Roles of Aryl Hydrocarbon Receptor in Aromatase-Dependent Cell Proliferation in Human Osteoblasts

 ,
,
Abstract
1. Introduction
2. Results
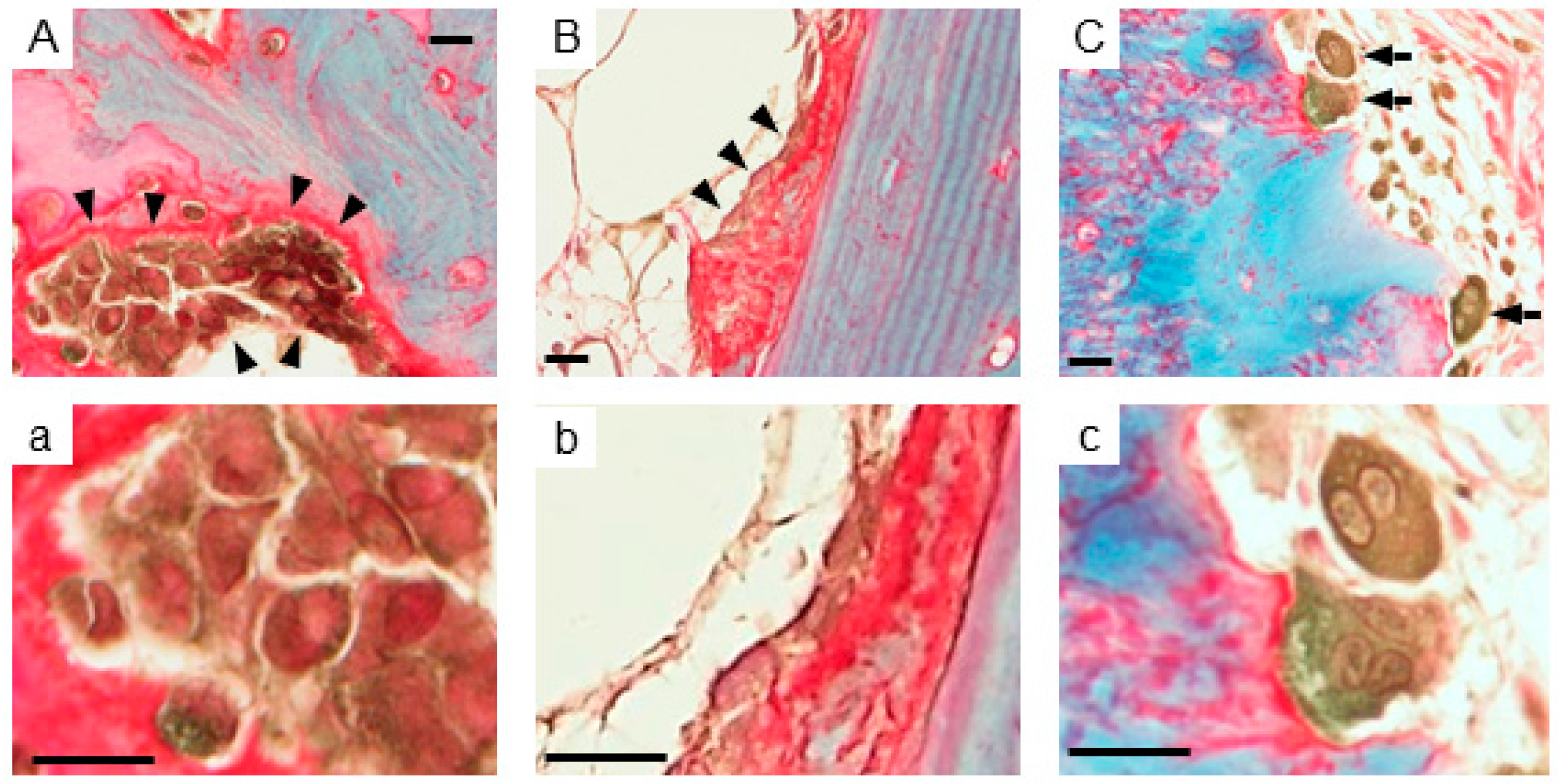
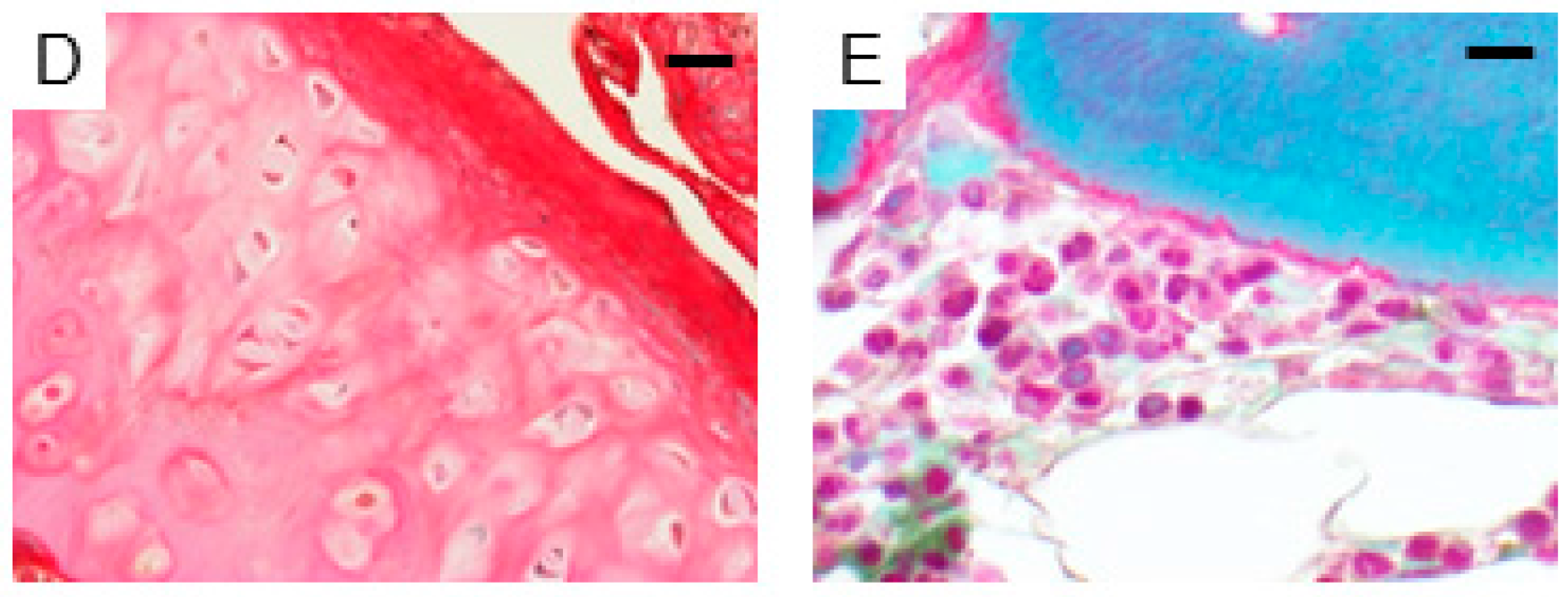
2.1. Expression of AhR in Human Bone Tissues
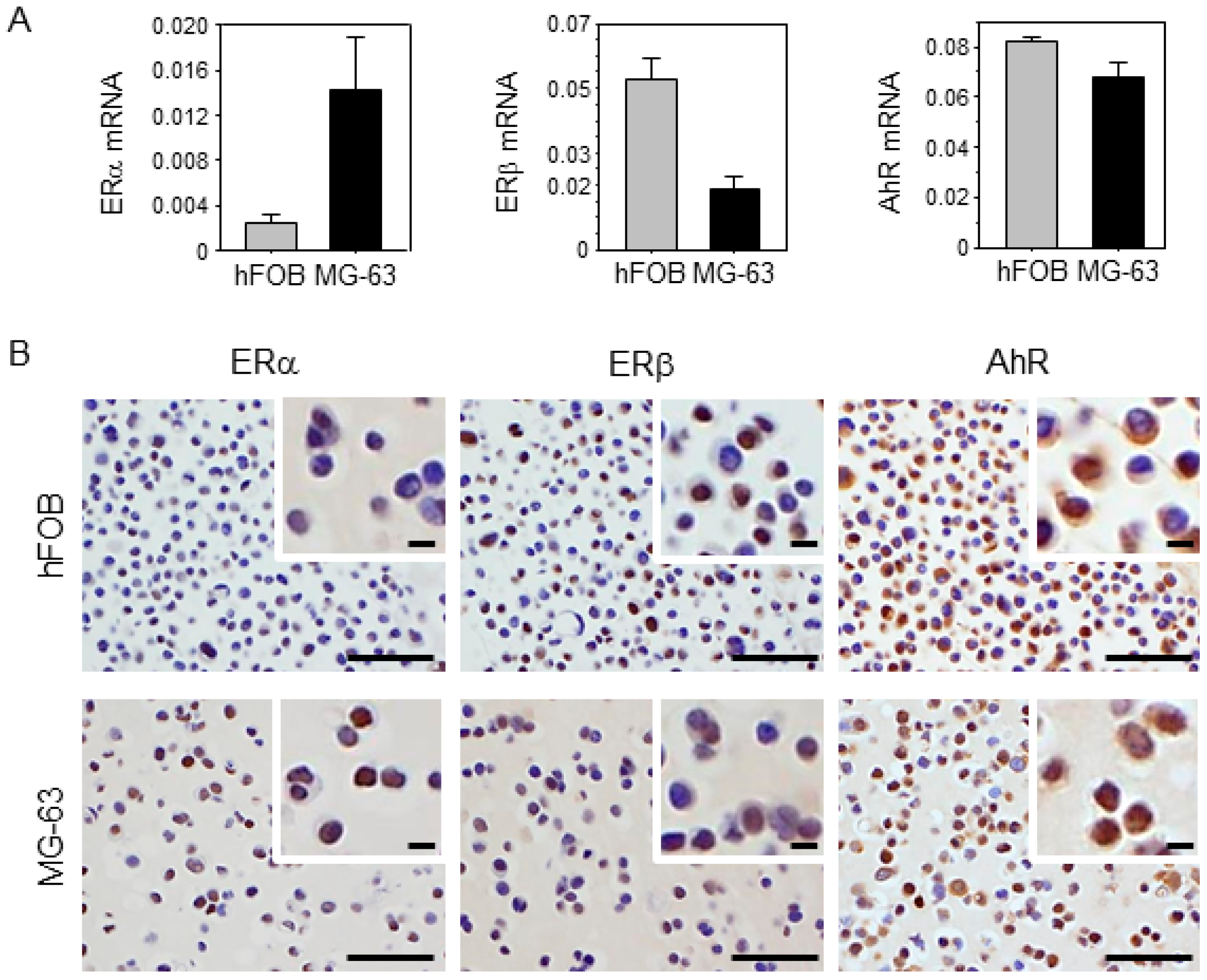
2.2. Characteristics of Osteoblast and Osteosarcoma Cell Lines Used in This Study
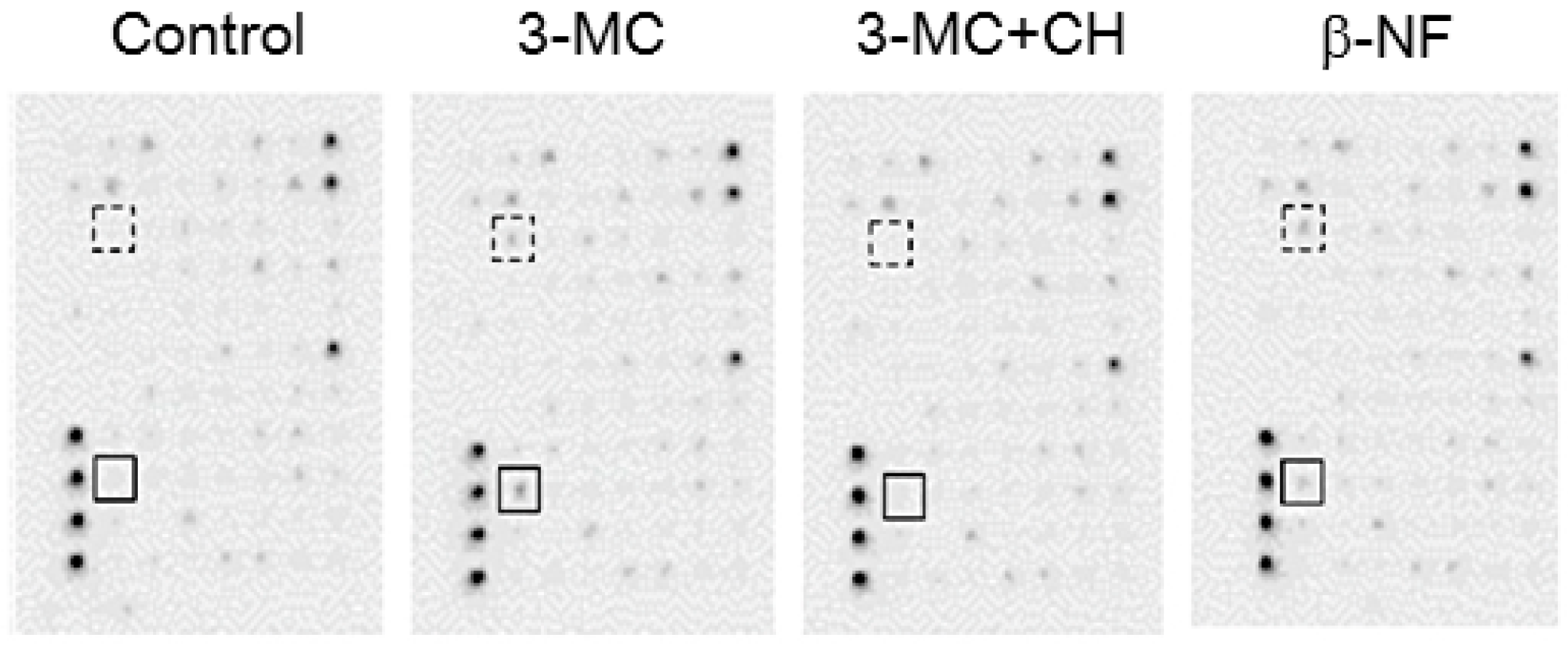
2.3. Gene Expression Induced by 3-MC Treatment in hFOB
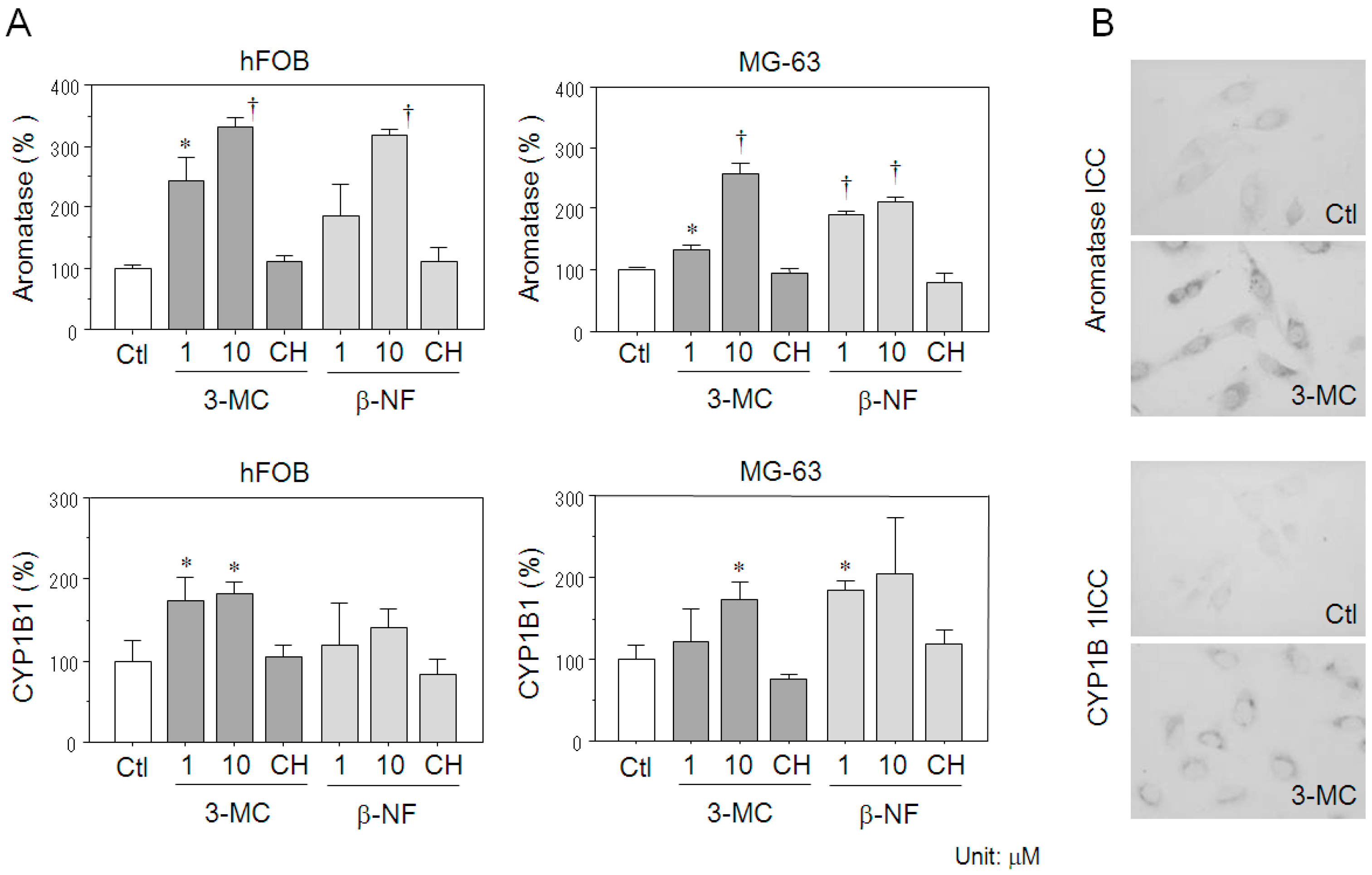
2.4. Effects of AhR Agonists on Aromatase Expression in Osteoblasts
2.5. Cytokines Secreted from hFOB Cells Stimulated by AhR Agonists
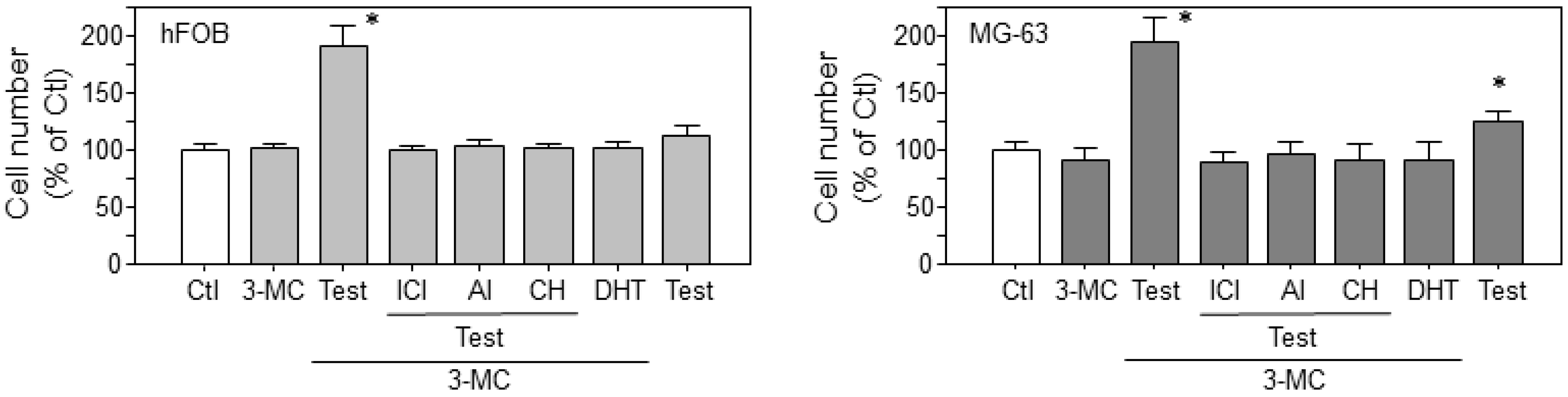
2.6. Estrogen-Dependent Cell Proliferation in Osteoblasts through the 3-MC-Induced Aromatase Pathway
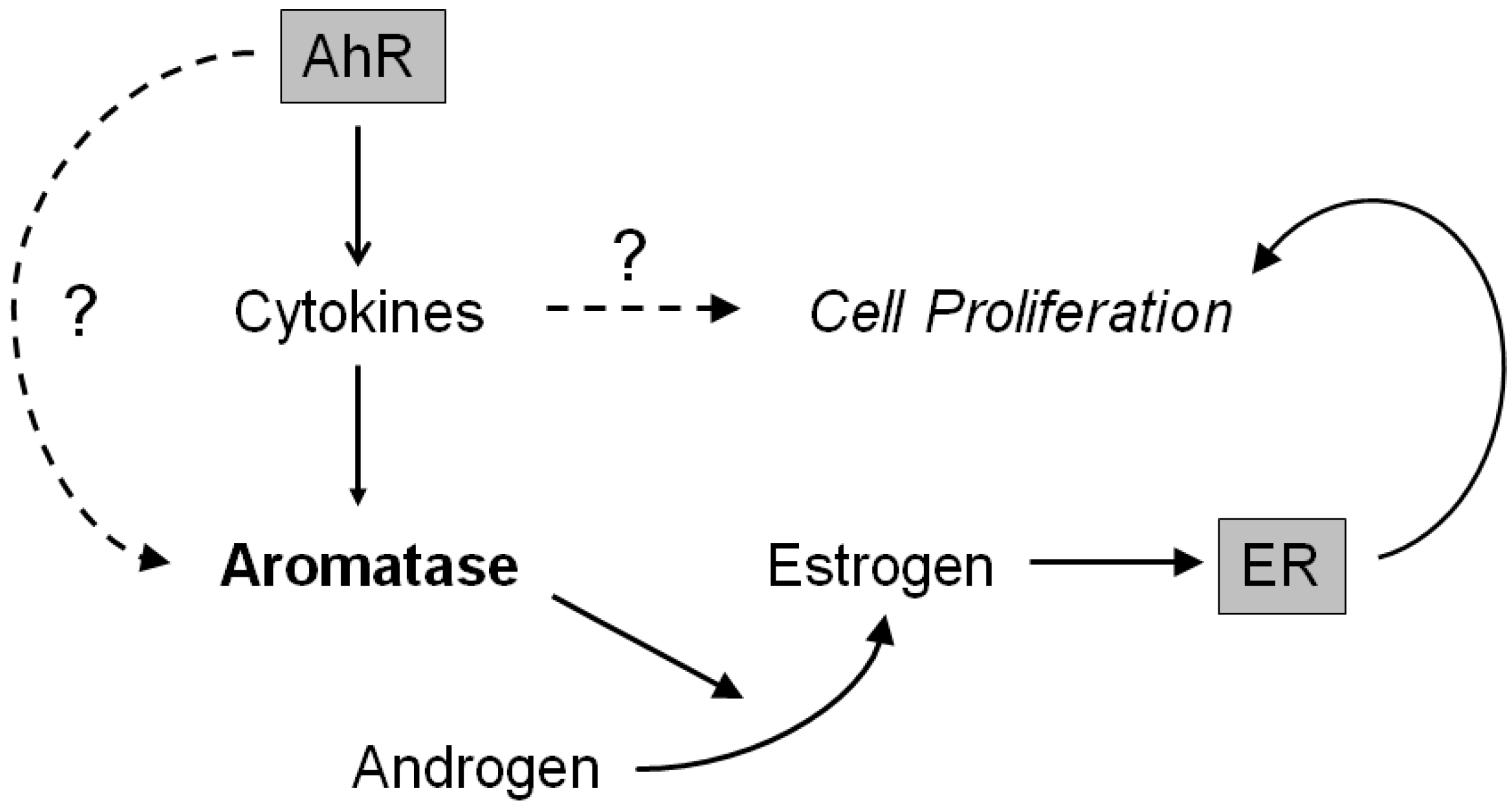
3. Discussion
4. Materials and Methods
4.1. Immunohistochemistry and Histochemistry
4.2. Chemicals
4.3. Osteoblast and Osteosarcoma Cell Lines and Culture Conditions
4.4. Characteristics of Osteoblast and Osteosarcoma Cell Lines
4.5. Microarray Analysis
4.6. Real-Time PCR
4.7. Immunocytochemistry
4.8. Cytokine Analysis
4.9. Cell Proliferation Assay
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hagiwara, H.; Sugizaki, T.; Tsukamoto, Y.; Senoh, E.; Goto, T.; Ishihara, Y. Effects of alkylphenols on bone metabolism in vivo and in vitro. Toxicol. Lett. 2008, 181, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Kiyama, R.; Wada-Kiyama, Y. Estrogenic endocrine disruptors: Molecular mechanisms of action. Environ. Int. 2015, 83, 11–40. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, T.; Kobayashi, N.; Mamiya, S.; Nakanishi, T.; Nishikawa, J. Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Mol. Pharmacol. 