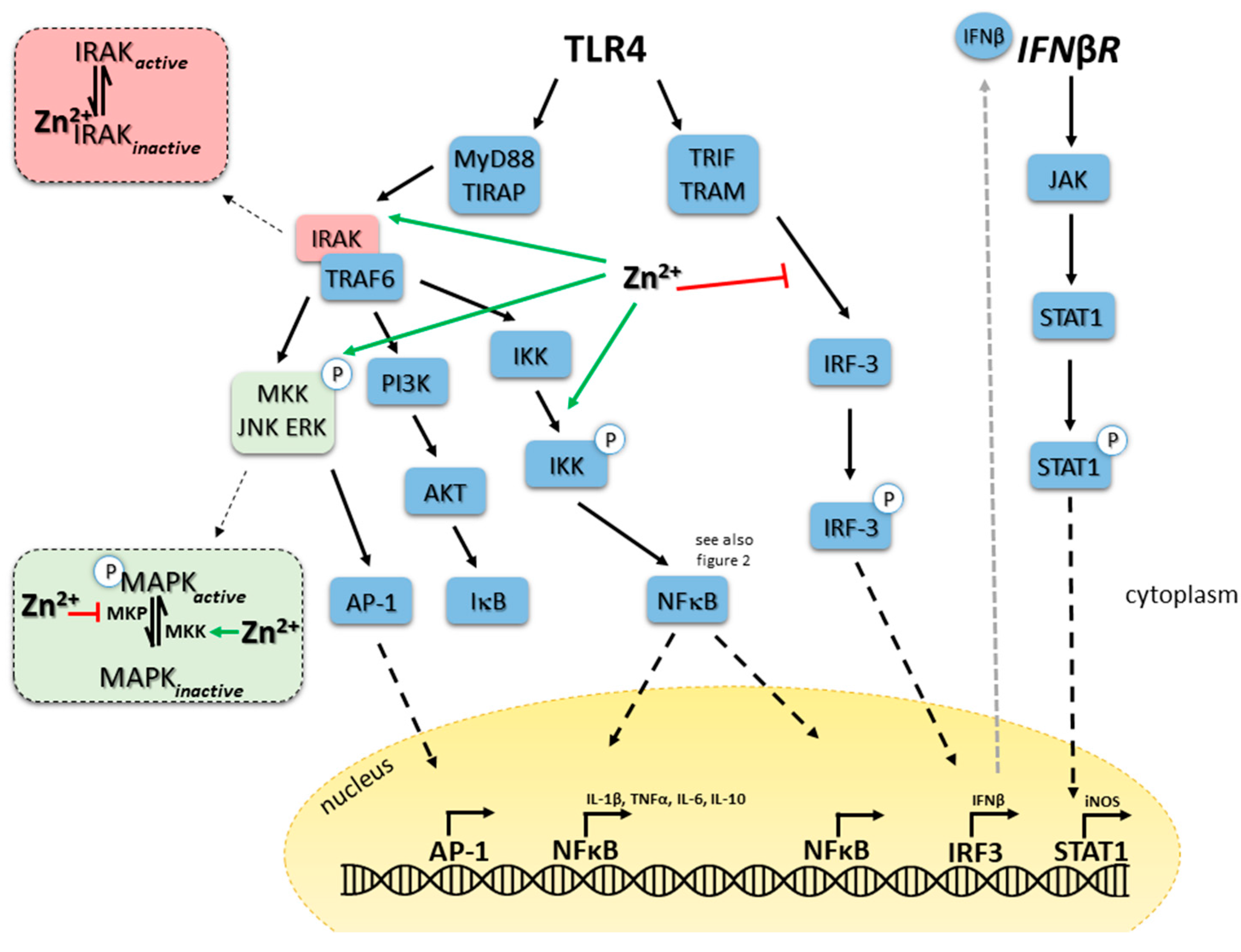
5.1. TLR4 Signaling in Monocytes
5.2. Zinc Homeostasis, Hematopoiesis and STAT3
5.3. Zinc Alters Killing Activity of Natural Killer Cells
5.4. Mast Cell Degranulation Depends on Zinc Levels
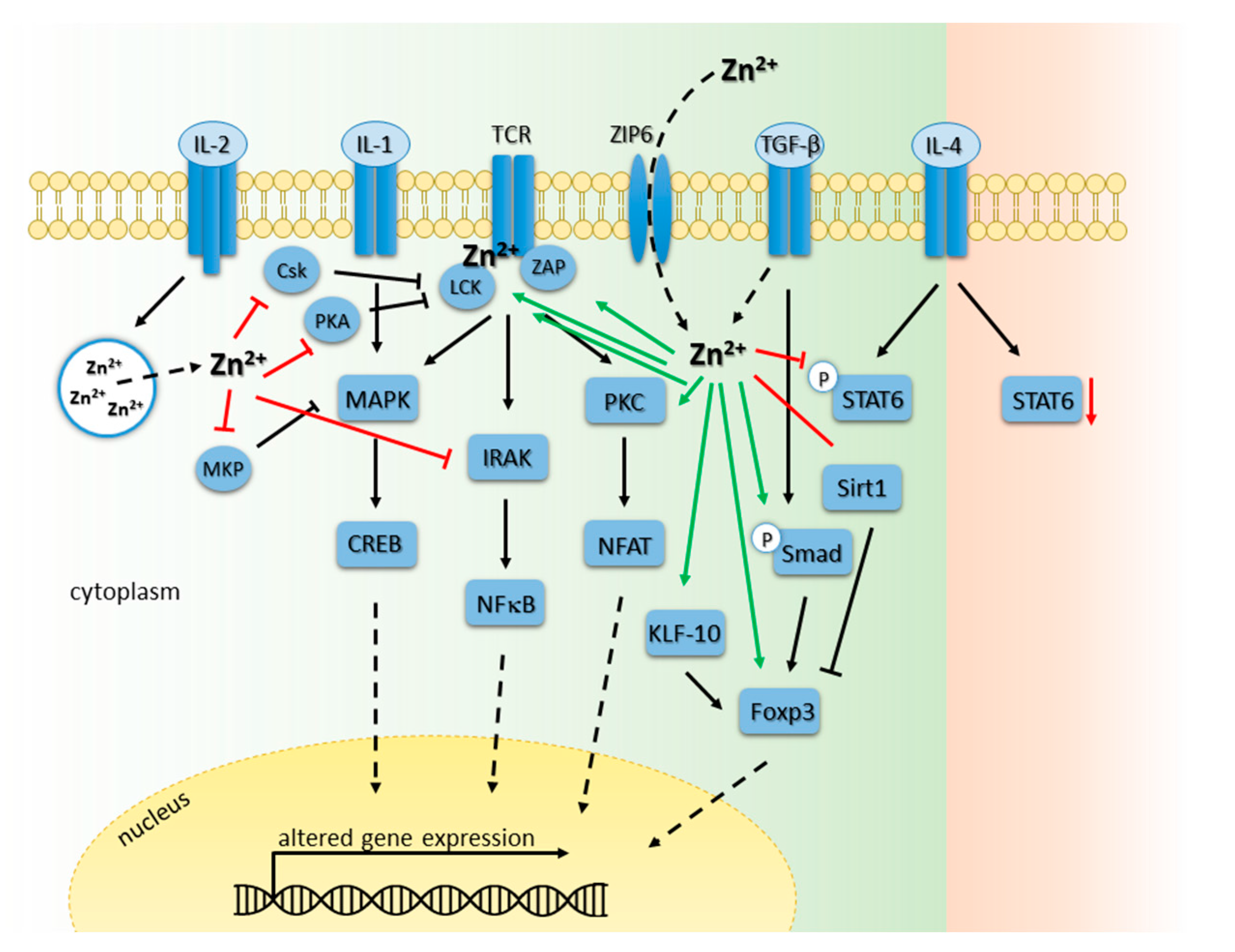
6. The Adaptive Immunity
6.1. Zinc Homeostasis in Development of T and B Cells
6.2. Zinc Signals in T Cell Receptor-Triggered Signaling Cascades
6.3. Zinc Signals in Interleukin Receptor Signaling Pathways in T Cells
6.4. Zinc Signals in B Cell Maturation, Survival and Function
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Prasad, A.S.; Miale, A., Jr.; Farid, Z.; Sandstead, H.H.; Schulert, A.R. Zinc metabolism in patients with the syndrome of iron deficiency anemia, hepatosplenomegaly, dwarfism, and hypognadism. J. Lab. Clin. Med. 1963, 61, 537–549. [Google Scholar] [PubMed]
- Krezel, A.; Maret, W. The biological inorganic chemistry of zinc ions. Arch. Biochem. Biophys. 2016, 611, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Pfaender, S.; Fohr, K.; Lutz, A.K.; Putz, S.; Achberger, K.; Linta, L.; Liebau, S.; Boeckers, T.M.; Grabrucker, A.M. Cellular zinc homeostasis contributes to neuronal differentiation in human induced pluripotent stem cells. Neural Plast. 2016, 2016, 3760702. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Rink, L. Multiple impacts of zinc on immune function. Metallomics Integr. Biomet. Sci. 2014, 6, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Brieger, A.; Rink, L.; Haase, H. Differential regulation of tlr-dependent myd88 and trif signaling pathways by free zinc ions. J. Immunol. 2013, 191, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Duke, R.C.; Chervenak, R.; Cohen, J.J. Endogenous endonuclease-induced DNA fragmentation: An early event in cell-mediated cytolysis. Proc. Natl. Acad. Sci. USA 1983, 80, 6361–6365. [Google Scholar] [CrossRef] [PubMed]
- Finamore, A.; Massimi, M.; Conti Devirgiliis, L.; Mengheri, E. Zinc deficiency induces membrane barrier damage and increases neutrophil transmigration in Caco-2 cells. J. Nutr. 2008, 138, 1664–1670. [Google Scholar] [PubMed]
- Miyoshi, Y.; Tanabe, S.; Suzuki, T. Cellular zinc is required for intestinal epithelial barrier maintenance via the regulation of claudin-3 and occludin expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G105–G116. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid. Redox Signal. 2006, 8, 1419–1441. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, E.; Hogstrand, C.; Maret, W. Redox and zinc signalling pathways converging on protein tyrosine phosphatases. Free Radic. Biol. Med. 2014, 75 (Suppl. S1), S9. [Google Scholar] [CrossRef] [PubMed]
- Truong-Tran, A.Q.; Carter, J.; Ruffin, R.E.; Zalewski, P.D. The role of zinc in caspase activation and apoptotic cell death. Biometals 2001, 14, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Sunderman, F.W., Jr. The influence of zinc on apoptosis. Ann. Clin. Lab. Sci. 1995, 25, 134–142. [Google Scholar] [PubMed]
- Maret, W. Zinc in pancreatic islet biology, insulin sensitivity, and diabetes. Prev. Nutr. Food Sci. 2017, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bredholt, M.; Frederiksen, J.L. Zinc in multiple sclerosis: A systematic review and meta-analysis. ASN Neuro 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Yang, X.; Cai, G.; Fan, D.; Xia, Q.; Liu, L.; Hu, Y.; Ding, N.; Xu, S.; Wang, L.; et al. Serum levels of copper and zinc in patients with rheumatoid arthritis: A meta-analysis. Biol. Trace Elem. Res. 2015, 168, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stelmashook, E.V.; Isaev, N.K.; Genrikhs, E.E.; Amelkina, G.A.; Khaspekov, L.G.; Skrebitsky, V.G.; Illarioshkin, S.N. Role of zinc and copper ions in the pathogenetic mechanisms of alzheimer’s and parkinson’s diseases. Biochem. Mosc. 2014, 79, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, B. Zinc homeostasis and neurodegenerative disorders. Front. Aging Neurosci. 2013, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Alder, H.; Taccioli, C.; Chen, H.; Jiang, Y.; Smalley, K.J.; Fadda, P.; Ozer, H.G.; Huebner, K.; Farber, J.L.; Croce, C.M.; et al. Dysregulation of miR-31 and miR-21 induced by zinc deficiency promotes esophageal cancer. Carcinogenesis 2012, 33, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Ressnerova, A.; Raudenska, M.; Holubova, M.; Svobodova, M.; Polanska, H.; Babula, P.; Masarik, M.; Gumulec, J. Zinc and copper homeostasis in head and neck cancer: Review and meta-analysis. Curr. Med. Chem. 2016, 23, 1304–1330. [Google Scholar] [CrossRef] [PubMed]
- Overbeck, S.; Rink, L.; Haase, H. Modulating the immune response by oral zinc supplementation: A single approach for multiple diseases. Arch. Immunol. Ther. Exp. Warsz. 2008, 56, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.Z.; Rink, L. Zinc in infection and inflammation. Nutrients 2017, 9. [Google Scholar] [CrossRef]
- World Health Organization (WHO). World Health Organization—The World Health Report; WHO: Geneva, Switzerland, 2002; Volume 83. [Google Scholar]
- Rink, L. Zinc in Human Health; IOS Press: Amsterdam, The Netherlands, 2011; p. 596. [Google Scholar]
- Lowe, N.M.; Dykes, F.C.; Skinner, A.L.; Patel, S.; Warthon-Medina, M.; Decsi, T.; Fekete, K.; Souverein, O.W.; Dullemeijer, C.; Cavelaars, A.E.; et al. Eurreca-estimating zinc requirements for deriving dietary reference values. Crit. Rev. Food Sci. Nutr. 2013, 53, 1110–1123. [Google Scholar] [CrossRef] [PubMed]
- Krebs, N.F. Overview of zinc absorption and excretion in the human gastrointestinal tract. J. Nutr. 2000, 130 (Suppl. 5S), 1374s–1377s. [Google Scholar] [PubMed]
- Cousins, R.J. Gastrointestinal factors influencing zinc absorption and homeostasis. Int. J. Vitam. Nutr. Res. 2010, 80, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Wojtkiewicz, J.; Makowska, K.; Bejer-Olenska, E.; Gonkowski, S. Zinc transporter 3 (znt3) as an active substance in the enteric nervous system of the porcine esophagus. J. Mol. Neurosci. 2017, 61, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Wojtkiewicz, J.; Rytel, L.; Makowska, K.; Gonkowski, S. Co-localization of zinc transporter 3 (Znt3) with sensory neuromediators and/or neuromodulators in the enteric nervous system of the porcine esophagus. Biometals 2017, 30, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Skrovanek, S.; DiGuilio, K.; Bailey, R.; Huntington, W.; Urbas, R.; Mayilvaganan, B.; Mercogliano, G.; Mullin, J.M. Zinc and gastrointestinal disease. World J. Gastrointest. Pathophysiol. 2014, 5, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Rink, L. Functional significance of zinc-related signaling pathways in immune cells. Annu. Rev. Nutr. 2009, 29, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Fukada, T. Roles of zinc signaling in the immune system. J. Immunol. Res. 2016, 2016, 6762343. [Google Scholar] [CrossRef] [PubMed]
