The Emerging Role of Zinc in the Pathogenesis of Multiple Sclerosis



Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Zinc in the Central Nervous System
3. The Molecular Mechanisms of Zinc-Induced Neurotoxicity
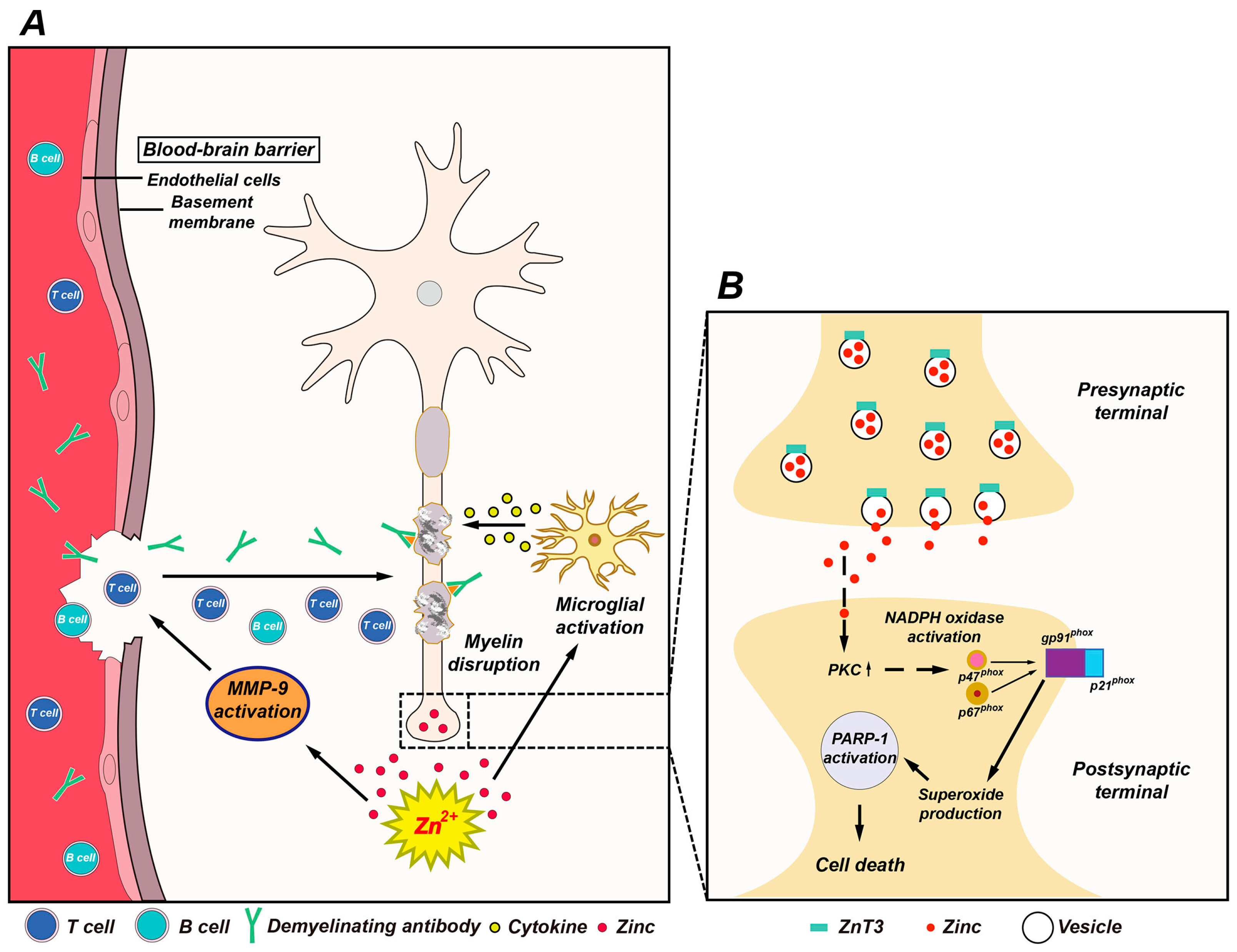
4. The Possible Role of Zinc in MS
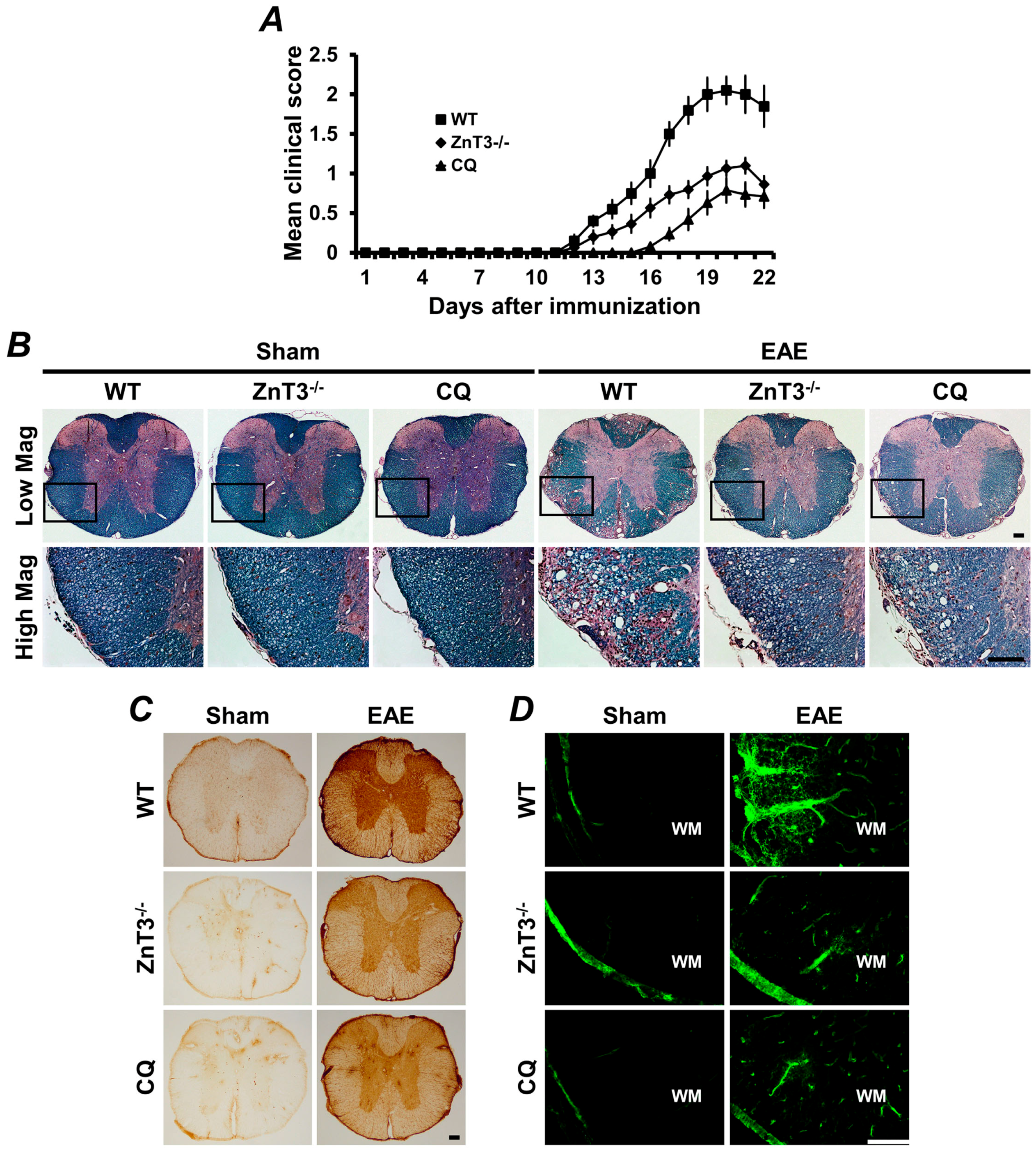
4.1. Vesicular Zinc as a Key Mediator of Neuronal Death in MS
4.2. Role of Zinc in Oligodendrocyte Death
5. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2002, 359, 1221–1231. [Google Scholar] [CrossRef]
- Baranzini, S.E.; Mudge, J.; van Velkinburgh, J.C.; Khankhanian, P.; Khrebtukova, I.; Miller, N.A.; Zhang, L.; Farmer, A.D.; Bell, C.J.; Kim, R.W.; et al. Genome, epigenome and rna sequences of monozygotic twins discordant for multiple sclerosis. Nature 2010, 464, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Mentis, A.A.; Dardiotis, E.; Grigoriadis, N.; Petinaki, E.; Hadjigeorgiou, G.M. Viruses and endogenous retroviruses in multiple sclerosis: From correlation to causation. Acta Neurol. Scand. 2017. [Google Scholar] [CrossRef] [PubMed]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantinescu, C.S.; Zhang, W.; Tench, C.; Gran, B. The association between human endogenous retroviruses and multiple sclerosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0172415. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Tumani, H.; Kiechle, U.; Muche, R.; Richards, G.; Lehmensiek, V.; Ludolph, A.C.; Otto, M. Igg antibodies against measles, rubella, and varicella zoster virus predict conversion to multiple sclerosis in clinically isolated syndrome. PLoS ONE 2009, 4, e7638. [Google Scholar] [CrossRef] [PubMed]
- Pormohammad, A.; Azimi, T.; Falah, F.; Faghihloo, E. Relationship of human herpes virus 6 and multiple sclerosis: A systematic review and meta-analysis. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Koskderelioglu, A.; Afsar, I.; Pektas, B.; Gedizlioglu, M. Is toxoplasma gondii infection protective against multiple sclerosis risk? Mult. Scler. Relat. Disord. 2017, 15, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.S.; Hasseldam, H.; Bacher, I.H.; Thamsborg, S.M.; Johansen, F.F.; Kringel, H. Trichuris suis secrete products that reduce disease severity in a multiple sclerosis model. Acta Parasitol. 2017, 62, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Belbasis, L.; Bellou, V.; Evangelou, E.; Ioannidis, J.P.; Tzoulaki, I. Environmental risk factors and multiple sclerosis: An umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 2015, 14, 263–273. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. The role of oxidative stress in the pathogenesis of multiple sclerosis: The need for effective antioxidant therapy. J. Neurol. 2004, 251, 261–268. [Google Scholar] [PubMed]