2005, 67, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Hata, S.; Nagasaki, S.; Suzuki, T.; Ito, K.; Kumamoto, H.; Sasano, H. Steroid and xenobiotic receptor-mediated effects of bisphenol A on human osteoblasts. Life Sci. 2016, 155, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Hata, S.; Saito, R.; Ono, K.; Sasano, H.; Kumamoto, H. Expression of Aryl Hydrocarbon Receptor in Bone Tissues. In Interface Oral Health Science 2011; Sasaki, K., Suzuki, O., Takahashi, N., Eds.; Springer: Tokyo, Japan, 2012. [Google Scholar]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999, 13, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Galván, N.; Teske, D.E.; Zhou, G.; Moorthy, B.; MacWilliams, P.S.; Czuprynski, C.J.; Jefcoate, C.R. Induction of CYP1A1 and CYP1B1 in liver and lung by benzo(a)pyrene and 7,12-dimethylbenz(a)anthracene do not affect distribution of polycyclic hydrocarbons to target tissue: Role of AhR and CYP1B1 in bone marrow cytotoxicity. Toxicol. Appl. Pharmacol. 2005, 202, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Lin, S.H.; Hou, Y.Y.; Chen, Y.H. Suppression of Lipid Accumulation by Indole-3-Carbinol Is Associated with Increased Expression of the Aryl Hydrocarbon Receptor and CYP1B1 Proteins in Adipocytes and with Decreased Adipocyte-Stimulated Endothelial Tube Formation. Int. J. Mol. Sci. 2016, 17, 1256. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, G.M.; Xi, X.; Kulkarni, A.A.; Olsen, K.C.; Pollock, S.J.; Baglole, C.J.; Gupta, S.; Casey, A.E.; Huxlin, K.R.; Sime, P.J.; et al. The aryl hydrocarbon receptor ligand ITE inhibits TGFβ1-induced human myofibroblast differentiation. Am. J. Pathol. 2011, 178, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- Poormasjedi-Meibod, M.S.; Salimi Elizei, S.; Leung, V.; Baradar Jalili, R.; Ko, F.; Ghahary, A. Kynurenine Modulates MMP-1 and Type-I Collagen Expression Via Aryl Hydrocarbon Receptor Activation in Dermal Fibroblasts. J. Cell. Physiol. 2016, 231, 2749–2760. [Google Scholar] [CrossRef] [PubMed]