- Brieger, A.; Rink, L. Zink und immunfunktionen. Ernährung Med. 2010, 25, 156–160. [Google Scholar] [CrossRef]
- Mocchegiani, E.; Costarelli, L.; Giacconi, R.; Cipriano, C.; Muti, E.; Tesei, S.; Malavolta, M. Nutrient–gene interaction in ageing and successful ageing: A single nutrient (Zinc) and some target genes related to inflammatory/immune response. Mech. Ageing Dev. 2006, 127, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Freeland-Graves, J.H.; Bodzy, P.W.; Eppright, M.A. Zinc status of vegetarians. J. Am. Diet. Assoc. 1980, 77, 655–661. [Google Scholar] [PubMed]
- Sandström, B.; Cederblad, Å.; Lindblad, B.S.; Lönnerdal, B. Acrodermatitis enteropathica, zinc metabolism, copper status, and immune function. Arch. Pediatr. Adolesc. Med. 1994, 148, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, M.; Olbert, M.; Wyszogrodzka, G.; Młyniec, K.; Librowski, T. Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-κB signaling. Inflammopharmacology 2017, 25, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Rink, L.; Haase, H. Zinc homeostasis and immunity. Trends Immunol. 2007, 28, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Dubben, S.; Honscheid, A.; Winkler, K.; Rink, L.; Haase, H. Cellular zinc homeostasis is a regulator in monocyte differentiation of HL-60 cells by 1 α,25-dihydroxyvitamin D3. J. Leukoc. Biol. 2010, 87, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Weston, W.L.; Huff, J.C.; Humbert, J.R.; Hambidge, K.M.; Neldner, K.H.; Walravens, P.A. Zinc correction of defective chemotaxis in acrodermatitis enteropathica. Arch. Dermatol. 1977, 113, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Rink, L.; Haase, H. Zinc signals in neutrophil granulocytes are required for the formation of neutrophil extracellular traps. Innate Immun. 2013, 19, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Rink, L.; Haase, H. Chelation of free Zn2+ impairs chemotaxis, phagocytosis, oxidative burst, degranulation, and cytokine production by neutrophil granulocytes. Biol. Trace Elem. Res. 2016, 171, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C. Zinc: Physiology, deficiency, and parenteral nutrition. Nutr. Clin. Pract. 2015, 30, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Malavolta, M. Zinc dyshomeostasis, ageing and neurodegeneration: Implications of A2M and inflammatory gene polymorphisms. J. Alzheimer’s Dis. 2007, 12, 101–109. [Google Scholar] [CrossRef]
- Kimura, T.; Kambe, T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; O’Bryant, Z.; Xiong, Z.-G. Zinc-Permeable Ion Channels: Effects on Intracellular Zinc Dynamics and Potential Physiological/Pathophysiological Significance. Curr. Med. Chem. 2015, 22, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Wessels, I.; Cousins, R.J. Zinc dyshomeostasis during polymicrobial sepsis in mice involves zinc transporter Zip14 and can be overcome by zinc supplementation. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G768–G778. [Google Scholar] [CrossRef] [PubMed]
- Knoell, D.L.; Julian, M.W.; Bao, S.; Besecker, B.; Macre, J.E.; Leikauf, G.D.; DiSilvestro, R.A.; Crouser, E.D. Zinc deficiency increases organ damage and mortality in a murine model of polymicrobial sepsis. Crit. Care Med. 2009, 37, 1380. [Google Scholar] [CrossRef] [PubMed]
- Wessels, I.; Haase, H.; Engelhardt, G.; Rink, L.; Uciechowski, P. Zinc deficiency induces production of the proinflammatory cytokines IL-1β and TNFα in promyeloid cells via epigenetic and redox-dependent mechanisms. J. Nutr. Biochem. 2013, 24, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Impact of the discovery of human zinc deficiency on health. J. Trace Elem. Med. Biol. 2014, 28, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Brophy, M.B.; Hayden, J.A.; Nolan, E.M. Calcium ion gradients modulate the zinc affinity and antibacterial activity of human calprotectin. J. Am. Chem. Soc. 2012, 134, 18089–18100. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Kuroda, E.; Yamashita, U. Dysfunction of macrophages in metallothionein-knock out mice. J. UOEH 2004, 26, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; R Cannon, B.; Sorci, G.; Riuzzi, F.; Hsu, K.; J Weber, D.; L Geczy, C. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Prasad, A.S.; Beck, F.W.; Fitzgerald, J.T.; Snell, D.; Bao, G.W.; Singh, T.; Cardozo, L.J. Zinc decreases c-reactive protein, lipid peroxidation, and inflammatory cytokines in elderly subjects: A potential implication of zinc as an atheroprotective agent. Am. J. Clin. Nutr. 2010, 91, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Lienau, S.; Engelardt, G.; Rink, L.; Weßels, I. The role of zinc in calprotectin expression in human monocytic cells. Unpublished work. 2017. [Google Scholar]
- Yamasaki, S.; Sakata-Sogawa, K.; Hasegawa, A.; Suzuki, T.; Kabu, K.; Sato, E.; Kurosaki, T.; Yamashita, S.; Tokunaga, M.; Nishida, K.; et al. Zinc is a novel intracellular second messenger. J. Cell Biol. 2007, 177, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Metals on the move: Zinc ions in cellular regulation and in the coordination dynamics of zinc proteins. Biometals 2011, 24, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Hasegawa, A.; Hojyo, S.; Ohashi, W.; Fukada, T.; Nishida, K.; Hirano, T. A novel role of the l-type calcium channel α1D subunit as a gatekeeper for intracellular zinc signaling: Zinc wave. PLoS ONE 2012, 7, e39654. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Morikawa, H.; Kamon, H.; Iguchi, M.; Hojyo, S.; Fukada, T.; Yamashita, S.; Kaisho, T.; Akira, S.; Murakami, M.; et al. Toll-like receptor-mediated regulation of zinc homeostasis influences dendritic cell function. Nat. Immunol. 2006, 7, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Kambe, T. Zinc Signals in Cellular Functions and Disorders; Springer: Tokyo, Japan, 2014. [Google Scholar]
- Von Bülow, V.; Rink, L.; Haase, H. Zinc-mediated inhibition of cyclic nucleotide phosphodiesterase activity and expression suppresses TNF-α and IL-1β production in monocytes by elevation of guanosine 3′,5′-cyclic monophosphate. J. Immunol. 2005, 175, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.; Samarasinghe, R.; Knoch, M.E.; Lewis, M.; Aizenman, E.; DeFranco, D.B. Selective inhibition of mitogen-activated protein kinase phosphatases by zinc accounts for extracellular signal-regulated kinase 1/2-dependent oxidative neuronal cell death. Mol. Pharmacol. 2008, 74, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Barford, D.; Das, A.K.; Egloff, M.P. The structure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 133–164. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Eck, M.J.; Harrison, S.C. A Zn2+ ion links the cytoplasmic tail of CD4 and the N-terminal region of Lck. J. Biol. Chem. 1998, 273, 18729–18733. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.S.; Rodriguez, C.; Veillette, A.; Lodish, H.F. Zinc is essential for binding of p56(lck) to CD4 and CD8α. J. Biol. Chem. 1998, 273, 32878–32882. [Google Scholar] [CrossRef] [PubMed]
