Cardiotoxic Effects of Short-Term Doxorubicin Administration: Involvement of Connexin 43 in Calcium Impairment


 ,
,  ,
,  ,
,  , and
, and
Abstract
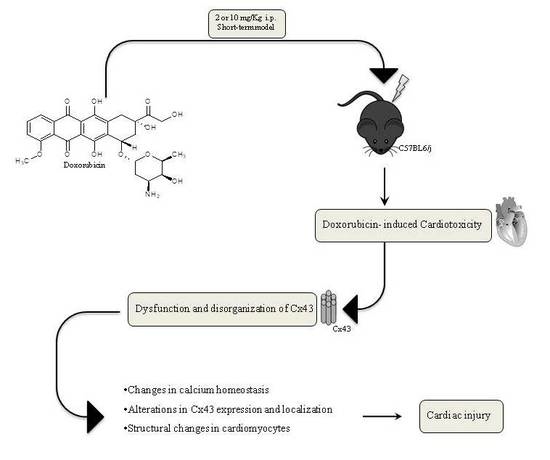
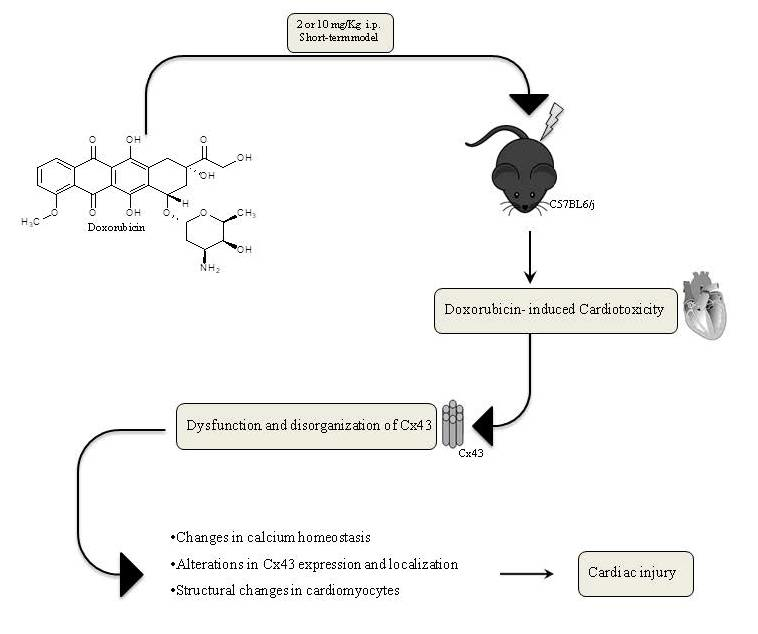
1. Introduction
2. Results
2.1. Cardiac Functions
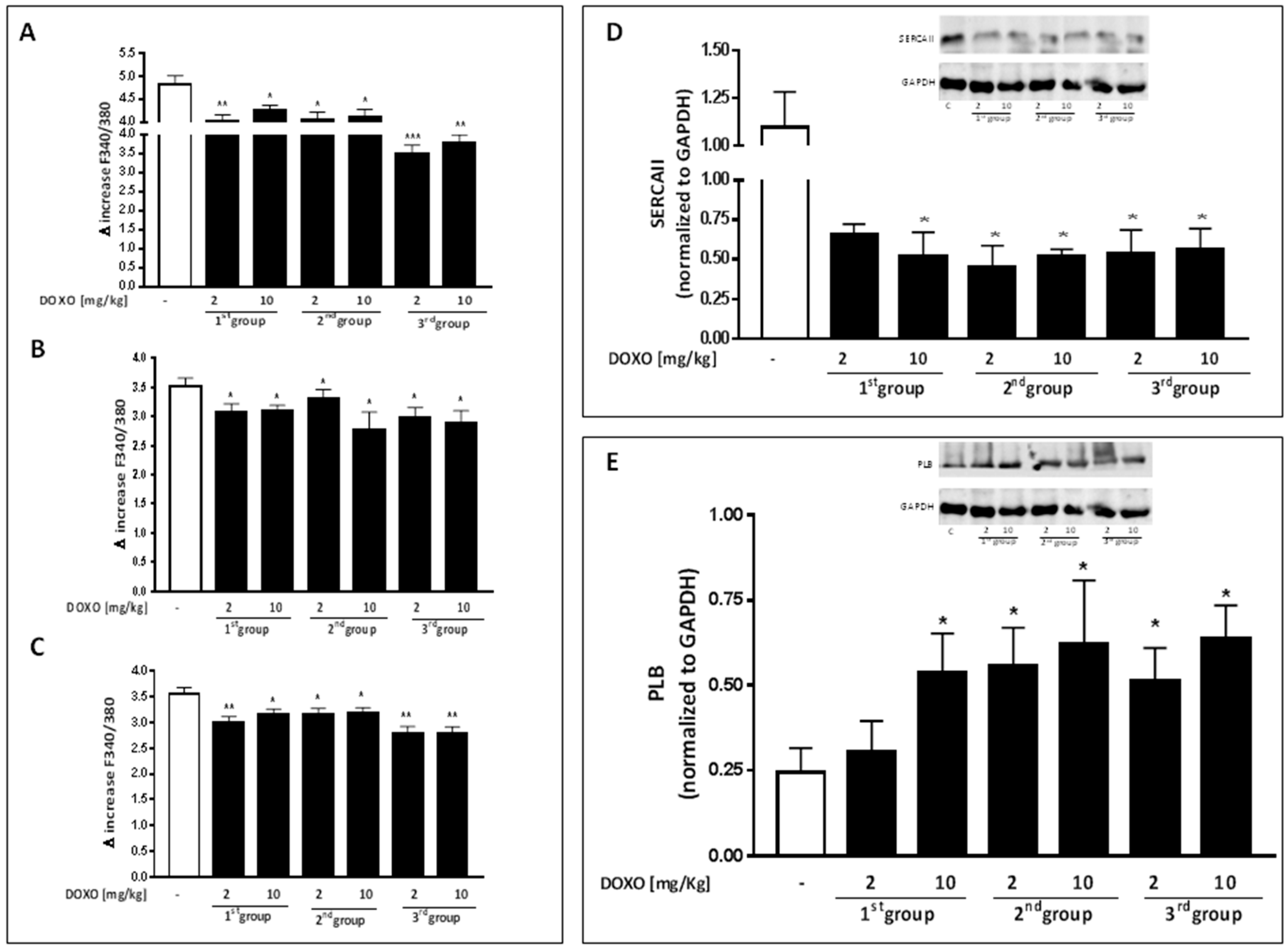
2.2. Doxorubicin Administration Alters Calcium Homeostasis
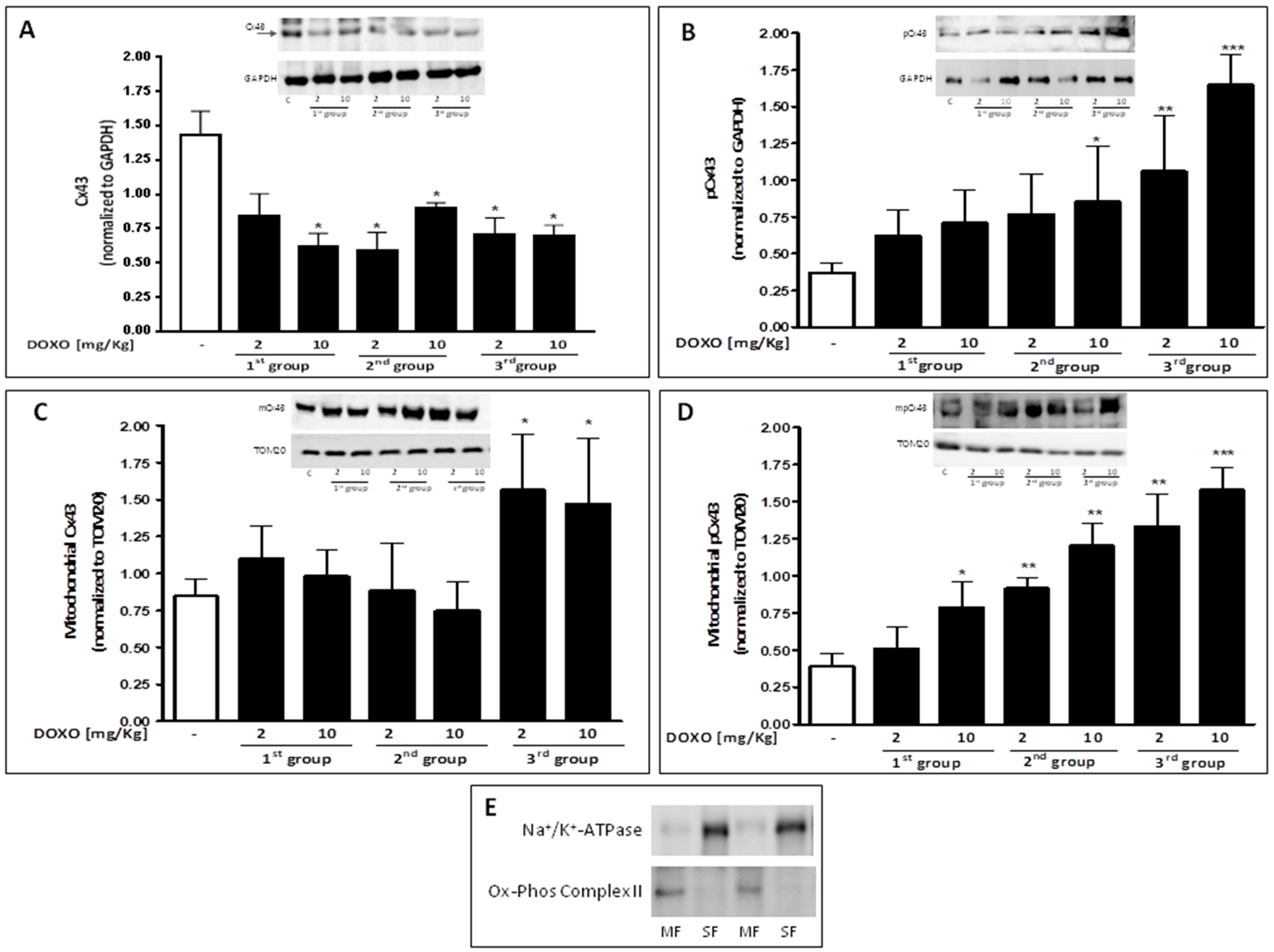
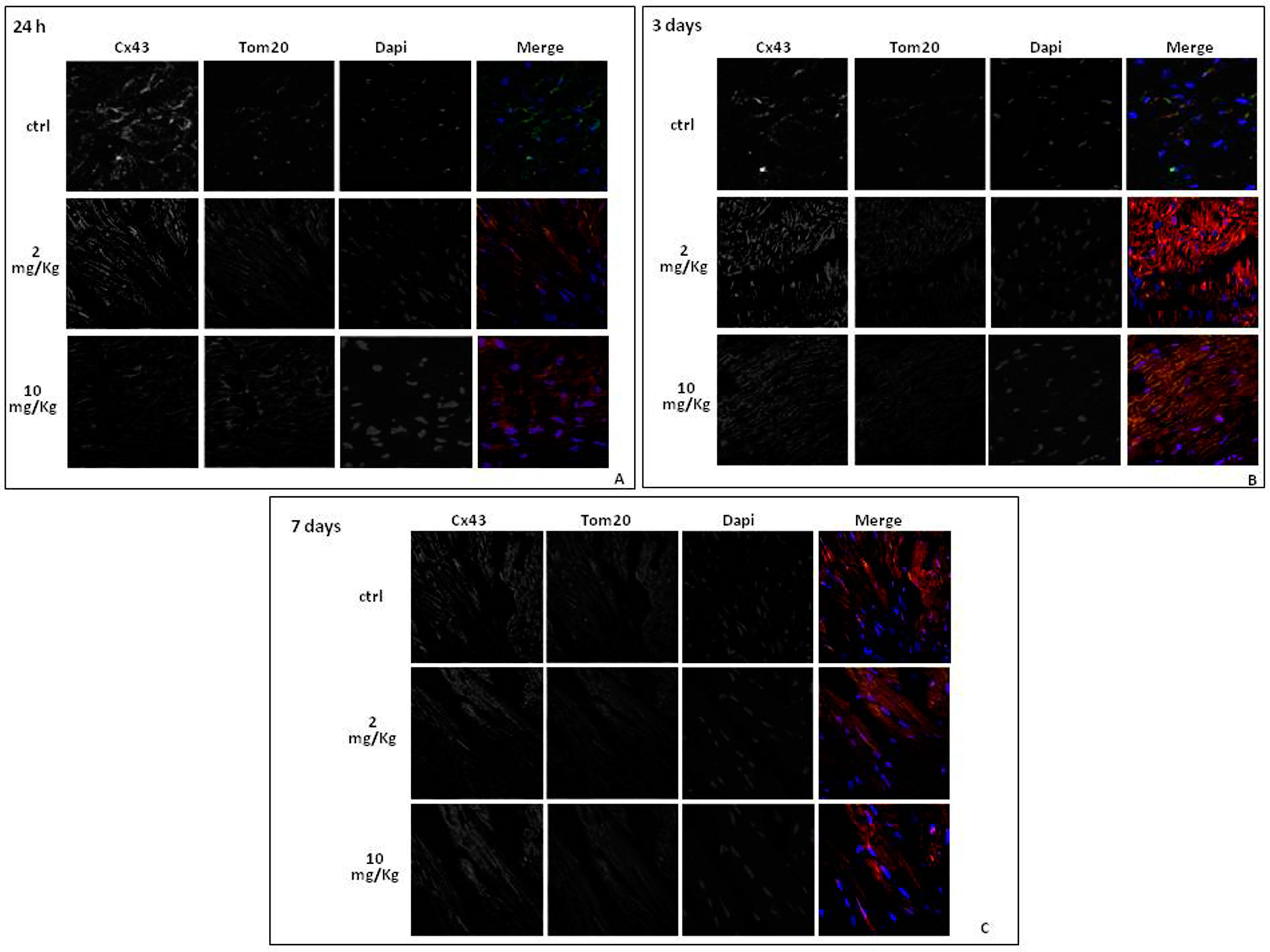
2.3. Doxorubicin Administration Affects Cx43 Expression and Localization
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Experimental Protocols
4.4. Echocardiogram
4.5. Protein Extraction and Western Blot Analysis
4.6. Mitochondrial Protein Extraction and Western Blot Analysis for Mitochondrial Cx43 and pCx43
4.7. Primary Cardiomyocytes Isolation and Measurement of Intracellular Ca2+ Signaling
4.8. Immunohistochemical Analysis
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wallace, K.B. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol. Toxicol. 2003, 93, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Ferrans, V.J.; Clark, J.R.; Zhang, J.; Yu, Z.X.; Herman, E.H. Pathogenesis and prevention of doxorubicin cardiomyopathy. Tsitologiya 1997, 39, 928–936. [Google Scholar]
- Medrano, F.L.; Munoz, A.S.; Sánchez, V.S.; Pérez-Herrero, J.R.C. Cardiotoxicity of 5 fluorouracil: Ischemia or myocardial toxicity? Rev. Clin. Esp. 2001, 201, 106–107. [Google Scholar]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin Cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Fujiwara, H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef] [PubMed]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid. Med. Cell Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef] [PubMed]