- Naruse, M.; Ishihara, Y.; Miyagawa-Tomita, S.; Koyama, A.; Hagiwara, H. 3-Methylcholanthrene, which binds to the arylhydrocarbon receptor, inhibits proliferation and differentiation of osteoblasts in vitro and ossification in vivo. Endocrinology 2002, 143, 3575–3581. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.P.; Holz, J.D.; Mulcahey, M.; Sheu, T.J.; Gasiewicz, T.A.; Puzas, J.E. Environmental toxicants may modulate osteoblast differentiation by a mechanism involving the aryl hydrocarbon receptor. J. Bone Miner. Res. 2007, 22, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Du, Y.; Zhang, X.; Sun, Y.; Li, S.; Dou, Y.; Li, Z.; Yuan, H.; Zhao, W. The aryl hydrocarbon receptor suppresses osteoblast proliferation and differentiation through the activation of the ERK signaling pathway. Toxicol. Appl. Pharmacol. 2014, 280, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.Y.; Kondo, T.; Matsumoto, T.; Fujii-Kuriyama, Y.; Imai, Y. Aryl hydrocarbon receptor catabolic activity in bone metabolism is osteoclast dependent in vivo. Biochem. Biophys. Res. Commun. 2014, 450, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, T.; Lind, P.M.; Sävendahl, L. Expression of the aryl hydrocarbon receptor in growth plate cartilage and the impact of its local modulation on longitudinal bone growth. Int. J. Mol. Sci. 2015, 16, 8059–8069. [Google Scholar] [CrossRef] [PubMed]