- Kaltenberg, J.; Plum, L.M.; Ober-Blobaum, J.L.; Honscheid, A.; Rink, L.; Haase, H. Zinc signals promote IL-2-dependent proliferation of T cells. Eur. J. Immunol. 2010, 40, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Lee, W.W.; Tomar, D.; Pryshchep, S.; Czesnikiewicz-Guzik, M.; Lamar, D.L.; Li, G.; Singh, K.; Tian, L.; Weyand, C.M.; et al. Regulation of T cell receptor signaling by activation-induced zinc influx. J. Exp. Med. 2011, 208, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Romir, J.; Lilie, H.; Egerer-Sieber, C.; Bauer, F.; Sticht, H.; Muller, Y.A. Crystal structure analysis and solution studies of human Lck-SH3; zinc-induced homodimerization competes with the binding of proline-rich motifs. J. Mol. Biol. 2007, 365, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Aude-Garcia, C.; Dalzon, B.; Ravanat, J.-L.; Collin-Faure, V.; Diemer, H.; Strub, J.M.; Cianferani, S.; Van Dorsselaer, A.; Carrière, M.; Rabilloud, T. A combined proteomic and targeted analysis unravels new toxic mechanisms for zinc oxide nanoparticles in macrophages. J. Proteom. 2016, 134, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Bao, B.; Beck, F.W.; Sarkar, F.H. Zinc-suppressed inflammatory cytokines by induction of A20-mediated inhibition of nuclear factor-κB. Nutrition 2011, 27, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Petris, M.J.; Peck, S.C. Separation of zinc-dependent and zinc-independent events during early LPS-stimulated TLR4 signaling in macrophage cells. FEBS Lett. 2014, 588, 2928–2935. [Google Scholar] [CrossRef] [PubMed]
- Wellinghausen, N.; Martin, M.; Rink, L. Zinc inhibits interleukin-1-dependent T cell stimulation. Eur. J. Immunol. 1997, 27, 2529–2535. [Google Scholar] [CrossRef] [PubMed]
- Daaboul, D.; Rosenkranz, E.; Uciechowski, P.; Rink, L. Repletion of zinc in zinc-deficient cells strongly up-regulates IL-1β-induced IL-2 production in T-cells. Metallomics 2012, 4, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Honscheid, A.; Dubben, S.; Rink, L.; Haase, H. Zinc differentially regulates mitogen-activated protein kinases in human T cells. J. Nutr. Biochem. 2012, 23, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Azriel-Tamir, H.; Sharir, H.; Schwartz, B.; Hershfinkel, M. Extracellular zinc triggers ERK-dependent activation of Na+/H+ exchange in colonocytes mediated by the zinc-sensing receptor. J. Biol. Chem. 2004, 279, 51804–51816. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Sunahara, R.K.; Hudson, T.Y.; Heyduk, T.; Howlett, A.C. Zinc inhibition of cAMP signaling. J. Biol. Chem. 2002, 277, 11859–11865. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.M.; Brieger, A.; Engelhardt, G.; Hebel, S.; Nessel, A.; Arlt, M.; Kaltenberg, J.; Schwaneberg, U.; Huber, M.; Rink, L.; et al. PTEN-inhibition by zinc ions augments interleukin-2-mediated Akt phosphorylation. Metallomics 2014, 6, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Gruber, K.; Maywald, M.; Rosenkranz, E.; Haase, H.; Plumakers, B.; Rink, L. Zinc deficiency adversely influences interleukin-4 and interleukin-6 signaling. J. Biol. Regul. Homeost. Agents 2013, 27, 661–671. [Google Scholar] [PubMed]
- Dierichs, L.; Kloubert, V.; Rink, L. Cellular zinc homeostasis modulates polarization of THP-1-derived macrophages. Eur. J. Nutr. 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, E.; Metz, C.H.; Maywald, M.; Hilgers, R.D.; Wessels, I.; Senff, T.; Haase, H.; Jager, M.; Ott, M.; Aspinall, R.; et al. Zinc supplementation induces regulatory T cells by inhibition of Sirt-1 deacetylase in mixed lymphocyte cultures. Mol. Nutr. Food Res. 2016, 60, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Maywald, M.; Meurer, S.K.; Weiskirchen, R.; Rink, L. Zinc supplementation augments TGF-β1-dependent regulatory T cell induction. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, E.; Maywald, M.; Hilgers, R.D.; Brieger, A.; Clarner, T.; Kipp, M.; Plumakers, B.; Meyer, S.; Schwerdtle, T.; Rink, L. Induction of regulatory T cells in Th1-/Th17-driven experimental autoimmune encephalomyelitis by zinc administration. J. Nutr. Biochem. 2016, 29, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Maywald, M.; Rink, L. Zinc supplementation dampens T helper 9 differentiation in allogeneic immune reactions in vitro. Unpublished work. 2017. [Google Scholar]
- Campo, C.A.; Wellinghausen, N.; Faber, C.; Fischer, A.; Rink, L. Zinc inhibits the mixed lymphocyte culture. Biol. Trace Elem. Res. 2001, 79, 15–22. [Google Scholar] [PubMed]
- Kumar, S.; Rajagopalan, S.; Sarkar, P.; Dorward, D.W.; Peterson, M.E.; Liao, H.-S.; Guillermier, C.; Steinhauser, M.L.; Vogel, S.S.; Long, E.O. Zinc-induced polymerization of killer-cell Ig-like receptor into filaments promotes its inhibitory function at cytotoxic immunological synapses. Mol. Cell 2016, 62, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Rolles, B.; Maywald, M.; Rink, L. Influence of zinc deficiency and supplementation on nk cell cytotoxicity. Unpublished work. 2017. [Google Scholar]
- Perry, D.K.; Smyth, M.J.; Stennicke, H.R.; Salvesen, G.S.; Duriez, P.; Poirier, G.G.; Hannun, Y.A. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J. Biol. Chem. 1997, 272, 18530–18533. [Google Scholar] [CrossRef] [PubMed]
- Kown, M.H.; van der Steenhoven, T.J.; Jahncke, C.L.; Mari, C.; Lijkwan, M.A.; Koransky, M.L.; Blankenberg, F.G.; Strauss, H.W.; Robbins, R.C. Zinc chloride-mediated reduction of apoptosis as an adjunct immunosuppressive modality in cardiac transplantation. J. Heart Lung Transpl. 2002, 21, 360–365. [Google Scholar] [CrossRef]
- Morgan, C.I.; Ledford, J.R.; Zhou, P.; Page, K. Zinc supplementation alters airway inflammation and airway hyperresponsiveness to a common allergen. J. Inflamm. 2011, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Wellinghausen, N.; Schromm, A.B.; Seydel, U.; Brandenburg, K.; Luhm, J.; Kirchner, H.; Rink, L. Zinc enhances lipopolysaccharide-induced monokine secretion by alteration of fluidity state of lipopolysaccharide. J. Immunol. 1996, 157, 3139–3145. [Google Scholar] [PubMed]
- Chuapil, M. Effect of zinc on cells and biomembranes. Med. Clin. N. Am. 1976, 60, 799–812. [Google Scholar] [CrossRef]