- Bitsch, A.; Bruck, W. Differentiation of multiple sclerosis subtypes. CNS Drugs 2002, 16, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (eae) as a model for multiple sclerosis (ms). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.D.; Karpus, W.J. Experimental autoimmune encephalomyelitis in the mouse. Curr. Protoc. Immunol. 2007, 15.1.1–15.1.18. [Google Scholar]
- Ellwardt, E.; Zipp, F. Molecular mechanisms linking neuroinflammation and neurodegeneration in ms. Exp. Neurol. 2014, 262 Pt A, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J. Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 1989, 31, 145–238. [Google Scholar] [PubMed]
- Frederickson, C.J.; Danscher, G. Zinc-containing neurons in hippocampus and related cns structures. Prog. Brain Res. 1990, 83, 71–84. [Google Scholar] [PubMed]
- Bonaventura, P.; Benedetti, G.; Albarede, F.; Miossec, P. Zinc and its role in immunity and inflammation. Autoimmun. Rev. 2015, 14, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Cousins, R.J.; Liuzzi, J.P.; Lichten, L.A. Mammalian zinc transport, trafficking, and signals. J. Biol. Chem. 2006, 281, 24085–24089. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the cns. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kambe, T. The functions of metallothionein and zip and znt transporters: An overview and perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tepaamorndech, S. The SLC30 family of zinc transporters—A review of current understanding of their biological and pathophysiological roles. Mol. Aspects Med. 2013, 34, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Eide, D.J. The SLC39 family of zinc transporters. Mol. Aspects Med. 2013, 34, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Maret, W. Thionein/metallothionein control Zn(II) availability and the activity of enzymes. J. Biol. Inorg. Chem. 2008, 13, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Guan, Z.; Franklin, R.B.; Feng, P. Metallothionein can function as a chaperone for zinc uptake transport into prostate and liver mitochondria. J. Inorg. Biochem. 2004, 98, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D. Molecular biology of metallothionein gene expression. Experientia. Suppl. 1987, 52, 63–80. [Google Scholar] [PubMed]