- Horling, K.; Santos, A.N.; Fischer, B. The AhR is constitutively activated and affects granulosa cell features in the human cell line KGN. Mol. Hum. Reprod. 2011, 17, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Brauze, D.; Zawierucha, P.; Kiwerska, K.; Bednarek, K.; Oleszak, M.; Rydzanicz, M.; Jarmuz-Szymczak, M. Induction of expression of aryl hydrocarbon receptor-dependent genes in human HepaRG cell line modified by shRNA and treated with β-naphthoflavone. Mol. Cell. Biochem. 2017, 425, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Morgan, M.; Yoo, C.; Deoraj, A.; Roy, S.; Yadav, V.K.; Garoub, M.; Assaggaf, H.; Doke, M. Integrated bioinformatics, environmental epidemiologic and genomic approaches to identify environmental and molecular links between endometriosis and breast cancer. Int. J. Mol. Sci. 2015, 16, 25285–25322. [Google Scholar] [CrossRef] [PubMed]
- Sasano, H.; Uzuki, M.; Sawai, T.; Nagura, H.; Matsunaga, G.; Kashimoto, O.; Harada, N. Aromatase in human bone tissue. J. Bone Miner. Res. 1997, 12, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Suzuki, T.; Hatori, M.; Igarashi, K.; Aisaki, K.-I.; Kanno, J.; Nakamura, Y.; Uzuki, M.; Sawai, T.; Sasano, H. Effects of aromatase inhibitors on human osteoblast and osteoblast-like cells: A possible androgenic bone protective effects induced by exemestane. Bone 2007, 40, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Miki, Y.; Hata, S.; Ishida, T.; Suzuki, T.; Ohuchi, N.; Sasano, H. Aryl hydrocarbon receptor induced intratumoral aromatase in breast cancer. Breast Cancer Res. Treat. 2017, 161, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, K.; Lagerquist, M.; Moverare-Skrtic, S.; Andersson, N.; Windahl, S.H.; Swanson, C.; Mohan, S.; Poutanen, M.; Ohlsson, C. Elevated aromatase expression in osteoblasts leads to increased bone mass without systemic adverse effects. J. Bone Miner. Res. 2009, 24, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Cai, M.X.; Thomas, P.E.; Conney, A.H.; Zhu, B.T. Characterization of the oxidative metabolites of 17beta-estradiol and estrone formed by 15 selectively expressed human cytochrome p450 isoforms. Endocrinology 2003, 144, 3382–3398. [Google Scholar] [CrossRef] [PubMed]