- Hansen, K.B.; Furukawa, H.; Traynelis, S.F. Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol. Pharmacol. 2010, 78, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Tartey, S.; Takeuchi, O. Pathogen recognition and Toll-like receptor targeted therapeutics in innate immune cells. Int. Rev. Immunol. 2017, 36, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Futosi, K.; Fodor, S.; Mócsai, A. Reprint of Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Aster, J.C.; Knutsen, C.A.; Kruckeberg, W.C. Zinc inhibition of calmodulin: A proposed molecular mechanism of zinc action on cellular functions. Am. J. Hematol. 1979, 7, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Wellinghausen, N.; Rink, L. The significance of zinc for leukocyte biology. J. Leukoc. Biol. 1998, 64, 571–577. [Google Scholar] [PubMed]
- Mustelin, T.; Vang, T.; Bottini, N. Protein tyrosine phosphatases and the immune response. Nat. Rev. Immunol. 2005, 5, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Medgyesi, D.; Hobeika, E.; Biesen, R.; Kollert, F.; Taddeo, A.; Voll, R.E.; Hiepe, F.; Reth, M. The protein tyrosine phosphatase PTP1B is a negative regulator of CD40 and BAFF-R signaling and controls B cell autoimmunity. J. Exp. Med. 2014, 211, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. Protein tyrosine phosphatases as targets of the combined insulinomimetic effects of zinc and oxidants. Biometals 2005, 18, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Jacob, C.; Vallee, B.L.; Fischer, E.H. Inhibitory sites in enzymes: Zinc removal and reactivation by thionein. Proc. Natl. Acad. Sci. USA 1999, 96, 1936–1940. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. Fluctuations of cellular, available zinc modulate insulin signaling via inhibition of protein tyrosine phosphatases. J. Trace Elem. Med. Biol. 2005, 19, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Sly, L.M.; Rauh, M.J.; Kalesnikoff, J.; Büchse, T.; Krystal, G. SHIP, SHIP2, and PTEN activities are regulated in vivo by modulation of their protein levels: SHIP is up-regulated in macrophages and mast cells by lipopolysaccharide. Exp. Hematol. 2003, 31, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Ober-Blobaum, J.L.; Engelhardt, G.; Hebel, S.; Heit, A.; Heine, H.; Rink, L. Zinc signals are essential for lipopolysaccharide-induced signal transduction in monocytes. J. Immunol. 2008, 181, 6491–6502. [Google Scholar] [CrossRef] [PubMed]
- Hennigar, S.R.; Seo, Y.A.; Sharma, S.; Soybel, D.I.; Kelleher, S.L. ZnT2 is a critical mediator of lysosomal-mediated cell death during early mammary gland involution. Sci. Rep. 2015, 5, 8033. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, T.D. Lysosomal metal, redox and proton cycles influencing the CysHis cathepsin reaction. Metallomics 2013, 5, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Vinkenborg, J.L.; Nicolson, T.J.; Bellomo, E.A.; Koay, M.S.; Rutter, G.A.; Merkx, M. Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat. Methods 2009, 6, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Vener, A.V.; Aksenova, M.V.; Burbaeva, G.S. Drastic reduction of the zinc-and magnesium-stimulated protein tyrosine kinase activities in Alzheimer’s disease hippocampus. FEBS Lett. 1993, 328, 6–8. [Google Scholar] [CrossRef]
- Bennasroune, A.; Mazot, P.; Boutterin, M.-C.; Vigny, M. Activation of the orphan receptor tyrosine kinase ALK by zinc. Biochem. Biophys. Res. Commun. 2010, 398, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, E.; Carugo, K.D.; Hyvönen, M.; Surdo, P.L.; Riley, A.M.; Potter, B.V.; O’Brien, R.; Ladbury, J.E.; Saraste, M. Structure of the PH domain from Bruton’s tyrosine kinase in complex with inositol 1,3,4,5-tetrakisphosphate. Structure 1999, 7, 449–460. [Google Scholar] [CrossRef]
- Arbibe, L.; Jean-Paul, M.; Teusch, N.; Kline, L.; Guha, M.; Mackman, N.; Godowski, P.J.; Ulevitch, R.J.; Knaus, U.G. Toll-like receptor 2-mediated NF-κB activation requires a Rac1-dependent pathway. Nat. Immunol. 2000, 1, 533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xing, F.; Zheng, H.; Xi, J.; Cui, X.; Xu, Z. Roles of mitochondrial Src tyrosine kinase and zinc in nitric oxide-induced cardioprotection against ischemia/reperfusion injury. Free Radic. Res. 2013, 47, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-J.; Bao, S.; Napolitano, J.R.; Burris, D.L.; Yu, L.; Tridandapani, S.; Knoell, D.L. Zinc regulates the acute phase response and serum amyloid A production in response to sepsis through JAK-STAT3 signaling. PLoS ONE 2014, 9, e94934. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yang, Z.; Wang, J.; Yu, J.; Guo, J.; Liu, S.; Qian, C.; Song, L.; Wu, Y.; Cheng, J. Zinc Chloride Transiently Maintains Mouse Embryonic Stem Cell Pluripotency by Activating Stat3 Signaling. PLoS ONE 2016, 11, e0148994. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, E.; Massarotti, A.; Hogstrand, C.; Maret, W. Zinc ions modulate protein tyrosine phosphatase 1B activity. Metallomics 2014, 6, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.; Hogstrand, C.; Maret, W. Picomolar concentrations of free zinc (II) ions regulate receptor protein-tyrosine phosphatase β activity. J. Biol. Chem. 2012, 287, 9322–9326. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Exp. Cell Res. 2003, 291, 289–298. [Google Scholar] [CrossRef]
- Takahashi, K.; Akaishi, E.; Abe, Y.; Ishikawa, R.; Tanaka, S.; Hosaka, K.; Kubohara, Y. Zinc inhibits calcineurin activity in vitro by competing with nickel. Biochem. Biophys. Res. Commun. 2003, 307, 64–68. [Google Scholar] [CrossRef]
- Lohmann, R.D.; Beyersmann, D. Cadmium and zinc mediated changes of the Ca2+-dependent endonuclease in apoptosis. Biochem. Biophys. Res. Commun. 1993, 190, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Percival, M.D.; Yeh, B.; Falgueyret, J.-P. Zinc dependent activation of cAMP-specific phosphodiesterase (PDE4A). Biochem. Biophys. Res. Commun. 1997, 241, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Colbran, J.L.; McAllister-Lucas, L.M.; Corbin, J.D. Zinc interactions and conserved motifs of the cGMP-binding cGMP-specific phosphodiesterase suggest that it is a zinc hydrolase. J. Biol. Chem. 1994, 269, 22477–22480. [Google Scholar] [PubMed]
- He, F.; Seryshev, A.B.; Cowan, C.W.; Wensel, T.G. Multiple zinc binding sites in retinal rod cGMP phosphodiesterase, PDE6αβ. J. Biol. Chem. 2000, 275, 20572–20577. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.I.; Jeong, J.Y.; Jue, D.M. Thiol-reactive metal compounds inhibit NF-κB activation by blocking I κB kinase. J. Immunol. 2000, 164, 5981–5989. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Hebel, S.; Engelhardt, G.; Rink, L. Flow cytometric measurement of labile zinc in peripheral blood mononuclear cells. Anal. Biochem. 2006, 352, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Krężel, A.; Hao, Q.; Maret, W. The zinc/thiolate redox biochemistry of metallothionein and the control of zinc ion fluctuations in cell signaling. Arch. Biochem. Biophys. 2007, 463, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Korichneva, I. Redox regulation of cardiac protein kinase C. Exp. Clin. Cardiol. 2005, 10, 256–261. [Google Scholar] [PubMed]
