Transforming Growth Factor-β Drives the Transendothelial Migration of Hepatocellular Carcinoma Cells


Abstract
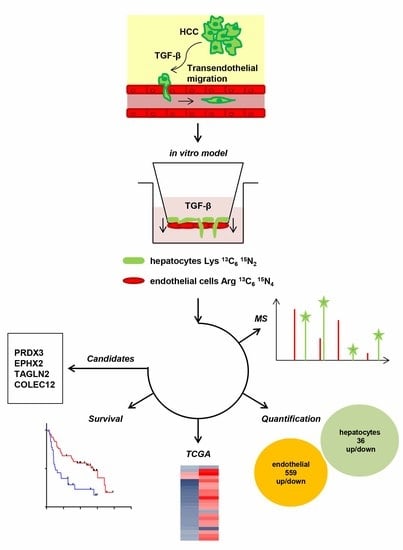
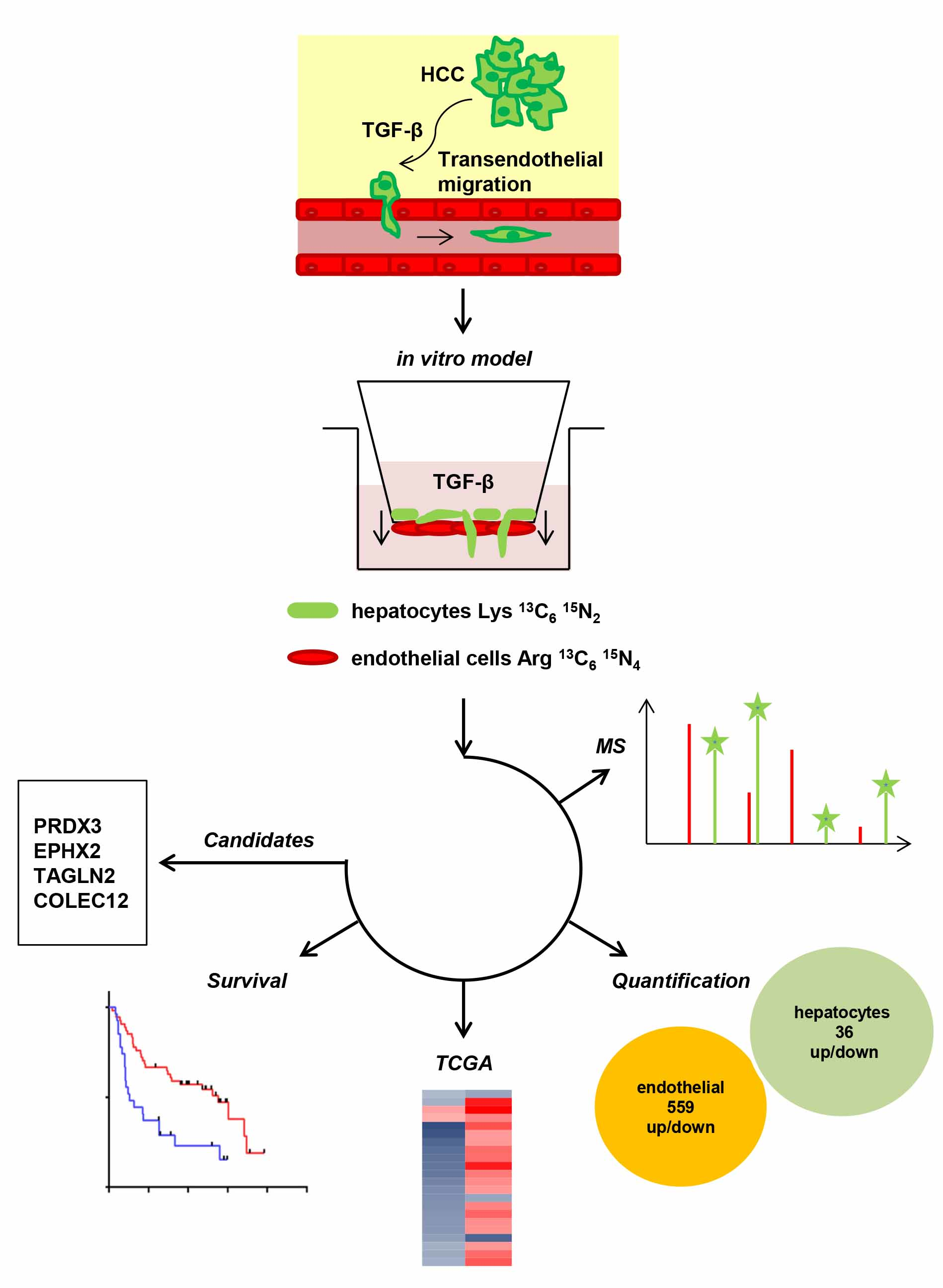
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
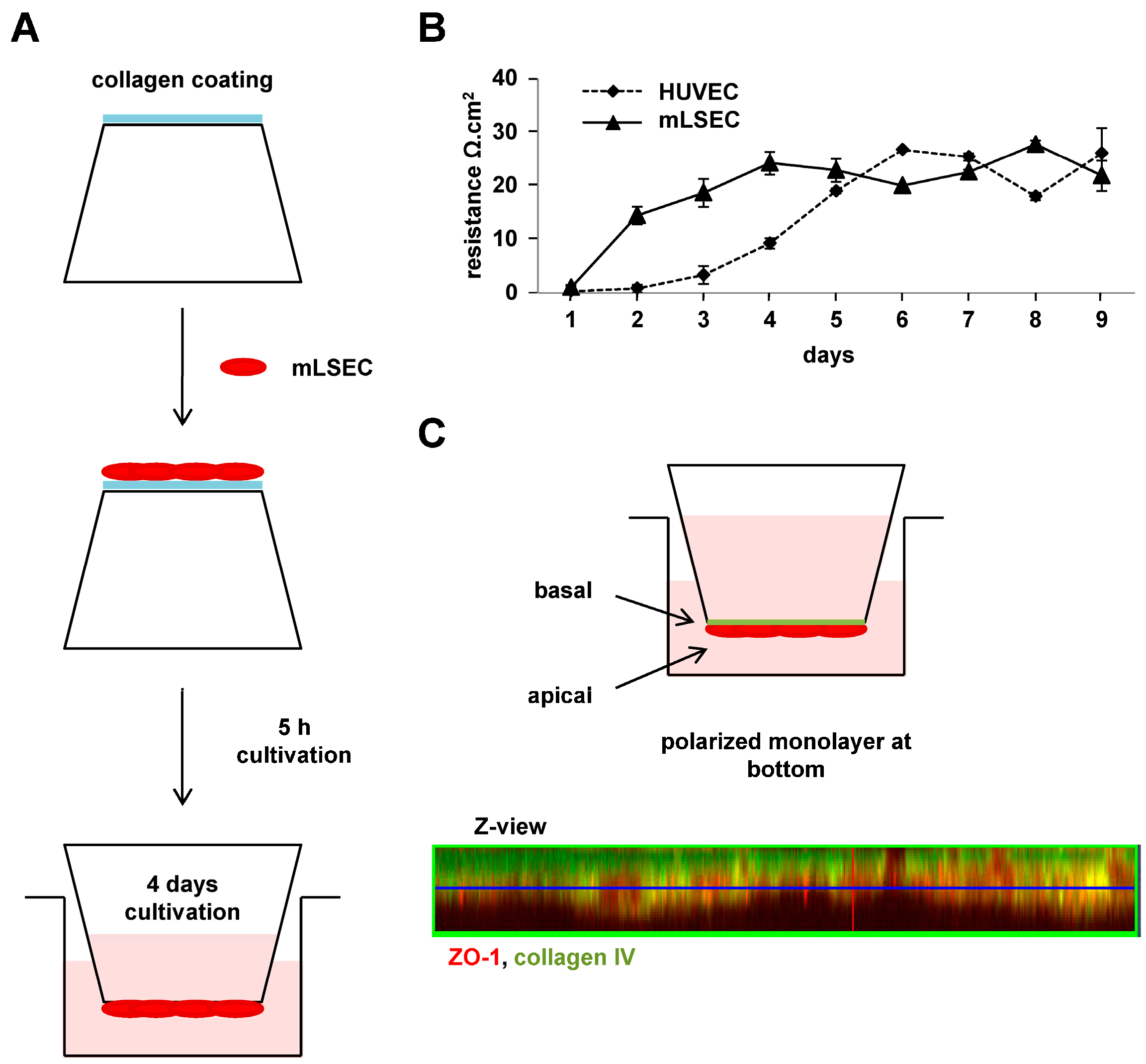
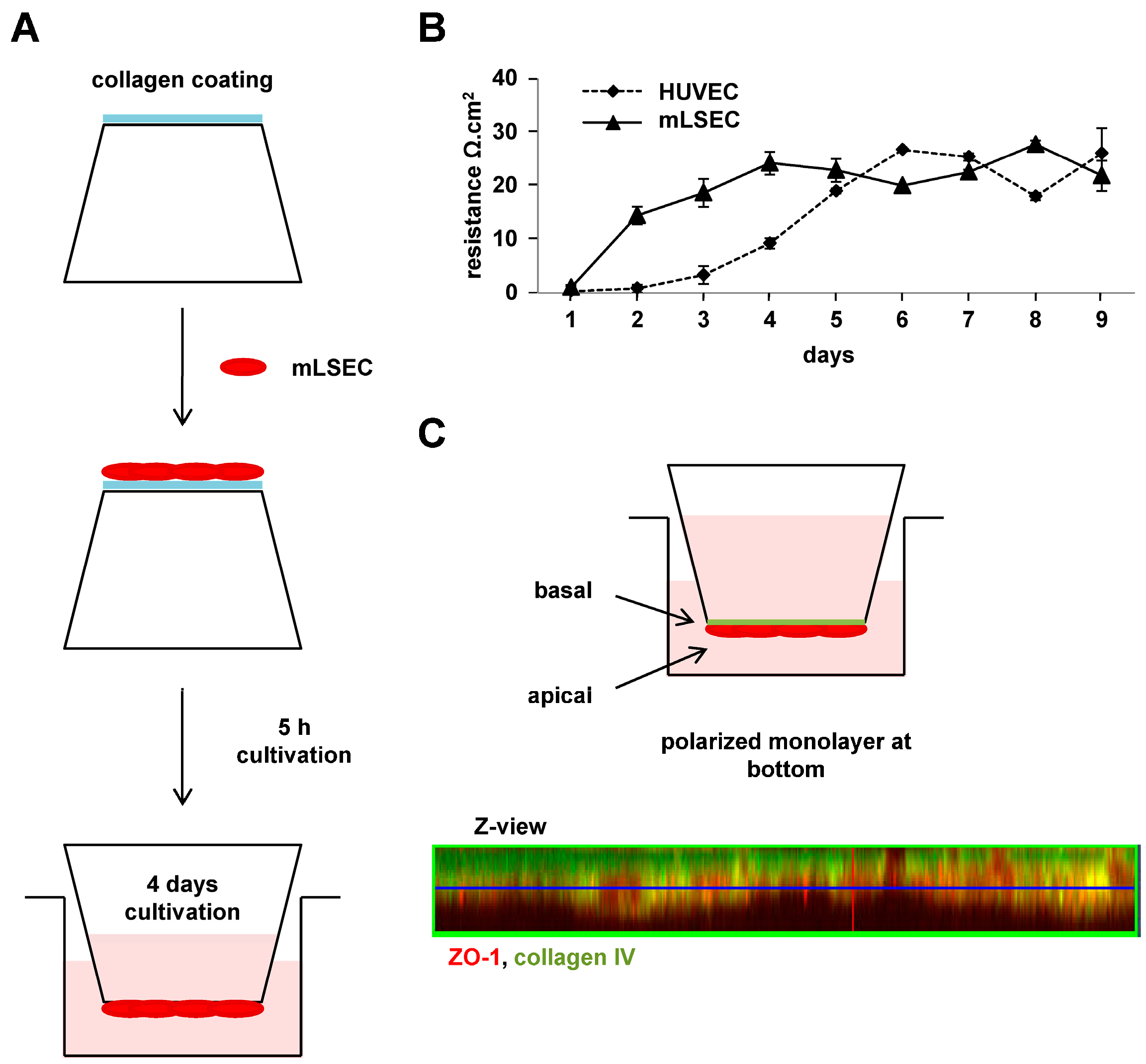
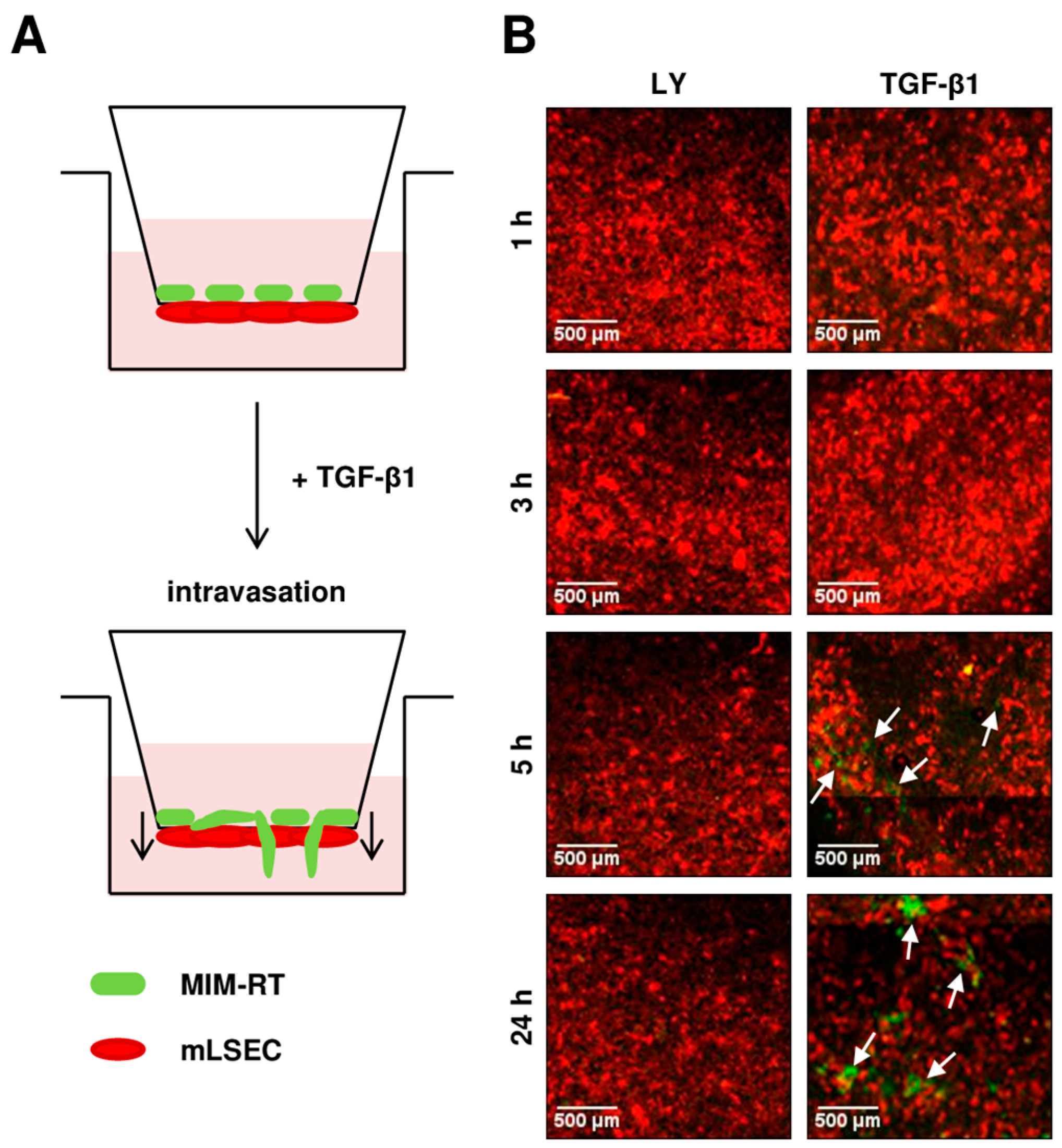
2.1. Integrity and Polarization of Liver Endothelial Cells for Vascular Invasion
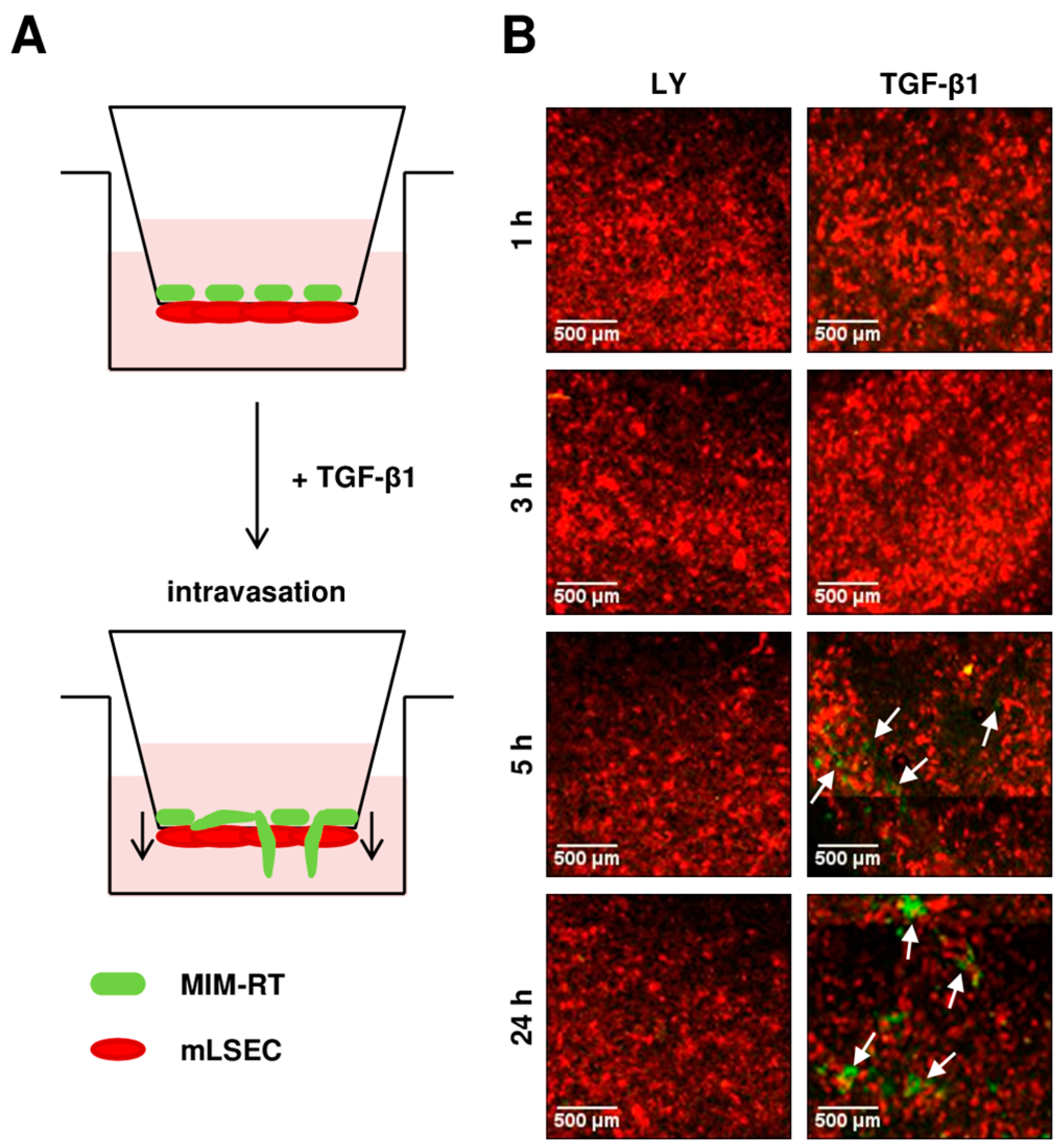
2.2. Transendothelial Migration Depends on TGF-β
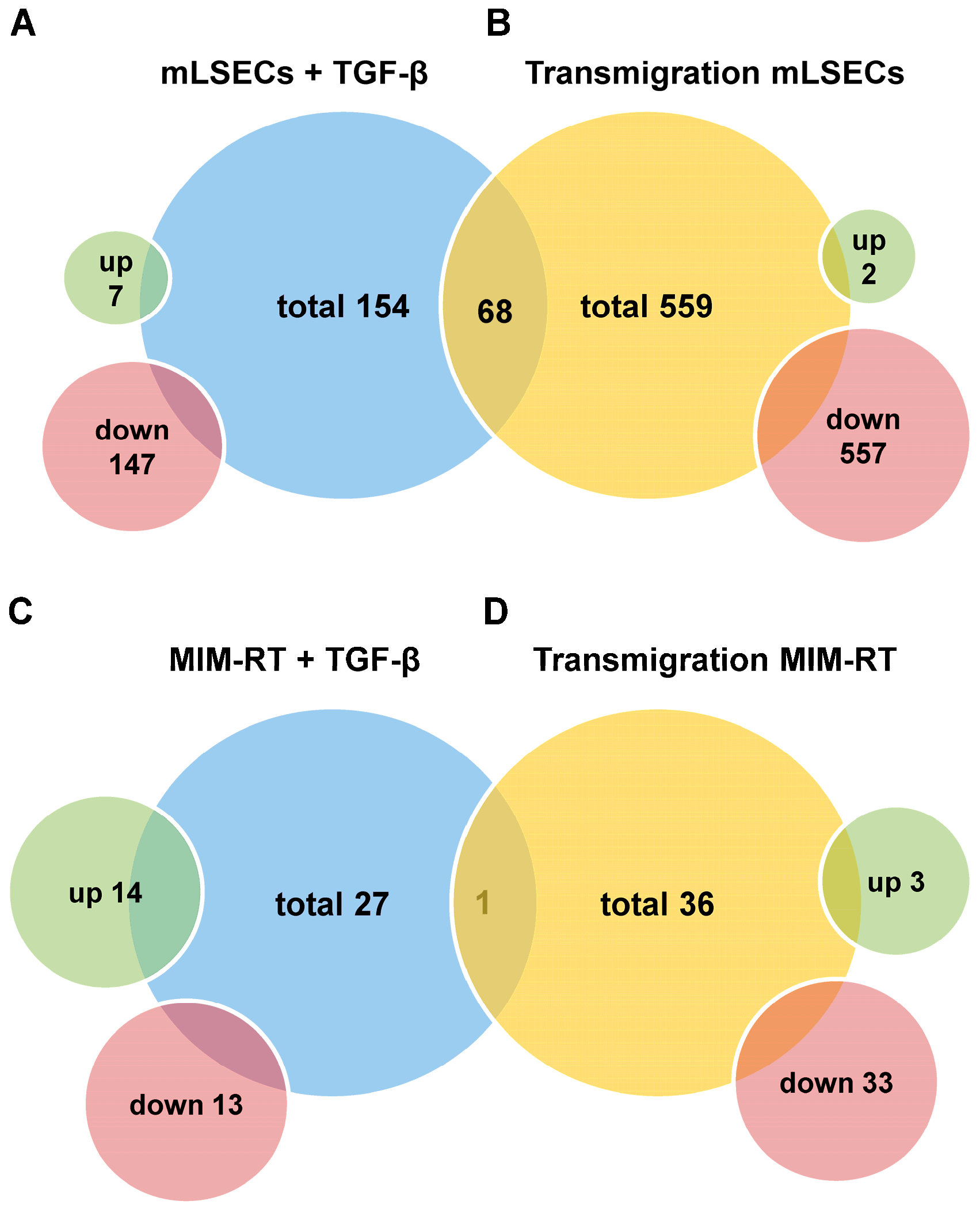
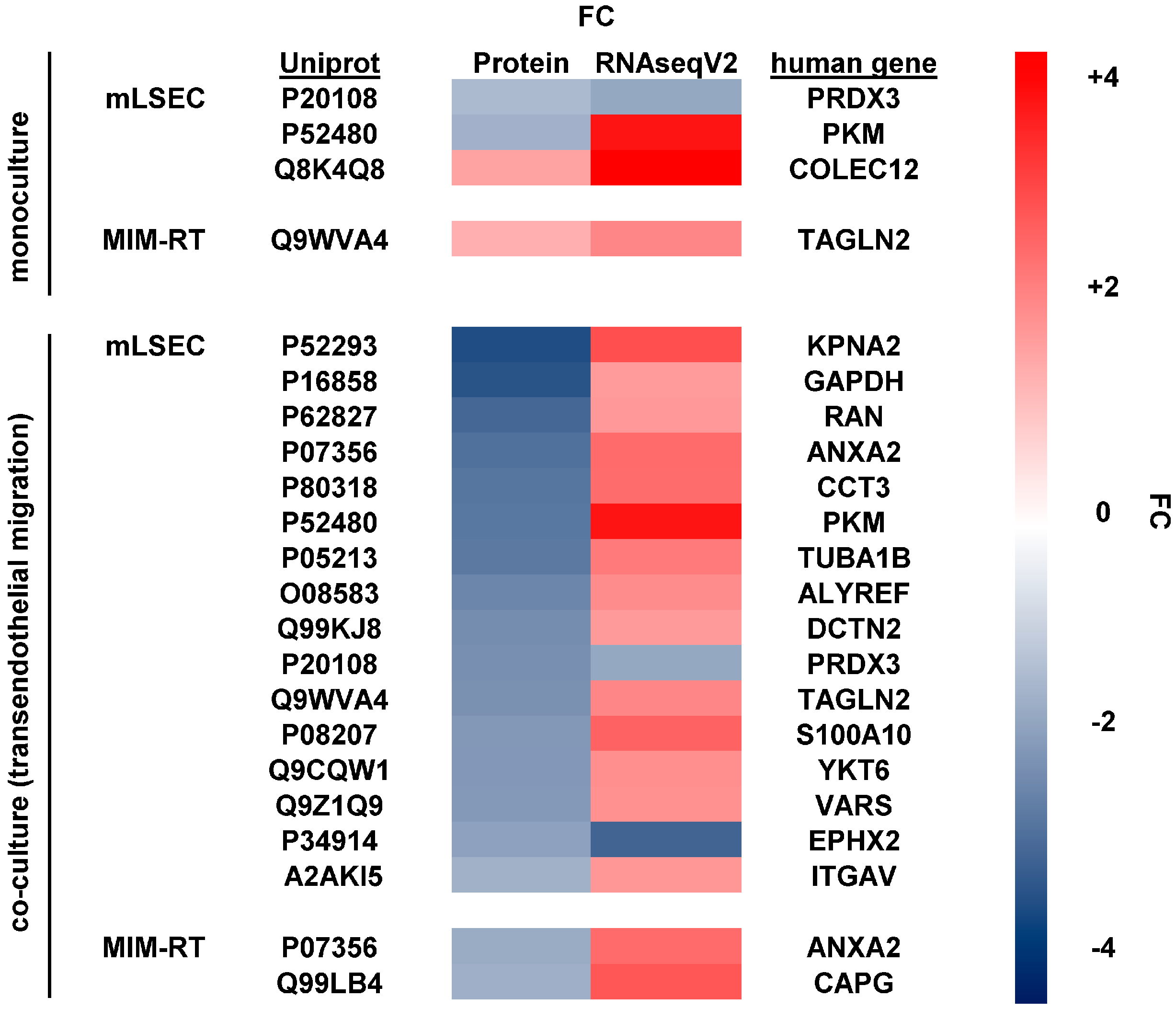
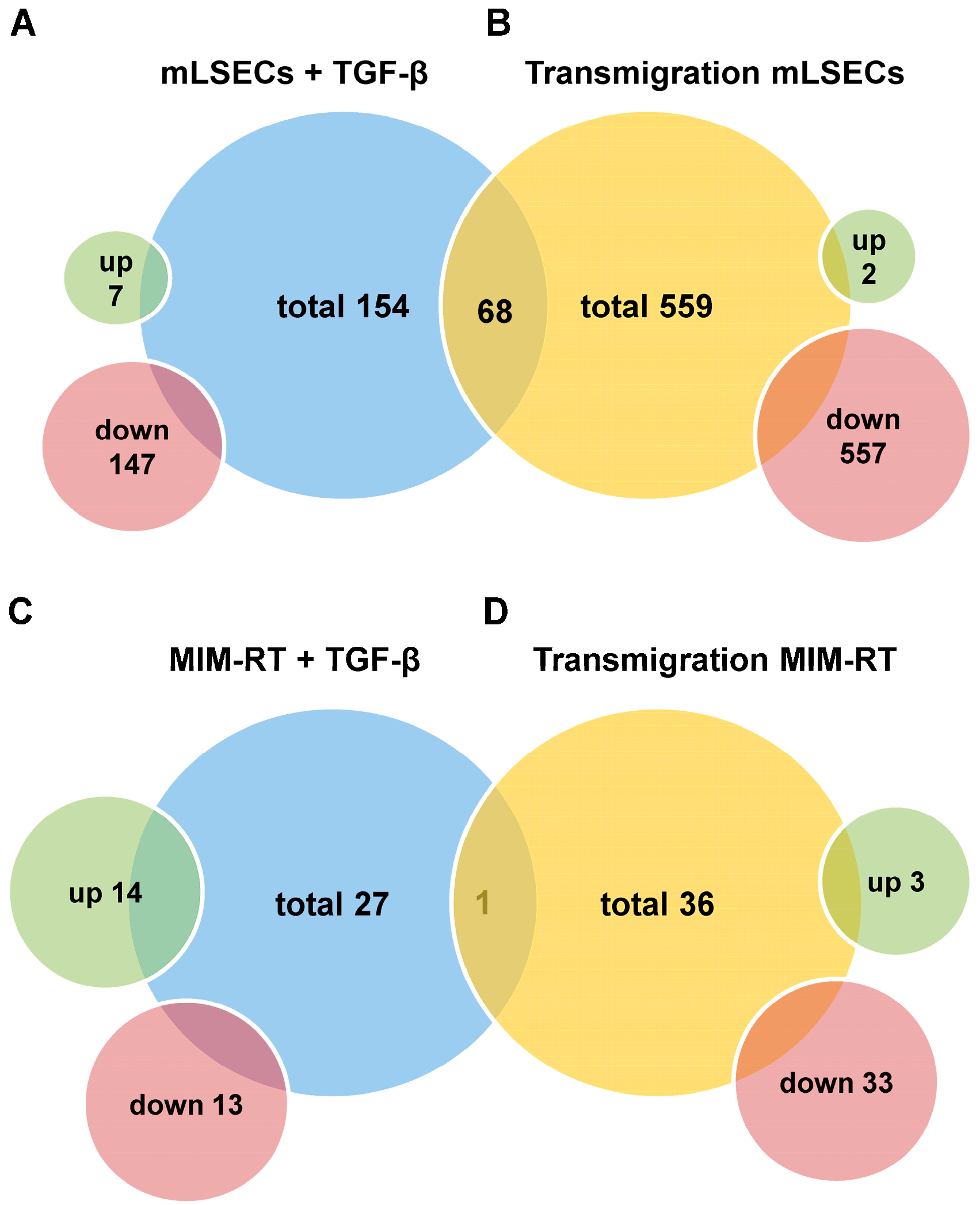
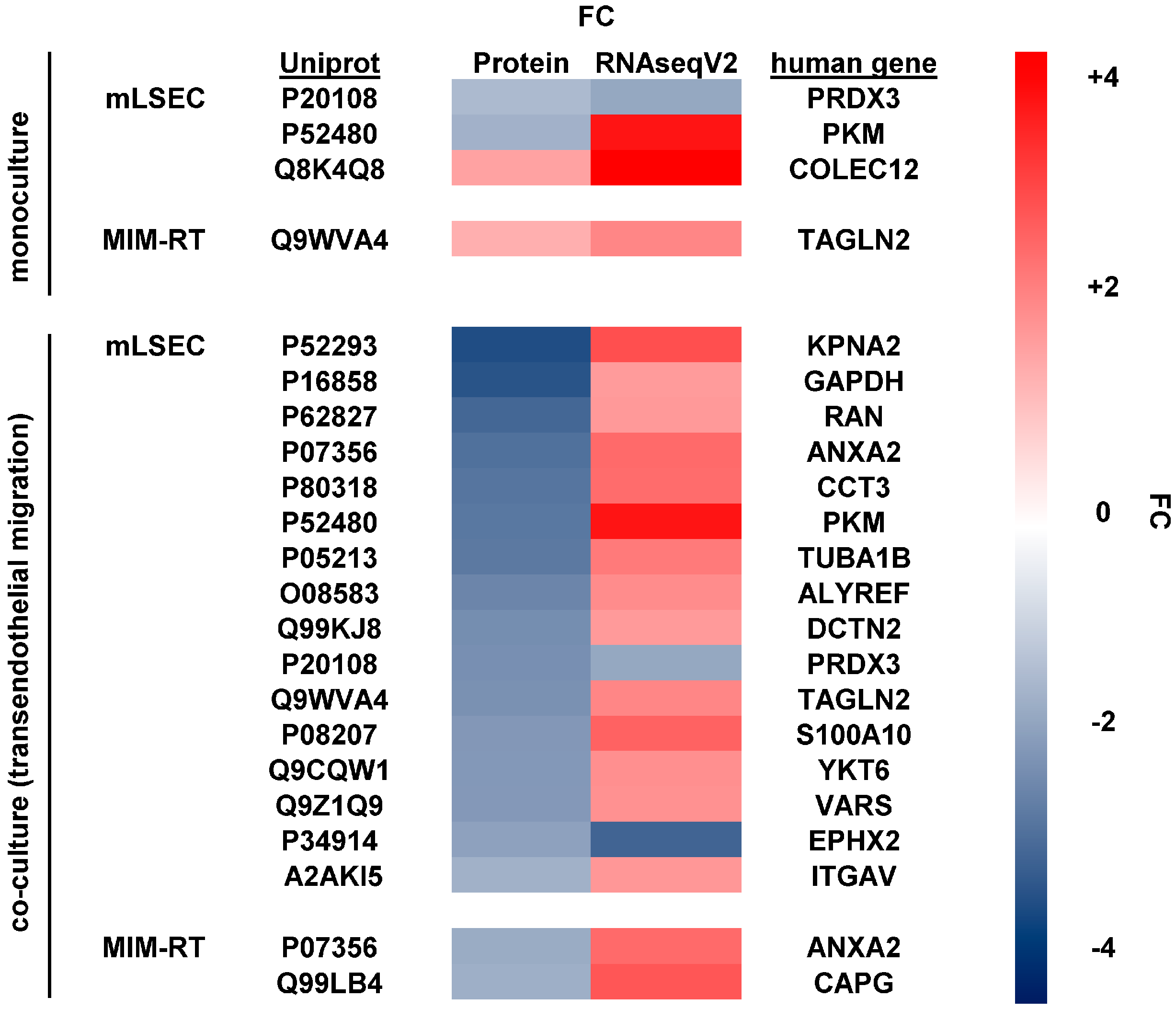
2.3. Molecular Alterations during Transmigration
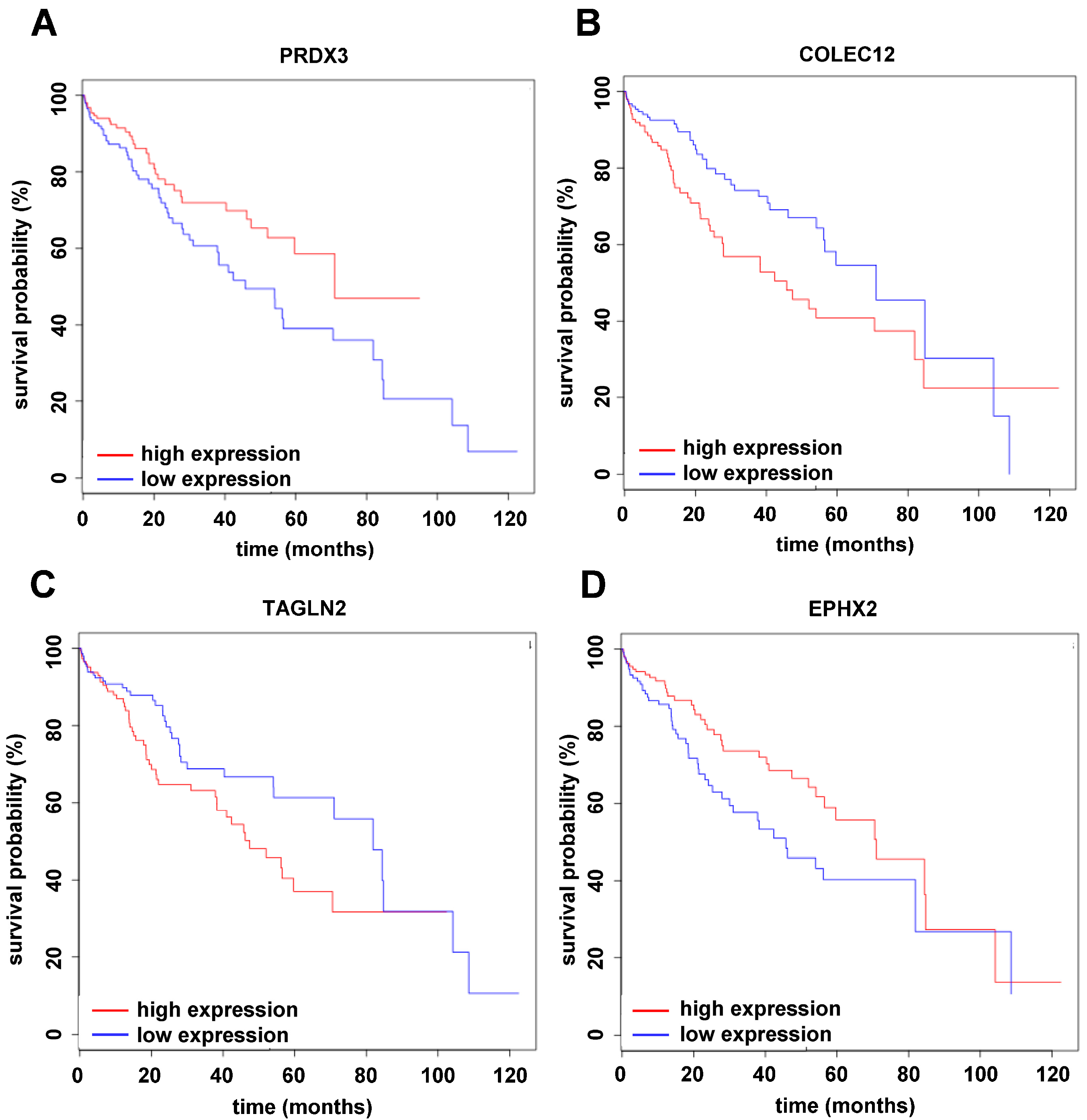
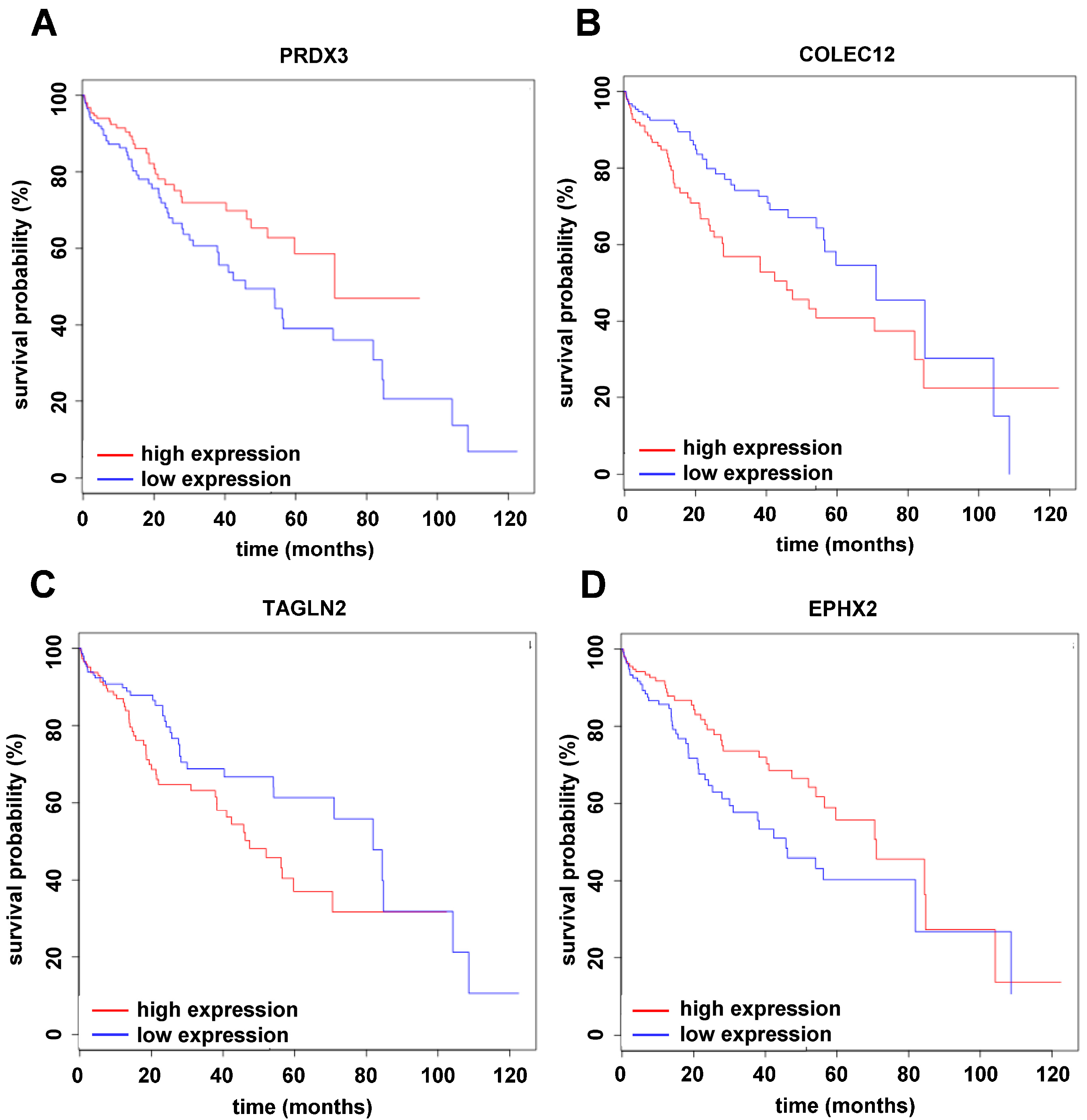
2.4. Translating Experimental Data to HCC Patients
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cultivation of Cells on Transwell Membrane
4.3. Transmigration Kinetics
4.4. Transendothelial Electrical Resistance
4.5. Confocal Immunofluoresecence Microscopy
4.6. Stable Isotope Labelling with Amino Acids (SILAC)
4.7. Electrophoresis and In-Gel Digestion
4.8. Mass Spectrometry
4.9. Comparison of Differentially Expressed Genes with TCGA Data
4.10. Survival Analysis Using Differentially Expressed Genes
4.11. Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BCLC | Barcelona Clinic Liver Cancer |
| EMT | Epithelial to mesenchymal transition |