- Menna, P.; Salvatorelli, E.; Minotti, G. Cardiotoxicity of antitumor drugs. Chem. Res. Toxicol. 2008, 21, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, M.; Del Pizzo, M.; Marzocco, S.; Sorrentino, R.; Ciccarelli, M.; Iaccarino, G.; Pinto, A.; Popolo, A. Inflammatory mediators in a short-time mouse model of doxorubicin-induced cardiotoxicity. Toxicol. Appl. Pharmacol. 2016, 293, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Li, Q.; Na, R.; Li, X.; Liu, B.; Meng, L.; Liutong, H.; Fang, W.; Zhu, N.; Zheng, X. Impact of repeated intravenous bone marrow mesenchymal stem cells infusion on myocardial collagen network remodeling in a rat model of doxorubicin-induced dilated cardiomyopathy. Mol. Cell. Biochem. 2014, 387, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, J.M.; Wallace, K.B. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell. Biol. Toxicol. 2007, 23, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, M.S.; Vander Heide, R.S.; L’Ecuyer, T.J. Molecular basis of anthracycline-induced cardiotoxicity and its prevention. Mol. Genet. Metab. 2000, 71, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Zhang, M.; Tang, Y.; Xie, Y.; Huang, X.; Li, Y. Doxorubicin induces sarcoplasmic reticulum calcium regulation dysfunction via the decrease of SERCA2 and phospholamban expressions in rats. Cell Biochem. Biophys. 2014, 70, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.L.; Romano, M.M.; Campos Pulici, E.C.; Carvalho, E.E.; de Souza, F.R.; Tanaka, D.M.; Maciel, B.C.; Salgado, H.C.; Fazan-Júnior, R.; Rossi, M.A.; et al. Short-term and long-term models of doxorubicin-induced cardiomyopathy in rats: A comparison of functional and histopathological changes. Exp. Toxicol. Pathol. 2017, 69, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, L.; Ma, J.; Lu, L.; Wang, X.; Ren, J.; Yang, J. Rutin attenuates doxorubicin-induced cardiotoxicity via regulating autophagy and apoptosis. Biochim. Biophys. Acta 2017, 1863, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Robert, J. Long-term and short-term models for studying anthracycline cardiotoxicity and protectors. Cardiovasc. Toxicol. 