- Aydemir, T.B.; Liuzzi, J.P.; McClellan, S.; Cousins, R.J. Zinc transporter ZIP8 (SLC39A8) and zinc influence IFN-γ expression in activated human T cells. J. Leukoc. Biol. 2009, 86, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Slepchenko, K.G.; Lu, Q.; Li, Y.V. Zinc wave during the treatment of hypoxia is required for initial reactive oxygen species activation in mitochondria. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 44. [Google Scholar] [PubMed]
- Zalewski, P.; Forbes, I.; Giannakis, C.; Cowled, P.; Betts, W. Synergy between zinc and phorbol ester in translocation of protein kinase C to cytoskeleton. FEBS Lett. 1990, 273, 131–134. [Google Scholar] [CrossRef]
- Beyersmann, D.; Haase, H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals 2001, 14, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, M.; Leanderson, P.; Tagesson, C. Novel aspect on metal fume fever: Zinc stimulates oxygen radical formation in human neutrophils. Hum. Exp. Toxicol. 1998, 17, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.R. The antioxidant properties of zinc. J. Nutr. 2000, 130 (Suppl. 5S), 1447S–1454S. [Google Scholar] [PubMed]
- Freitas, M.; Porto, G.; Lima, J.L.; Fernandes, E. Zinc activates neutrophils’ oxidative burst. Biometals 2010, 23, 31. [Google Scholar] [CrossRef] [PubMed]
- Londesborough, J.; Suoranta, K. Zinc-containing cyclic nucleotide phosphodiesterases from bakers’ yeast. Methods Enzymol. 1988, 159, 777–785. [Google Scholar] [PubMed]
- Von Bülow, V.; Dubben, S.; Engelhardt, G.; Hebel, S.; Plümäkers, B.; Heine, H.; Rink, L.; Haase, H. Zinc-dependent suppression of TNF-α production is mediated by protein kinase A-induced inhibition of Raf-1, IκB Kinase β, and NF-κB. J. Immunol. 2007, 179, 4180–4186. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Heyduk, T.; Sunahara, R.K. Zinc inhibition of adenylyl cyclase correlates with conformational changes in the enzyme. Cell Signal. 2004, 16, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Du, Z.; Patel, T.B. Copper and Zinc Inhibit Gαs Function A NUCLEOTIDE-FREE STATE OF Gαs INDUCED BY Cu2+ AND Zn2+. J. Biol. Chem. 2005, 280, 2579–2586. [Google Scholar] [CrossRef] [PubMed]
- Uzzo, R.G.; Crispen, P.L.; Golovine, K.; Makhov, P.; Horwitz, E.M.; Kolenko, V.M. Diverse effects of zinc on NF-κB and AP-1 transcription factors: Implications for prostate cancer progression. Carcinogenesis 2006, 27, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-J.; Bao, S.; Gálvez-Peralta, M.; Pyle, C.J.; Rudawsky, A.C.; Pavlovicz, R.E.; Killilea, D.W.; Li, C.; Nebert, D.W.; Wewers, M.D. ZIP8 regulates host defense through zinc-mediated inhibition of NF-κB. Cell Rep. 2013, 3, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Bao, B.; Beck, F.W.; Kucuk, O.; Sarkar, F.H. Antioxidant effect of zinc in humans. Free Radic. Biol. Med. 2004, 37, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-W.; Fan, J.; Bai, S.-L.; Hou, W.-J.; Li, X.; Tong, H. Zinc prevents abdominal aortic aneurysm formation by induction of A20-mediated suppression of NF-κB pathway. PLoS ONE 2016, 11, e0148536. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Guo, S.; Gao, J.; Guo, Y.; Du, E.; Lv, Z.; Zhang, B. Maternal high-zinc diet attenuates intestinal inflammation by reducing DNA methylation and elevating h3k9 acetylation in the a20 promoter of offspring chicks. J. Nutri. Biochem. 2015, 26, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Staitieh, B.S.; Fan, X.; Neveu, W.; Guidot, D.M. Nrf2 regulates PU. 1 expression and activity in the alveolar macrophage. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1086–L1093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Laouar, A.; Denzin, L.K.; Sant’Angelo, D.B. Zbtb16 (PLZF) is stably suppressed and not inducible in non-innate T cells via T cell receptor-mediated signaling. Sci. Rep. 2015, 5, 12113. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D. The zinc finger-containing transcription factors GATA-4,-5, and-6 ubiquitously expressed regulators of tissue-specific gene expression. J. Biol. Chem. 2000, 275, 38949–38952. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.M.; Nandan, M.O.; Chanchevalap, S.; Dalton, W.B.; Hisamuddin, I.M. Krüppel-like factors 4 and 5: The yin and yang regulators of cellular proliferation. Cell Res. 2005, 15, 92. [Google Scholar] [CrossRef] [PubMed]
- Laity, J.H.; Andrews, G.K. Understanding the mechanisms of zinc-sensing by metal-response element binding transcription factor-1 (MTF-1). Arch. Biochem. Biophys. 2007, 463, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.L.; Johnson, P. Tyrosine phosphorylation in immune cells: Direct and indirect effects on toll-like receptor-induced proinflammatory cytokine production. Crit. Rev. Immunol. 2009, 29, 347–367. [Google Scholar]
- Cho, J.; Tsichlis, P.N. Phosphorylation at Thr-290 regulates Tpl2 binding to NF-κB1/p105 and Tpl2 activation and degradation by lipopolysaccharide. Proc. Natl. Acad. Sci. USA 2005, 102, 2350–2355. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair 2010, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Read, S.A.; O’Connor, K.S.; Suppiah, V.; Ahlenstiel, C.L.; Obeid, S.; Cook, K.M.; Cunningham, A.; Douglas, M.W.; Hogg, P.J.; Booth, D. Zinc is a potent and specific inhibitor of IFN-γ3 signalling. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Denk, A.; Wirth, T.; Baumann, B. NF-κB transcription factors: Critical regulators of hematopoiesis and neuronal survival. Cytokine Growth Factor Rev. 2000, 11, 303–320. [Google Scholar] [CrossRef]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.-I.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346. [Google Scholar] [CrossRef] [PubMed]
- Boone, D.L.; Turer, E.E.; Lee, E.G.; Regina-Celeste, A.; Wheeler, M.T.; Tsui, C.; Hurley, P.; Chien, M.; Chai, S.; Hitotsumatsu, O. The ubiquitin-modifying enzyme A20 is required for termination of toll-like receptor responses. Nat. Immunol. 2004, 5, 1052. [Google Scholar] [CrossRef] [PubMed]
- Tillmann, N.; Engelhardt, G.; Rink, L.; Weßels, I. Zinc in granulopoiesis. Unpublished work. 2017. [Google Scholar]
- Bellomo, E.; Birla Singh, K.; Massarotti, A.; Hogstrand, C.; Maret, W. The metal face of protein tyrosine phosphatase 1B. Coord. Chem. Rev. 2016, 327–328, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Supasai, S.; Aimo, L.; Adamo, A.; Mackenzie, G.; Oteiza, P. Zinc deficiency affects the STAT1/3 signaling pathways in part through redox-mediated mechanisms. Redox Biol. 2017, 11, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Aster, I.; Engelhardt, G.; Rink, L.; Weßels, I. The influence of zinc on granulocyte-macrophage colony stimulating factor-induced signaling in U937 cells. Unpublished work. 2017. [Google Scholar]