| COLEC12 | Collectin 12 |
| EPHX2 | Epoxide hydrolase |
| GFP | Green fluorescent protein |
| HCC | Hepatocellular carcinoma |
| HUVEC | Human umbilical vein endothelial cell |
| MLC | Myosin light chain |
| mLSEC | Murine liver sinusoidal endothelial cell |
| PRDX3 | Peroxiredoxin-3 |
| RFP | Red fluorescent protein |
| ROS | Reactive oxygen species |
| SILAC | Stable isotope labelling with amino acids |
| TCGA | The cancer genome atlas |
| TEER | Transendothelial electrical resistance |
| TGF | Transforming growth factor |
| TAGLN2 | Transgelin-2 |
| VEGF | Vascular endothelial growth factor |
| ZO-1 | Zonula occludens 1 |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. Biomed. Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gonzalez, A.; Reig, M.; Bruix, J. Treatment of Hepatocellular Carcinoma. Dig. Dis. 2016, 34, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Pons, F.; Varela, M.; Llovet, J.M. Staging systems in hepatocellular carcinoma. HPB 2005, 7, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhu, M.; Li, W.; Lin, B.; Dong, X.; Chen, Y.; Xie, X.; Guo, J.; Li, M. Alpha fetoprotein plays a critical role in promoting metastasis of hepatocellular carcinoma cells. J. Cell. Mol. Med. 2016, 20, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Peterse, J.L.; van’t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Chang, S.H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Mierke, C.T. Cancer cells regulate biomechanical properties of human microvascular endothelial cells. J. Biol. Chem. 2011, 286, 40025–40037. [Google Scholar] [CrossRef] [PubMed]