- Palmiter, R.D.; Findley, S.D.; Whitmore, T.E.; Durnam, D.M. Mt-iii, a brain-specific member of the metallothionein gene family. Proc. Natl. Acad. Sci. USA 1992, 89, 6333–6337. [Google Scholar] [CrossRef] [PubMed]
- Masters, B.A.; Quaife, C.J.; Erickson, J.C.; Kelly, E.J.; Froelick, G.J.; Zambrowicz, B.P.; Brinster, R.L.; Palmiter, R.D. Metallothionein III is expressed in neurons that sequester zinc in synaptic vesicles. J. Neurosci. 1994, 14, 5844–5857. [Google Scholar] [PubMed]
- Quaife, C.J.; Findley, S.D.; Erickson, J.C.; Froelick, G.J.; Kelly, E.J.; Zambrowicz, B.P.; Palmiter, R.D. Induction of a new metallothionein isoform (MT-IV) occurs during differentiation of stratified squamous epithelia. Biochemistry 1994, 33, 7250–7259. [Google Scholar] [CrossRef] [PubMed]
- Penkowa, M.; Espejo, C.; Martinez-Caceres, E.M.; Poulsen, C.B.; Montalban, X.; Hidalgo, J. Altered inflammatory response and increased neurodegeneration in metallothionein I+II deficient mice during experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2001, 119, 248–260. [Google Scholar] [CrossRef]
- McAllister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Cole, T.B.; Quaife, C.J.; Findley, S.D. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc. Natl. Acad. Sci. USA 1996, 93, 14934–14939. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, H.J.; Cole, T.B.; Born, D.E.; Schwartzkroin, P.A.; Palmiter, R.D. Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc Natl. Acad. Sci. USA 1997, 94, 12676–12681. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W. Detection of zinc translocation into apical dendrite of ca1 pyramidal neuron after electrical stimulation. J. Neurosci. Methods 2009, 177, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Linkous, D.H.; Flinn, J.M.; Koh, J.Y.; Lanzirotti, A.; Bertsch, P.M.; Jones, B.F.; Giblin, L.J.; Frederickson, C.J. Evidence that the ZnT3 protein controls the total amount of elemental zinc in synaptic vesicles. J. Histochem. Cytochem. 2008, 56, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.A.; Welch, M.G.; Frederickson, C.J. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature 1984, 308, 736–738. [Google Scholar] [CrossRef] [PubMed]
- Assaf, S.Y.; Chung, S.H. Release of endogenous Zn2+ from brain tissue during activity. Nature 1984, 308, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Yin, H.Z.; Carriedo, S.G.; Rao, S.S.; Weiss, J.H. Preferential Zn2+ influx through Ca2+-permeable ampa/kainate channels triggers prolonged mitochondrial superoxide production. Proc. Natl. Acad. Sci. USA 1999, 96, 2414–2419. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.H.; Sensi, S.L.; Koh, J.Y. Zn2+: A novel ionic mediator of neural injury in brain disease. Trends Pharmacol. Sci. 2000, 21, 395–401. [Google Scholar] [CrossRef]