- Rajagopalan, S.; Long, E.O. Zinc bound to the killer cell-inhibitory receptor modulates the negative signal in human NK cells. J. Immunol. 1998, 161, 1299–1305. [Google Scholar] [PubMed]
- Rajagopalan, S.; Winter, C.C.; Wagtmann, N.; Long, E.O. The Ig-related killer cell inhibitory receptor binds zinc and requires zinc for recognition of HLA-C on target cells. J. Immunol. 1995, 155, 4143–4146. [Google Scholar] [PubMed]
- Ho, L.H.; Ruffin, R.E.; Murgia, C.; Li, L.; Krilis, S.A.; Zalewski, P.D. Labile zinc and zinc transporter ZnT4 in mast cell granules: Role in regulation of caspase activation and NF-κB translocation. J. Immunol. 2004, 172, 7750–7760. [Google Scholar] [CrossRef] [PubMed]
- Kabu, K.; Yamasaki, S.; Kamimura, D.; Ito, Y.; Hasegawa, A.; Sato, E.; Kitamura, H.; Nishida, K.; Hirano, T. Zinc is required for Fc epsilon RI-mediated mast cell activation. J. Immunol. 2006, 177, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Ollig, J.; Kloubert, V.; Weßels, I.; Haase, H.; Rink, L. Parameters Influencing Zinc in Experimental Systems in Vivo and in Vitro. Metals 2016, 6, 71. [Google Scholar] [CrossRef]
- Osati-Ashtiani, F.; King, L.E.; Fraker, P.J. Variance in the resistance of murine early bone marrow B cells to a deficiency in zinc. Immunology 1998, 94, 94–100. [Google Scholar] [CrossRef] [PubMed]
- King, L.E.; Frentzel, J.W.; Mann, J.J.; Fraker, P.J. Chronic zinc deficiency in mice disrupted T cell lymphopoiesis and erythropoiesis while B cell lymphopoiesis and myelopoiesis were maintained. J. Am. Coll. Nutr. 2005, 24, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, M.; Savino, W.; Wade, S.; Kaiserlian, D.; Lemonnier, D.; Bach, J.F. In vivo and in vitro studies of thymulin in marginally zinc-deficient mice. Eur. J. Immunol. 1984, 14, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.R.; Hadden, E.M.; Hadden, J.W. Zinc induces thymulin secretion from human thymic epithelial cells in vitro and augments splenocyte and thymocyte responses in vivo. Int. J. Immunopharmacol. 1995, 17, 729–733. [Google Scholar] [CrossRef]
- Beck, F.W.; Kaplan, J.; Fine, N.; Handschu, W.; Prasad, A.S. Decreased expression of CD73 (ecto-5′-nucleotidase) in the CD8+ subset is associated with zinc deficiency in human patients. J. Lab. Clin. Med. 1997, 130, 147–156. [Google Scholar] [CrossRef]
- Fraker, P.J. Roles for cell death in zinc deficiency. J. Nutr. 2005, 135, 359–362. [Google Scholar] [PubMed]
- Chai, F.; Truong-Tran, A.Q.; Evdokiou, A.; Young, G.P.; Zalewski, P.D. Intracellular zinc depletion induces caspase activation and p21 Waf1/Cip1 cleavage in human epithelial cell lines. J. Infect. Dis. 2000, 182 (Suppl. S1), S85–S92. [Google Scholar] [CrossRef] [PubMed]
- King, K.L.; Cidlowski, J.A. Cell cycle regulation and apoptosis. Annu. Rev. Physiol. 1998, 60, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Meftah, S.; Abdallah, J.; Kaplan, J.; Brewer, G.J.; Bach, J.F.; Dardenne, M. Serum thymulin in human zinc deficiency. J. Clin. Investig. 1988, 82, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Coto, J.A.; Hadden, E.M.; Sauro, M.; Zorn, N.; Hadden, J.W. Interleukin 1 regulates secretion of zinc-thymulin by human thymic epithelial cells and its action on T-lymphocyte proliferation and nuclear protein kinase C. Proc. Natl. Acad. Sci. USA 1992, 89, 7752–7756. [Google Scholar] [CrossRef] [PubMed]
- Dowd, P.S.; Kelleher, J.; Guillou, P.J. T-lymphocyte subsets and interleukin-2 production in zinc-deficient rats. Br. J. Nutr. 1986, 55, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Effects of zinc deficiency on Th1 and Th2 cytokine shifts. J. Infect. Dis. 2000, 182 (Suppl. S1), S62–S68. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Bonneau, R.; Girard, M.A.; Beaulieu, C.; Larivee, P. Zinc status modulates bronchopulmonary eosinophil infiltration in a murine model of allergic inflammation. Chest 2003, 123 (Suppl. S3), 446S. [Google Scholar] [CrossRef]
- Honscheid, A.; Rink, L.; Haase, H. T-lymphocytes: A target for stimulatory and inhibitory effects of zinc ions. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Socha, K.; Karpinska, E.; Kochanowicz, J.; Soroczynska, J.; Jakoniuk, M.; Wilkiel, M.; Mariak, Z.D.; Borawska, M.H. Dietary habits; concentration of copper, zinc, and Cu-to-Zn ratio in serum and ability status of patients with relapsing-remitting multiple sclerosis. Nutrition 2017, 39–40, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.; Frischer, J.M.; Webb, S.M.; Tham, M.; Adiele, R.C.; Robinson, C.A.; Fitz-Gibbon, P.D.; Weigand, S.D.; Metz, I.; Nehzati, S.; et al. Pathogenic implications of distinct patterns of iron and zinc in chronic MS lesions. Acta Neuropathol. 2017, 134, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Kuroki, T.; Adachi, T.; Ono, S.; Hayashi, T.; Tajima, Y.; Eguchi, S.; Kanematsu, T. Effect of zinc on early graft failure following intraportal islet transplantation in rat recipients. Ann. Transpl. 2011, 16, 114–120. [Google Scholar] [CrossRef]
- Chimienti, F.; Seve, M.; Richard, S.; Mathieu, J.; Favier, A. Role of cellular zinc in programmed cell death: Temporal relationship between zinc depletion, activation of caspases, and cleavage of Sp family transcription factors. Biochem. Pharmacol. 2001, 62, 51–62. [Google Scholar] [CrossRef]
- Hanidziar, D.; Koulmanda, M. Inflammation and the balance of Treg and Th17 cells in transplant rejection and tolerance. Curr. Opin. Organ Transpl. 2010, 15, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Graca, L.; Thompson, S.; Lin, C.Y.; Adams, E.; Cobbold, S.P.; Waldmann, H. Both CD4+CD25+ and CD4+CD25− regulatory cells mediate dominant transplantation tolerance. J. Immunol. 2002, 168, 5558–5565. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Huter, E.N.; Stummvoll, G.H.; DiPaolo, R.J.; Glass, D.D.; Shevach, E.M. Cutting edge: Antigen-specific TGF β-induced regulatory T cells suppress Th17-mediated autoimmune disease. J. Immunol. 2008, 181, 8209–8213. [Google Scholar] [CrossRef] [PubMed]
- Davidson, T.S.; DiPaolo, R.J.; Andersson, J.; Shevach, E.M. Cutting Edge: IL-2 is essential for TGF-β-mediated induction of Foxp3+ T regulatory cells. J. Immunol. 2007, 178, 4022–4026. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C.; von Boehmer, H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Kasagi, S.; Zhang, P.; Che, L.; Abbatiello, B.; Maruyama, T.; Nakatsukasa, H.; Zanvit, P.; Jin, W.; Konkel, J.E.; Chen, W. In vivo-generated antigen-specific regulatory T cells treat autoimmunity without compromising antibacterial immune response. Sci. Transl. Med. 2014, 6, 241ra78. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, D.A.; Zheng, S.G.; Wang, J.; Gray, J.D. Critical role of IL-2 and TGF-β in generation, function and stabilization of Foxp3+CD4+ Treg. Eur. J. Immunol. 2008, 38, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Takaki, H.; Ichiyama, K.; Koga, K.; Chinen, T.; Takaesu, G.; Sugiyama, Y.; Kato, S.; Yoshimura, A.; Kobayashi, T. STAT6 Inhibits TGFβ1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J. Biol. Chem. 2008, 283, 14955–14962. [Google Scholar] [CrossRef] [PubMed]