- Napoli, N.; Rini, G.B.; Serber, D.; Giri, T.; Yarramaneni, J.; Bucchieri, S.; Camarda, L.; Di Fede, G.; Camarda, M.R.; Jain, S.; et al. The Val432Leu polymorphism of the CYP1B1 gene is associated with differences in estrogen metabolism and bone density. Bone 2009, 44, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Yahata, T.; Tamura, N.; Nagata, H.; Tanaka, K. Relationship between single nucleotide polymorphisms in CYP1A1 and CYP1B1 genes and the bone mineral density and serum lipid profiles in postmenopausal Japanese women taking hormone therapy. Menopause 2009, 16, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Hurh, Y.J.; Na, H.K.; Kim, J.H.; Chun, Y.J.; Kim, D.H.; Kang, K.S.; Cho, M.H.; Surh, Y.J. Resveratrol inhibits TCDD-induced expression of CYP1A1 and CYP1B1 and catechol estrogen-mediated oxidative DNA damage in cultured human mammary epithelial cells. Carcinogenesis 2004, 25, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Schroeder, J.C.; Francey, L.J.; Kusnadi, A.; Perdew, G.H. Mechanistic insights into the events that lead to synergistic induction of interleukin 6 transcription upon activation of the aryl hydrocarbon receptor and inflammatory signaling. J. Biol. Chem. 2010, 285, 24388–24397. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Sciullo, E.; Matsumura, F. Activation of inflammatory mediators and potential role of ah-receptor ligands in foam cell formation. Cardiovasc. Toxicol. 2004, 4, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Morioka, M.; Shimodaira, K.; Kuwano, Y.; Fujikawa, H.; Saito, H.; Yanaihara, T. Effect of interleukin-1beta on aromatase activity and cell proliferation in human osteoblast-like cells (HOS). Biochem. Biophys. Res. Commun. 