- Minder, P.; Zajac, E.; Quigley, J.P.; Deryugina, E.I. EGFR regulates the development and microarchitecture of intratumoral angiogenic vasculature capable of sustaining cancer cell intravasation. Neoplasia 2015, 17, 634–649. [Google Scholar] [CrossRef] [PubMed]
- Roh-Johnson, M.; Bravo-Cordero, J.J.; Patsialou, A.; Sharma, V.P.; Guo, P.; Liu, H.; Hodgson, L.; Condeelis, J. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene 2014, 33, 4203–4212. [Google Scholar] [CrossRef] [PubMed]
- Haidari, M.; Zhang, W.; Wakame, K. Disruption of endothelial adherens junction by invasive breast cancer cells is mediated by reactive oxygen species and is attenuated by AHCC. Life Sci. 2013, 93, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [PubMed]
- Khuon, S.; Liang, L.; Dettman, R.W.; Sporn, P.H.; Wysolmerski, R.B.; Chew, T.L. Myosin light chain kinase mediates transcellular intravasation of breast cancer cells through the underlying endothelial cells: A three-dimensional FRET study. J. Cell Sci. 2010, 123, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Massague, J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Padua, D.; Zhang, X.H.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massague, J. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.R.; Tai, Y.; Jin, Y.; Hammell, M.C.; Wilkinson, J.E.; Roe, J.S.; Vakoc, C.R.; Van Aelst, L. TGF-β/Smad signaling through DOCK4 facilitates lung adenocarcinoma metastasis. Genes Dev. 2015, 29, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Clements, R.T.; Minnear, F.L.; Singer, H.A.; Keller, R.S.; Vincent, P.A. RhoA and Rho-kinase dependent and independent signals mediate TGF-β-induced pulmonary endothelial cytoskeletal reorganization and permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L294–L306. [Google Scholar] [CrossRef] [PubMed]
- Fransvea, E.; Mazzocca, A.; Antonaci, S.; Giannelli, G. Targeting transforming growth factor (TGF)-βRI inhibits activation of β1 integrin and blocks vascular invasion in hepatocellular carcinoma. Hepatology 2009, 49, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.N.; Herrera, B.; Mikula, M.; Proell, V.; Fuchs, E.; Gotzmann, J.; Schulte-Hermann, R.; Beug, H.; Mikulits, W. Integration of Ras subeffector signaling in TGF-β mediated late stage hepatocarcinogenesis. Carcinogenesis 2005, 26, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.N.; Fuchs, E.; Mikula, M.; Huber, H.; Beug, H.; Mikulits, W. PDGF essentially links TGF-β signaling to nuclear β-catenin accumulation in hepatocellular carcinoma progression. Oncogene 2007, 26, 3395–3405. [Google Scholar] [CrossRef] [PubMed]
- Koudelkova, P.; Weber, G.; Mikulits, W. Liver Sinusoidal Endothelial Cells Escape Senescence by Loss of p19ARF. PLoS ONE 2015, 10, e0142134. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.E.; Ballermann, B.J. Inhibition of capillary morphogenesis and associated apoptosis by dominant negative mutant transforming growth factor-β receptors. J. Biol. Chem. 1995, 270, 21144–21150. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming growth factor-β 1 (TGF-β1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 2009, 219, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Moon, A. Identification of Biomarkers for Breast Cancer Using Databases. J. Cancer Prev. 2016, 21, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Guell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.Z.; Kim, H.J.; Kang, S.W.; Rhee, S.G. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res. Clin. Pract. 1999, 45, 101–112. [Google Scholar] [CrossRef]
- Choi, J.H.; Kim, T.N.; Kim, S.; Baek, S.H.; Kim, J.H.; Lee, S.R.; Kim, J.R. Overexpression of mitochondrial thioredoxin reductase and peroxiredoxin III in hepatocellular carcinomas. Anticancer Res. 2002, 22, 3331–3335. [Google Scholar] [PubMed]
- Qiao, B.; Wang, J.; Xie, J.; Niu, Y.; Ye, S.; Wan, Q.; Ye, Q. Detection and identification of peroxiredoxin 3 as a biomarker in hepatocellular carcinoma by a proteomic approach. Int. J. Mol. Med. 2012, 29, 832–840. [Google Scholar] [PubMed]
- Li, L.; Yu, A.Q. The functional role of peroxiredoxin 3 in reactive oxygen species, apoptosis, and chemoresistance of cancer cells. J. Cancer Res. Clin. Oncol. 2015, 141, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.G.; Li, L.; Liu, C.H.; Hong, S.; Zhang, M.J. Peroxiredoxin 3 is resistant to oxidation-induced apoptosis of Hep-3b cells. Clin. Transl. Oncol. 