- Tone, Y.; Furuuchi, K.; Kojima, Y.; Tykocinski, M.L.; Greene, M.I.; Tone, M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 2008, 9, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Hogstrand, C.; Kille, P.; Nicholson, R.I.; Taylor, K.M. Zinc transporters and cancer: A potential role for ZIP7 as a hub for tyrosine kinase activation. Trends Mol. Med. 2009, 15, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Raaf, J.; Issinger, O.G. Protein kinase CK2 in health and disease: Protein kinase CK2: From structures to insights. Cell. Mol. Life Sci. 2009, 66, 1800–1816. [Google Scholar] [CrossRef] [PubMed]
- St-Denis, N.A.; Litchfield, D.W. Protein kinase CK2 in health and disease: From birth to death: The role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell. Mol. Life Sci. 2009, 66, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Ulges, A.; Klein, M.; Reuter, S.; Gerlitzki, B.; Hoffmann, M.; Grebe, N.; Staudt, V.; Stergiou, N.; Bohn, T.; Bruhl, T.J.; et al. Protein kinase CK2 enables regulatory T cells to suppress excessive Th2 responses in vivo. Nat. Immunol. 2015, 16, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Fragale, A.; Gabriele, L.; Stellacci, E.; Borghi, P.; Perrotti, E.; Ilari, R.; Lanciotti, A.; Remoli, A.L.; Venditti, M.; Belardelli, F.; et al. IFN regulatory factor-1 negatively regulates CD4+ CD25+ regulatory T cell differentiation by repressing Foxp3 expression. J. Immunol. 2008, 181, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Maywald, M.; Rink, L. Zinc supplementation induces CD4+CD25+Foxp3+ antigen-specific regulatory T cells and suppresses IFN-γ production by upregulation of Foxp3 and KLF-10 and downregulation of IRF-1. Eur. J. Nutr. 2017, 56, 1859–1869. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Wara, A.K.; Icli, B.; Sun, X.; Packard, R.R.; Esen, F.; Stapleton, C.J.; Subramaniam, M.; Kretschmer, K.; Apostolou, I.; et al. Kruppel-like factor KLF10 targets transforming growth factor-β1 to regulate CD4+CD25− T cells and T regulatory cells. J. Biol. Chem. 2009, 284, 24914–24924. [Google Scholar] [CrossRef] [PubMed]
- Schutze, N.; Trojandt, S.; Kuhn, S.; Tomm, J.M.; von Bergen, M.; Simon, J.C.; Polte, T. Allergen-Induced IL-6 Regulates IL-9/IL-17A Balance in CD4+ T Cells in Allergic Airway Inflammation. J. Immunol. 2016, 197, 2653–2664. [Google Scholar] [CrossRef] [PubMed]
- Elyaman, W.; Khoury, S.J. Th9 cells in the pathogenesis of EAE and multiple sclerosis. Semin. Immunopathol. 2017, 39, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, C.; Fukada, T.; Kanamoto, M.; Ohashi, W.; Hojyo, S.; Atsumi, T.; Ueda, N.; Azuma, I.; Hirota, H.; Murakami, M.; et al. Zinc suppresses Th17 development via inhibition of STAT3 activation. Int. Immunol. 2010, 22, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.J.; Hodes, R.J. T-cell development is regulated by the coordinated function of proximal and distal Lck promoters active at different developmental stages. Eur. J. Immunol. 2016, 46, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.W.; Sun, Z.Y.; Blacklow, S.C.; Wagner, G.; Eck, M.J. A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science 2003, 301, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- Mustelin, T.; Tasken, K. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem. J. 2003, 371, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Furlan, G.; Minowa, T.; Hanagata, N.; Kataoka-Hamai, C.; Kaizuka, Y. Phosphatase CD45 both positively and negatively regulates T cell receptor phosphorylation in reconstituted membrane protein clusters. J. Biol. Chem. 2014, 289, 28514–28525. [Google Scholar] [CrossRef] [PubMed]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, D.; Xing, W.; Ma, X.; Yin, Y.; Wei, Q.; Li, G. An approach to assay calcineurin activity and the inhibitory effect of zinc ion. Anal. Biochem. 2008, 375, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Yang, M.; Guo, B.; Cao, J.; Yang, L.; Guo, X.; Li, Y.; Gao, Z. Zinc inhibits H2O2-induced MC3T3-E1 cells apoptosis via MAPK and PI3K/AKT pathways. Biol. Trace Elem. Res. 2012, 148, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Kim, M.Y.; Mo, J.S.; Ann, E.J.; Lee, K.S.; Park, J.H.; Kim, J.Y.; Seo, M.S.; Choi, E.J.; Park, H.S. Zinc-induced downregulation of Notch signaling is associated with cytoplasmic retention of Notch1-IC and RBP-Jk via PI3k-Akt signaling pathway. Cancer Lett. 2007, 255, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Eom, S.J.; Kim, E.Y.; Lee, J.E.; Kang, H.J.; Shim, J.; Kim, S.U.; Gwag, B.J.; Choi, E.J. Zn2+ induces stimulation of the c-Jun N-terminal kinase signaling pathway through phosphoinositide 3-Kinase. Mol. Pharmacol. 2001, 59, 981–986. [Google Scholar] [PubMed]
- Tang, X.; Shay, N.F. Zinc has an insulin-like effect on glucose transport mediated by phosphoinositol-3-kinase and Akt in 3T3-L1 fibroblasts and adipocytes. J. Nutr. 2001, 131, 1414–1420. [Google Scholar] [PubMed]
- Wu, W.; Wang, X.; Zhang, W.; Reed, W.; Samet, J.M.; Whang, Y.E.; Ghio, A.J. Zinc-induced PTEN protein degradation through the proteasome pathway in human airway epithelial cells. J. Biol. Chem. 2003, 278, 28258–28263. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.D.; Wang, B.; Pan, W.; Xu, H.; Jiang, X.; Liao, F.F. Functional interaction of phosphatase and tensin homologue (PTEN) with the E3 ligase NEDD4-1 during neuronal response to zinc. J. Biol. Chem. 2010, 285, 9847–9857. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Szamel, M.; Resch, K.; Somogyi, J. Zinc can increase the activity of protein kinase C and contributes to its binding to plasma membranes in T lymphocytes. J. Biol. Chem. 1988, 263, 6487–6490. [Google Scholar] [PubMed]
- Tan, S.L.; Parker, P.J. Emerging and diverse roles of protein kinase C in immune cell signalling. Biochem. J. 2003, 376, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Quest, A.F.; Bloomenthal, J.; Bardes, E.S.; Bell, R.M. The regulatory domain of protein kinase C coordinates four atoms of zinc. J. Biol. Chem. 1992, 267, 10193–10197. [Google Scholar] [PubMed]
- Hubbard, S.R.; Bishop, W.R.; Kirschmeier, P.; George, S.J.; Cramer, S.P.; Hendrickson, W.A. Identification and characterization of zinc binding sites in protein kinase C. Science 1991, 254, 1776–1779. [Google Scholar] [CrossRef] [PubMed]
- Forbes, I.J.; Zalewski, P.D.; Giannakis, C.; Petkoff, H.S.; Cowled, P.A. Interaction between protein kinase C and regulatory ligand is enhanced by a chelatable pool of cellular zinc. Biochim. Biophys. Acta 1990, 1053, 113–117. [Google Scholar] [CrossRef]