2000, 268, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Suzuki, T.; Abe, K.; Suzuki, S.; Niikawa, H.; Iida, S.; Hata, S.; Akahira, J.; Mori, K.; Evans, D.B.; et al. Intratumoral localization of aromatase and interaction between stromal and parenchymal cells in the non-small cell lung carcinoma microenvironment. Cancer Res. 2010, 70, 6659–6669. [Google Scholar] [CrossRef] [PubMed]
- Onoe, Y.; Miyaura, C.; Ohta, H.; Nozawa, S.; Suda, T. Expression of estrogen receptor beta in rat bone. Endocrinology 1997, 138, 4509–4512. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Suzuki, T.; Nagasaki, S.; Hata, S.; Akahira, J.; Sasano, H. Comparative effects of raloxifene, tamoxifen and estradiol on human osteoblasts in vitro: Estrogen receptor dependent or independent pathways of raloxifene. J. Steroid Biochem. Mol. Biol. 2009, 113, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Shanle, E.K.; Xu, W. Endocrine disrupting chemicals targeting estrogen receptor signaling: Identification and mechanisms of action. Chem. Res. Toxicol. 2011, 24, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Endler, A.; Chen, L.; Shibasaki, F. Coactivator recruitment of AhR/ARNT1. Int. J. Mol. Sci. 2014, 15, 11100–11110. [Google Scholar] [CrossRef] [PubMed]
- Rüegg, J.; Swedenborg, E.; Wahlström, D.; Escande, A.; Balaguer, P.; Pettersson, K.; Pongratz, I. The transcription factor aryl hydrocarbon receptor nuclear translocator functions as an estrogen receptor beta-selective coactivator, and its recruitment to alternative pathways mediates antiestrogenic effects of dioxin. Mol. Endocrinol. 