2007, 7, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, M.; Sorrentino, R.; Franceschelli, S.; Del Pizzo, M.; Pinto, A.; Popolo, A. Doxorubicin-Mediated Cardiotoxicity: Role of Mitochondrial Connexin 43. Cardiovasc. Toxicol. 2015, 15, 366–376. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Li, N.; Zhao, Z.; Han, F.; Wang, X.; Zeng, Y. Ischemic postconditioning improves the expression of cellular membrane connexin 43 and attenuates the reperfusion injury in rat acute myocardial infarction. Biomed. Rep. 2015, 3, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexion expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Allen, A. The cardiotoxicity of chemotherapeutic drugs. Semin. Oncol. 1992, 19, 529–542. [Google Scholar] [PubMed]
- Maron, B.J. Hypertrophic cardiomyopathy: A systematic review. JAMA 2002, 287, 1308–1320. [Google Scholar] [CrossRef] [PubMed]
- Johansen, D.; Cruciani, V.; Sundset, R.; Ytrehus, K.; Mikalsen, S.O. Ischemia induces closure of gap junctional channels and opening of hemichannels in heart-derived cells and tissue. Cell. Physiol. Biochem. 2011, 28, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Salas, D.; Puebla, C.; Lampe, P.D.; Lavandero, S.; Sáez, J.C. Role of Akt and Ca2+ on cell permeabilization via connexin43 hemichannels induced by metabolic inhibition. Biochim. Biophys. Acta 2015, 1852, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Popolo, A.; Morello, S.; Sorrentino, R.; Pinto, A. Antiadrenergic effect of adenosine involves connexin 43 turn-over in H9c2 cells. Eur. J. Pharmacol. 2013, 715, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Spray, D.C.; Burt, J.M. Structure-activity relations of the cardiac gap junction channel. Am. J. Physiol. 1990, 258, 195–205. [Google Scholar]