- Capasso, M.; Jeng, J.M.; Malavolta, M.; Mocchegiani, E.; Sensi, S.L. Zinc dyshomeostasis: A key modulator of neuronal injury. J. Alzheimers Dis. 2005, 8, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Garnier, P.; Aoyama, K.; Chen, Y.; Swanson, R.A. Zinc release contributes to hypoglycemia-induced neuronal death. Neurobiol. Dis. 2004, 16, 538–545. [Google Scholar] [PubMed]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal nadph oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Hernandez, M.D.; Goik, S.A.; Morton, J.D.; McGinty, J.F. Loss of zinc staining from hippocampal mossy fibers during kainic acid induced seizures: A histofluorescence study. Brain Res. 1988, 446, 383–386. [Google Scholar] [CrossRef]
- Tonder, N.; Johansen, F.F.; Frederickson, C.J.; Zimmer, J.; Diemer, N.H. Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci. Lett. 1990, 109, 247–252. [Google Scholar] [CrossRef]
- Suh, S.W.; Chen, J.W.; Motamedi, M.; Bell, B.; Listiak, K.; Pons, N.F.; Danscher, G.; Frederickson, C.J. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 2000, 852, 268–273. [Google Scholar] [CrossRef]
- Suh, S.W.; Hamby, A.M.; Gum, E.T.; Shin, B.S.; Won, S.J.; Sheline, C.T.; Chan, P.H.; Swanson, R.A. Sequential release of nitric oxide, zinc, and superoxide in hypoglycemic neuronal death. J. Cereb. Blood Flow Metab. 2008, 28, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.H.; Sensi, S.L. Ca2+-Zn2+ permeable ampa or kainate receptors: Possible key factors in selective neurodegeneration. Trends Neurosci. 2000, 23, 365–371. [Google Scholar] [CrossRef]
- Sensi, S.L.; Ton-That, D.; Sullivan, P.G.; Jonas, E.A.; Gee, K.R.; Kaczmarek, L.K.; Weiss, J.H. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc. Natl. Acad. Sci. USA 2003, 100, 6157–6162. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.M.; Koh, J.Y. Induction and activation by zinc of nadph oxidase in cultured cortical neurons and astrocytes. J. Neurosci. 2000, 20, RC111. [Google Scholar] [PubMed]
- Virag, L.; Szabo, C. The therapeutic potential of poly(adp-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef] [PubMed]