2014, 16, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Jiang, B.; Zhang, A.; Qian, Y.; Tan, H.; Gao, J.; Shao, C.; Gong, Y. Accelerated hepatocellular carcinoma development in CUL4B transgenic mice. Oncotarget 2015, 6, 15209–15221. [Google Scholar] [CrossRef] [PubMed]
- Isohookana, J.; Haapasaari, K.M.; Soini, Y.; Karihtala, P. Loss of Peroxiredoxin Expression Is Associated with an Aggressive Phenotype in Pancreatic Adenocarcinoma. Anticancer Res. 2016, 36, 427–433. [Google Scholar] [PubMed]
- Adebayo Michael, A.O.; Ahsan, N.; Zabala, V.; Francois-Vaughan, H.; Post, S.; Brilliant, K.E.; Salomon, A.R.; Sanders, J.A.; Gruppuso, P.A. Proteomic analysis of laser capture microdissected focal lesions in a rat model of progenitor marker-positive hepatocellular carcinoma. Oncotarget 2017, 8, 26041–26056. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.A.; McKay, M.J.; Molloy, M.P. Proteomics of Smad4 regulated transforming growth factor-β signalling in colon cancer cells. Mol. BioSyst. 2010, 6, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.Y.; Wang, H.C.; Yin, Y.H.; Sun, W.S.; Li, Y.; Zhang, C.Q.; Wang, Y.; Wang, S.; Chen, W.F. Identification and analysis of tumour-associated antigens in hepatocellular carcinoma. Br. J. Cancer 2005, 92, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.J.; Hein, E.; Munthe-Fog, L.; Skjoedt, M.O.; Bayarri-Olmos, R.; Romani, L.; Garred, P. Soluble Collectin-12 (CL-12) Is a Pattern Recognition Molecule Initiating Complement Activation via the Alternative Pathway. J. Immunol. 