2008, 22, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Madak-Erdogan, Z.; Katzenellenbogen, B.S. Aryl hydrocarbon receptor modulation of estrogen receptor α-mediated gene regulation by a multimeric chromatin complex involving the two receptors and the coregulator RIP140. Toxicol. Sci. 2012, 125, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Izawa, T.; Arakaki, R.; Mori, H.; Tsunematsu, T.; Kudo, Y.; Tanaka, E.; Ishimaru, N. The Nuclear Receptor AhR Controls Bone Homeostasis by Regulating Osteoclast Differentiation via the RANK/c-Fos Signaling Axis. J. Immunol. 2016, 197, 4639–4650. [Google Scholar] [CrossRef] [PubMed]
- Bord, S.; Horner, A.; Beavan, S.; Compston, J. Estrogen receptors alpha and beta are differentially expressed in developing human bone. J. Clin. Endocrinol. Metab. 2001, 86, 2309–2314. [Google Scholar] [PubMed]
- Batra, G.S.; Hainey, L.; Freemont, A.J.; Andrew, G.; Saunders, P.T.; Hoyland, J.A.; Braidman, I.P. Evidence for cell-specific changes with age in expression of oestrogen receptor (ER) alpha and beta in bone fractures from men and women. J. Pathol. 2003, 200, 65–73. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ratio | Common | Description |
|---|---|---|
| 23.8 | COL18A1 | Collagen, type XVIII, alpha 1 |
| 10.9 | OSTalpha | Organic solute transporter alpha |
| 7.2 | CABP5 | Calcium binding protein 5 |
| 7.1 | GPR34 | G protein-coupled receptor |
| 6.8 | ESR2 | Estrogen receptor 2 (ER beta) |
| 5.2 | COL23A1 | Collagen, type XXIII, alpha 1 |
| 4.9 | SLC25A24 | Solute carrier family 25, member 24 |
| 4.8 | GRB2 | Growth factor receptor-bound protein 2 |
| 4.5 | RUNX1 | Runt-related transcription factor 1 |
| 3.7 | IL1F8 | Interleukin 1 family, member 8 |
| 3.7 | FGF5 | Fibroblast growth factor 5 |
| 3.6 | IL1F7 | Interleukin 1 family, member 7 |
| 3.4 | CYP21A2 | Cytochrome P450, family 21, subfamily A, polypeptide 2 |