- Alex, J.; Cale, A.R.J.; Griffin, S.C.; Cowen, M.E.; Guvendik, L. Connexins: The basis of functional coupling of myocyte. J. Clin. Basic Cardiol. 2005, 8, 19–22. [Google Scholar]
- Rodriguez-Sinovas, A.; Boengler, K.; Cabestrero, A.; Gres, P.; Morente, M.; Ruiz-Meana, M.; Konietzka, I.; Miró, E.; Totzeck, A.; Heusch, G.; et al. Translocation of connexin43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein90-dependent TOM pathway and its importance for cardioprotection. Circ. Res. 2006, 99, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Thimm, J.; Mechler, A.; Lin, H.; Rhee, S.; Lal, R. Calcium-dependent open/closed conformations and interfacial energy maps of reconstituted hemichannels. J. Biol. Chem. 2005, 280, 10646–10654. [Google Scholar] [CrossRef] [PubMed]
- Bol, M.; Wang, N.; De Bock, M.; Wacquier, B.; Decrock, E.; Gadicherla, A.; Decaluwé, K.; Vanheel, B.; van Rijen, H.V.; Krysko, D.V.; et al. At the cross-point of connexins, calcium, and ATP: Blocking hemichannels inhibits vasoconstriction of rat small mesenteric arteries. Cardiovasc. Res. 2017, 113, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, M.; Tsujii, E.; Tanaka, H.; Matsushita, T.; Takamatsu, T. Abnormalities in gap junctions and Ca2+ dynamics in cardiomyocytes at the border zone of myocardial infarcts. Cell Commun. Adhes. 2001, 8, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Bhupathy, P.; Babu, G.J. Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc. Res. 2008, 77, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Fontes, M.S.; vanVeen, T.A.; deBakker, J.M.; vanRijen, H.V. Functional consequences of abnormal Cx43 expression in the heart. Biochim. Biophys. Acta 2012, 1818, 2020–2029. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraman, M.M.; Srisakuldee, W.; Nickel, B.E.; Kardami, E. Connexin43 phosphorylation and cytoprotection in the heart. Biochim. Biophys. Acta 2012, 1818, 2009–2013. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Schulz, R.; Heusch, G. Connexin43 signalling and cardioprotection. Heart 2006, 92, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Tsuchida, M.; Lampe, P.D.; Murakami, M. Cardiomyocyte FGF signaling is required for Cx43 phosphorylation and cardiac gap junction maintenance. Exp. Cell Res. 2013, 319, 2152–2165. [Google Scholar] [CrossRef] [PubMed]
- Kalvelyte, A.; Imbrasaite, A.; Bukauskiene, A.; Verselis, V.K.; Bukauskas, F.F. Connexins and apoptotic transformation. Biochem. Pharmacol. 2003, 66, 1661–1672. [Google Scholar] [CrossRef]
- Ascensão, A.; Oliveira, P.J.; Magalhães, J. Exercise as a beneficial adjunct therapy during Doxorubicin treatment—Role of mitochondria in cardioprotection. Int. J. Cardiol. 2012, 156, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Mercuro, G.; Cadeddu, C.; Piras, A.; Dessì, M.; Madeddu, C.; Deidda, M.; Serpe, R.; Massa, E.; Mantovani, G. Early epirubicin-induced myocardial dysfunction revealed by serial tissue Doppler echocardiography: Correlation with inflammatory and oxidative stress markers. Oncologist 2007, 12, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Xujie, L.; Xinggang, W.; Xian, Z.; Yeqing, X.; Ruizhen, C.; Haozhu, C. C57BL/6 Mice are More Appropriate than BALB/C Mice in Inducing Dilated Cardiomyopathy with Short-Term Doxorubicin Treatment. Acta Cardiol. Sin. 2012, 28, 236–240. [Google Scholar]
- Wided, K.; Hassiba, R.; Mesbah, L. Polyphenolic fraction of Algerian propolis reverses doxorubicin induced oxidative stress in liver cells and mitochondria. Pak. J. Pharm. Sci. 2014, 27, 1891–1897. [Google Scholar] [PubMed]