2015, 195, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Coombs, P.J.; Graham, S.A.; Drickamer, K.; Taylor, M.E. Selective binding of the scavenger receptor C-type lectin to Lewisx trisaccharide and related glycan ligands. J. Biol. Chem. 2005, 280, 22993–22999. [Google Scholar] [CrossRef] [PubMed]
- Worthley, D.L.; Bardy, P.G.; Gordon, D.L.; Mullighan, C.G. Mannose-binding lectin and maladies of the bowel and liver. World J. Gastroenterol. 2006, 12, 6420–6428. [Google Scholar] [CrossRef] [PubMed]
- Hensler, M.; Vancurova, I.; Becht, E.; Palata, O.; Strnad, P.; Tesarova, P.; Cabinakova, M.; Svec, D.; Kubista, M.; Bartunkova, J.; et al. Gene expression profiling of circulating tumor cells and peripheral blood mononuclear cells from breast cancer patients. Oncoimmunology 2016, 5, e1102827. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Teo, G.; Krueger, S.; Rock, T.M.; Koh, H.W.; Choi, H.; Vogel, C. Differential dynamics of the mammalian mRNA and protein expression response to misfolding stress. Mol. Syst. Biol. 2016, 12, 855. [Google Scholar] [CrossRef] [PubMed]
- Steen, H.; Jebanathirajah, J.A.; Rush, J.; Morrice, N.; Kirschner, M.W. Phosphorylation analysis by mass spectrometry: Myths, facts, and the consequences for qualitative and quantitative measurements. Mol. Cell. Proteom. 