- Knapp, L.T.; Klann, E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J. Biol. Chem. 2000, 275, 24136–24145. [Google Scholar] [CrossRef] [PubMed]
- Korichneva, I.; Hoyos, B.; Chua, R.; Levi, E.; Hammerling, U. Zinc release from protein kinase C as the common event during activation by lipid second messenger or reactive oxygen. J. Biol. Chem. 2002, 277, 44327–44331. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Gueth, S.; Somogyi, J. The tumor promoter tetradecanoyl-phorbol-acetate (TPA) elicits the redistribution of zinc in subcellular fractions of rabbit thymocytes measured by X-ray fluorescence. Biochem. Biophys. Res. Commun. 1987, 144, 863–868. [Google Scholar] [CrossRef]
- Chow, L.M.; Fournel, M.; Davidson, D.; Veillette, A. Negative regulation of T-cell receptor signalling by tyrosine protein kinase p50csk. Nature 1993, 365, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Vang, T.; Torgersen, K.M.; Sundvold, V.; Saxena, M.; Levy, F.O.; Skalhegg, B.S.; Hansson, V.; Mustelin, T.; Tasken, K. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J. Exp. Med. 2001, 193, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK signalling in cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Suzuki, S.; Millar, D.G.; Unno, M.; Hara, H.; Calzascia, T.; Yamasaki, S.; Yokosuka, T.; Chen, N.J.; Elford, A.R.; et al. A critical role for the innate immune signaling molecule IRAK-4 in T cell activation. Science 2006, 311, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, T.; Sato, S.; Jung, A.; Yamamoto, M.; Matsui, K.; Kato, H.; Uematsu, S.; Takeuchi, O.; Akira, S. Essential role of IRAK-4 protein and its kinase activity in Toll-like receptor-mediated immune responses but not in TCR signaling. J. Exp. Med. 2007, 204, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Mocchegiani, E.; Rink, L. Correlation between zinc status and immune function in the elderly. Biogerontology 2006, 7, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.; Gabriel, P.; Ibs, K.H.; Rink, L. Zinc in pharmacological doses suppresses allogeneic reaction without affecting the antigenic response. Bone Marrow Transpl. 2004, 33, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, E.; Hilgers, R.D.; Uciechowski, P.; Petersen, A.; Plumakers, B.; Rink, L. Zinc enhances the number of regulatory T cells in allergen-stimulated cells from atopic subjects. Eur. J. Nutr. 2017, 56, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.J.; Telford, W.G. A reappraisal of the role of zinc in life and death decisions of cells. Proc. Soc. Exp. Biol. Med. 1997, 215, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.J.; King, L.E. Reprogramming of the immune system during zinc deficiency. Annu. Rev. Nutr. 2004, 24, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.J.; King, L.E.; Laakko, T.; Vollmer, T.L. The dynamic link between the integrity of the immune system and zinc status. J. Nutr. 2000, 130 (Suppl. 5S), 1399S–1406S. [Google Scholar] [PubMed]
- DePasquale-Jardieu, P.; Fraker, P.J. Interference in the development of a secondary immune response in mice by zinc deprivation: Persistence of effects. J. Nutr. 1984, 114, 1762–1769. [Google Scholar] [PubMed]
- Cakman, I.; Rohwer, J.; Schutz, R.M.; Kirchner, H.; Rink, L. Dysregulation between Th1 and Th2 T cell subpopulations in the elderly. Mech. Ageing Dev. 1996, 87, 197–209. [Google Scholar] [CrossRef]
- Maywald, M.; Rink, L. Zinc homeostasis and immunosenescence. J. Trace Elem. Med. Biol. 2015, 29, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, M.; Di Paolo, B.; De Risio, F.; Niri, L.; Klinkmann, H.; Ivanovich, P.; Albertazzi, A. Effects of zinc supplementation in chronic haemodialysis patients. Nephrol. Dial. Transpl. 1993, 8, 1166–1168. [Google Scholar]
- Wessels, I. Epigenetics and Metal Deficiencies. Curr. Nutr. Rep. 2014, 3, 196–203. [Google Scholar] [CrossRef]
- Kopf, M.; Le Gros, G.; Bachmann, M.; Lamers, M.C.; Bluethmann, H.; Kohler, G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature 1993, 362, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Moulder, K.; Steward, M.W. Experimental zinc deficiency: Effects on cellular responses and the affinity of humoral antibody. Clin. Exp. Immunol. 1989, 77, 269–274. [Google Scholar] [PubMed]
- Fraker, P.J.; Gershwin, M.E.; Good, R.A.; Prasad, A. Interrelationships between zinc and immune function. Fed. Proc. 1986, 45, 1474–1479. [Google Scholar] [PubMed]
- Stennicke, H.R.; Salvesen, G.S. Biochemical characteristics of caspases-3, -6, -7, and -8. J. Biol. Chem. 1997, 272, 25719–25723. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.M.; Fraker, P.J. Functional capacity of the residual lymphocytes from zinc-deficient adult mice. Br. J. Nutr. 1993, 69, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Miyai, T.; Hojyo, S.; Ikawa, T.; Kawamura, M.; Irie, T.; Ogura, H.; Hijikata, A.; Bin, B.H.; Yasuda, T.; Kitamura, H.; et al. Zinc transporter SLC39A10/ZIP10 facilitates antiapoptotic signaling during early B-cell development. Proc. Natl. Acad. Sci. USA 2014, 111, 11780–11785. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Miyai, T.; Fujishiro, H.; Kawamura, M.; Yasuda, T.; Hijikata, A.; Bin, B.H.; Irie, T.; Tanaka, J.; Atsumi, T.; et al. Zinc transporter SLC39A10/ZIP10 controls humoral immunity by modulating B-cell receptor signal strength. Proc. Natl. Acad. Sci. USA 2014, 111, 11786–11791. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Zinc Signal | Duration | Effect |
|---|---|---|
| Zinc Flux | Seconds/minutes |
|
| Zinc wave | Minutes |
|
| homeostatic Zinc Signal | Hours |
|
| Signaling Molecule | IC50 | Reference |
|---|---|---|
| PTPRB | 98 pM | [114] |
| PTEN | 0.6 nM | [76] |
| PTP1B | 3–17 nM | [100] |
| SHP-1 | 92 nM | [115] |
| SHP-2 | 1–2 µM | [100] |
| TC PTP | 200 mM | [99] |
| Calcineurin | 250 nM–7 µM | [116] |
| Ca-dependent endonuclease | 1 µM (in the presence of 10 µM Ca) | [117] |
| LAR | 20 µM | [100] |
| PDE4A | >5 µM | [118] |
| PDE 5 | >1 µM | [119] |
| PDE 6 | >1 µM | [120] |
| IKK | 8.7 µM | [121] |
| Caspase 3 | <10 nM, 100 nM | [86,99] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maywald, M.; Wessels, I.; Rink, L. Zinc Signals and Immunity. Int. J. Mol. Sci. 2017, 18, 2222. https://doi.org/10.3390/ijms18102222
Maywald M, Wessels I, Rink L. Zinc Signals and Immunity. International Journal of Molecular Sciences. 2017; 18(10):2222. https://doi.org/10.3390/ijms18102222
Chicago/Turabian StyleMaywald, Martina, Inga Wessels, and Lothar Rink. 2017. "Zinc Signals and Immunity" International Journal of Molecular Sciences 18, no. 10: 2222. https://doi.org/10.3390/ijms18102222
APA StyleMaywald, M., Wessels, I., & Rink, L. (2017). Zinc Signals and Immunity. International Journal of Molecular Sciences, 18(10), 2222. https://doi.org/10.3390/ijms18102222