- Mattila, M.; Koskenvuo, J.; Söderström, M.; Eerola, K.; Savontaus, M. Intramyocardial injection of SERCA2a-expressing lentivirus improves myocardial function in doxorubicin-induced heart failure. J. Gene Med. 2016, 18, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Eisner, D.A.; O’Neill, S.C. Do calcium waves propagate between cells and synchronize alternating calcium release in rat ventricular myocytes? J. Physiol. 2012, 590, 6353–6361. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, M.; Verrilli, V.; Pinto, A.; Popolo, A. Role of connexin 43 in cardiovascular diseases. Eur. J. Pharmacol. 2015, 768, 71–76. [Google Scholar]
- Hervé, J.C.; Derangeon, M.; Sarrouilhe, D.; Giepmans, B.N.; Bourmeyster, N. Gap junctional channels are parts of multiprotein complexes. Biochim. Biophys. Acta 2012, 1818, 1844–1865. [Google Scholar] [CrossRef] [PubMed]
- Spray, D.C.; Hanstein, R.; Lopez-Quintero, S.V.; Stout, R.F., Jr.; Suadicani, S.O.; Thi, M.M. Gap junctions and Bystander Effects: Good Samaritans and executioners. Wiley Interdiscip. Rev. Membr. Transp. Signal 2013, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gadicherla, A.K.; Wang, N.; Bulic, M.; Agullo-Pascual, E.; Lissoni, A.; De Smet, M.; Delmar, M.; Bultynck, G.; Krysko, D.V.; Camara, A.; et al. Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res. Cardiol. 2017, 112, 27. [Google Scholar] [CrossRef] [PubMed]
- Azarashvili, T.; Baburina, Y.; Grachev, D.; Krestinina, O.; Evtodienko, Y.; Stricker, R.; Reiser, G. Calcium-induced permeability transition in rat brain mitochondria is promoted by carbenoxolone through targeting connexin43. Am. J. Physiol. Cell Physiol. 2011, 300, C707–C720. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Ungefug, E.; Heusch, G.; Leybaert, L.; Schulz, R. Connexin 43 impacts on mitochondrial potassium uptake. Front. Pharmacol. 2013, 4, 73. [Google Scholar] [CrossRef] [PubMed]