- Shuttleworth, C.W.; Weiss, J.H. Zinc: New clues to diverse roles in brain ischemia. Trends Pharmacol. Sci. 2011, 32, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Aizenman, E.; Stout, A.K.; Hartnett, K.A.; Dineley, K.E.; McLaughlin, B.; Reynolds, I.J. Induction of neuronal apoptosis by thiol oxidation: Putative role of intracellular zinc release. J. Neurochem. 2000, 75, 1878–1888. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Talantova, M.V.; Lee, W.D.; Scholzke, M.N.; Harrop, A.; Mathews, E.; Gotz, T.; Han, J.; Ellisman, M.H.; Perkins, G.A.; et al. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004, 41, 351–365. [Google Scholar] [CrossRef]
- Zhang, Y.; Aizenman, E.; DeFranco, D.B.; Rosenberg, P.A. Intracellular zinc release, 12-lipoxygenase activation and mapk dependent neuronal and oligodendroglial death. Mol. Med. 2007, 13, 350–355. [Google Scholar] [PubMed]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Koh, J.Y. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: Evidence against synaptic vesicle origin. J. Neurosci. 2000, 20, RC79. [Google Scholar] [PubMed]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Seo, J.N.; Wu, G.; Sohn, M.; Chung, T.N.; Suh, S.W. Copper/zinc chelation by clioquinol reduces spinal cord white matter damage and behavioral deficits in a murine MOG-induced multiple sclerosis model. Neurobiol. Dis. 2013, 54, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, J.H.; Kho, A.R.; Kim, I.Y.; Lee, S.H.; Lee, B.E.; Choi, E.; Sohn, M.; Stevenson, M.; Chung, T.N.; et al. Inhibition of nadph oxidase activation reduces eae-induced white matter damage in mice. J. Neuroinflam. 2015, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Koh, J.Y. The role of nadph oxidase and neuronal nitric oxide synthase in zinc-induced poly(adp-ribose) polymerase activation and cell death in cortical culture. Exp. Neurol. 2002, 177, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, I.Y.; Kim, J.H.; Kho, A.R.; Lee, S.H.; Lee, B.E.; Sohn, M.; Koh, J.Y.; Suh, S.W. Zinc transporter 3 (ZnT3) gene deletion reduces spinal cord white matter damage and motor deficits in a murine mog-induced multiple sclerosis model. Neurobiol. Dis. 2016, 94, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.K.; Fehlings, M.G. Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J. Neurosci. 1997, 17, 1055–1063. [Google Scholar] [PubMed]
- Mato, S.; Sanchez-Gomez, M.V.; Bernal-Chico, A.; Matute, C. Cytosolic zinc accumulation contributes to excitotoxic oligodendroglial death. Glia 2013, 61, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Kiedrowski, L. Cytosolic acidification and intracellular zinc release in hippocampal neurons. J. Neurochem. 