2006, 5, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Gotzmann, J.; Huber, H.; Thallinger, C.; Wolschek, M.; Jansen, B.; Schulte-Hermann, R.; Beug, H.; Mikulits, W. Hepatocytes convert to a fibroblastoid phenotype through the cooperation of TGF-β1 and Ha-Ras: Steps towards invasiveness. J. Cell Sci. 2002, 115, 1189–1202. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koudelkova, P.; Costina, V.; Weber, G.; Dooley, S.; Findeisen, P.; Winter, P.; Agarwal, R.; Schlangen, K.; Mikulits, W. Transforming Growth Factor-β Drives the Transendothelial Migration of Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2017, 18, 2119. https://doi.org/10.3390/ijms18102119
Koudelkova P, Costina V, Weber G, Dooley S, Findeisen P, Winter P, Agarwal R, Schlangen K, Mikulits W. Transforming Growth Factor-β Drives the Transendothelial Migration of Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences. 2017; 18(10):2119. https://doi.org/10.3390/ijms18102119
Chicago/Turabian StyleKoudelkova, Petra, Victor Costina, Gerhard Weber, Steven Dooley, Peter Findeisen, Peter Winter, Rahul Agarwal, Karin Schlangen, and Wolfgang Mikulits. 2017. "Transforming Growth Factor-β Drives the Transendothelial Migration of Hepatocellular Carcinoma Cells" International Journal of Molecular Sciences 18, no. 10: 2119. https://doi.org/10.3390/ijms18102119
APA StyleKoudelkova, P., Costina, V., Weber, G., Dooley, S., Findeisen, P., Winter, P., Agarwal, R., Schlangen, K., & Mikulits, W. (2017). Transforming Growth Factor-β Drives the Transendothelial Migration of Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences, 18(10), 2119. https://doi.org/10.3390/ijms18102119