- Miro-Casas, E.; Ruiz-Meana, M.; Agullo, E.; Stahlhofen, S.; Rodríguez-Sinovas, A.; Cabestrero, A.; Jorge, I.; Torre, I.; Vazquez, J.; Boengler, K.; et al. Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc. Res. 2009, 83, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Waza, A.A.; Andrabi, K.; Hussain, M.U. Protein kinase C (PKC) mediated interaction between conexin43 (Cx43) and K(+)(ATP) channel subunit (Kir6.1) in cardiomyocyte mitochondria: Implications in cytoprotection against hypoxia induced cell apoptosis. Cell Signal. 2014, 26, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Freireich, E.J. The future of clinical cancer research in the next millennium. Clin. Cancer Res. 1997, 3, 2563–2570. [Google Scholar] [PubMed]
- Sardão, V.A.; Oliveira, P.J.; Holy, J.; Oliveira, C.R.; Wallace, K.B. Doxorubicin-induced mitochondrial dysfunction is secondary to nuclear p53 activation in H9c2 cardiomyoblasts. Cancer Chemother. Pharmacol. 2009, 64, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Popolo, A.; Bianco, G.; Pinto, A.; Autore, G. Pro-apoptotic effect of methylguanidine on hydrogen peroxide-treated rat glioma cell line. Neurochem. Int. 2010, 57, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Dodoni, G.; Rodriguez-Sinovas, A.; Cabestrero, A.; Ruiz-Meana, M.; Gres, P.; Konietzka, I.; Lopez-Iglesias, C.; Garcia-Dorado, D.; Di Lisa, F.; et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Popolo, A.; Piccinelli, A.L.; Morello, S.; Sorrentino, R.; Osmany, C.R.; Rastrelli, L.; Pinto, A. Cytotoxic activity of nemorosone in human MCF-7 breast cancer cells. Can. J. Physiol. Pharmacol. 2011, 89, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Hurt, C.; Neviere, R. Mitochondria death/survival signaling pathways in cardiotoxicity induced by anthracyclines and anticancer-targeted therapies. Biochem. Res. Int. 2012, 2012, 951539. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Parameters | Control | 2 mg/kg | 10 mg/kg |
|---|---|---|---|---|
| 1st group | LVEDD | 3.97 ± 0.11 | 3.92 ± 0.11 | 4.09 ± 0.10 |
| LVESD | 2.62 ± 0.17 | 2.77 ± 0.09 | 3.00 ± 0.06 * | |
| % EF | 62.17 ± 4.1 | 58.39 ± 1.12 * | 52.7 ± 1.38 ** | |