| 3.2 | CYP19A1 | Aromatase |
| 3.2 | OSMR | Oncostatin M receptor |
| 3.2 | MAPK8IP3 | Mitogen-activated protein kinase 8 interacting protein 3 |
| 2.8 | ARSB | Arylsulfatase B |
| 2.8 | CYP1B1 | Cytochrome P450, family 1, subfamily B, polypeptide 1 |
| 2.7 | NR3C2 | Nuclear receptor subfamily 3, group C, member 2 |
| 2.7 | SULT1A4 | Sulfotransferase family, cytosolic, 1A, phenol-preferring, member 4 |
| 2.5 | CYP4F3 | Cytochrome P450, family 4, subfamily F, polypeptide 3 |
| 2.4 | EPHX1 | Epoxide hydrolase 1, microsomal (xenobiotic) |
| Ratio, a fold change against the vehicle control. | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miki, Y.; Hata, S.; Ono, K.; Suzuki, T.; Ito, K.; Kumamoto, H.; Sasano, H. Roles of Aryl Hydrocarbon Receptor in Aromatase-Dependent Cell Proliferation in Human Osteoblasts. Int. J. Mol. Sci. 2017, 18, 2159. https://doi.org/10.3390/ijms18102159
Miki Y, Hata S, Ono K, Suzuki T, Ito K, Kumamoto H, Sasano H. Roles of Aryl Hydrocarbon Receptor in Aromatase-Dependent Cell Proliferation in Human Osteoblasts. International Journal of Molecular Sciences. 2017; 18(10):2159. https://doi.org/10.3390/ijms18102159
Chicago/Turabian StyleMiki, Yasuhiro, Shuko Hata, Katsuhiko Ono, Takashi Suzuki, Kiyoshi Ito, Hiroyuki Kumamoto, and Hironobu Sasano. 2017. "Roles of Aryl Hydrocarbon Receptor in Aromatase-Dependent Cell Proliferation in Human Osteoblasts" International Journal of Molecular Sciences 18, no. 10: 2159. https://doi.org/10.3390/ijms18102159
APA StyleMiki, Y., Hata, S., Ono, K., Suzuki, T., Ito, K., Kumamoto, H., & Sasano, H. (2017). Roles of Aryl Hydrocarbon Receptor in Aromatase-Dependent Cell Proliferation in Human Osteoblasts. International Journal of Molecular Sciences, 18(10), 2159. https://doi.org/10.3390/ijms18102159