| % FS | 30.41 ± 0.72 | 33.26 ± 2.93 | 26.76 ± 0.92 * | |
| 2nd group | LVEDD | 3.94 ± 0.05 | 3.87 ± 0.05 | 3.99 ± 0.06 |
| LVESD | 2.78 ± 0.054 | 2.78 ± 0.05 | 2.90 ± 0.06* | |
| % EF | 57.2 ± 1.25 | 54063 ± 1.6 * | 53.73 ± 1.61 * | |
| % FS | 30.41 ± 0.85 | 27.95 ± 1.06 * | 27.43 ± 1.02 * | |
| 3rd group | LVEDD | 3.86 ± 0.04 | 3.94 ± 0.07 | 3.96 ± 0.06 * |
| LVESD | 2.73 ± 0.15 | 2.85 ± 0.06 | 2.9 ± 0.14 * | |
| % EF | 59.00 ± 4.17 | 54.4 ± 2.6 * | 50.49 ± 4.79 ** | |
| % FS | 31.76 ± 1.68 | 30.97 ± 2.91 | 25.37 ± 3.02 * |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecoraro, M.; Rodríguez-Sinovas, A.; Marzocco, S.; Ciccarelli, M.; Iaccarino, G.; Pinto, A.; Popolo, A. Cardiotoxic Effects of Short-Term Doxorubicin Administration: Involvement of Connexin 43 in Calcium Impairment. Int. J. Mol. Sci. 2017, 18, 2121. https://doi.org/10.3390/ijms18102121
Pecoraro M, Rodríguez-Sinovas A, Marzocco S, Ciccarelli M, Iaccarino G, Pinto A, Popolo A. Cardiotoxic Effects of Short-Term Doxorubicin Administration: Involvement of Connexin 43 in Calcium Impairment. International Journal of Molecular Sciences. 2017; 18(10):2121. https://doi.org/10.3390/ijms18102121
Chicago/Turabian StylePecoraro, Michela, Antonio Rodríguez-Sinovas, Stefania Marzocco, Michele Ciccarelli, Guido Iaccarino, Aldo Pinto, and Ada Popolo. 2017. "Cardiotoxic Effects of Short-Term Doxorubicin Administration: Involvement of Connexin 43 in Calcium Impairment" International Journal of Molecular Sciences 18, no. 10: 2121. https://doi.org/10.3390/ijms18102121
APA StylePecoraro, M., Rodríguez-Sinovas, A., Marzocco, S., Ciccarelli, M., Iaccarino, G., Pinto, A., & Popolo, A. (2017). Cardiotoxic Effects of Short-Term Doxorubicin Administration: Involvement of Connexin 43 in Calcium Impairment. International Journal of Molecular Sciences, 18(10), 2121. https://doi.org/10.3390/ijms18102121