2012, 121, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Mato, S.; Soria, F.N.; Sanchez-gomez, M.V.; Alberdi, E.; Matute, C. Zn2+-induced erk activation mediates parp-1-dependent ischemic-reoxygenation damage to oligodendrocytes. Glia 2013, 61, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.; Li, J.; Dong, L.; Xu, P.; Chen, W.; Neve, R.L.; Volpe, J.J.; Rosenberg, P.A. Intracellular zinc release and erk phosphorylation are required upstream of 12-lipoxygenase activation in peroxynitrite toxicity to mature rat oligodendrocytes. J. Biol. Chem. 2006, 281, 9460–9470. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Vana, A.C.; Ribeiro, R.; Zhang, Y. Distinct role of nitric oxide and peroxynitrite in mediating oligodendrocyte toxicity in culture and in experimental autoimmune encephalomyelitis. Neuroscience 2011, 184, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.S.; Zhao, M.L.; Brosnan, C.F.; Lee, S.C. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am. J. Pathol. 2001, 158, 2057–2066. [Google Scholar] [CrossRef]
- Calabrese, V.; Scapagnini, G.; Ravagna, A.; Bella, R.; Foresti, R.; Bates, T.E.; Giuffrida Stella, A.M.; Pennisi, G. Nitric oxide synthase is present in the cerebrospinal fluid of patients with active multiple sclerosis and is associated with increases in cerebrospinal fluid protein nitrotyrosine and S-nitrosothiols and with changes in glutathione levels. J. Neurosci. Res. 2002, 70, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lin, W.; Tchantchou, F.; Lai, R.; Wen, J.; Zhang, Y. Protein kinase C mediates peroxynitrite toxicity to oligodendrocytes. Mol. Cell. Neurosci. 2011, 48, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.M.; Kim, Y.H.; Koh, J.Y. Mediation by membrane protein kinase C of zinc-induced oxidative neuronal injury in mouse cortical cultures. J. Neurochem. 1999, 72, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, B.Y.; Jung, J.W.; Suh, S.W. The Emerging Role of Zinc in the Pathogenesis of Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 2070. https://doi.org/10.3390/ijms18102070
Choi BY, Jung JW, Suh SW. The Emerging Role of Zinc in the Pathogenesis of Multiple Sclerosis. International Journal of Molecular Sciences. 2017; 18(10):2070. https://doi.org/10.3390/ijms18102070
Chicago/Turabian StyleChoi, Bo Young, Jong Won Jung, and Sang Won Suh. 2017. "The Emerging Role of Zinc in the Pathogenesis of Multiple Sclerosis" International Journal of Molecular Sciences 18, no. 10: 2070. https://doi.org/10.3390/ijms18102070
APA StyleChoi, B. Y., Jung, J. W., & Suh, S. W. (2017). The Emerging Role of Zinc in the Pathogenesis of Multiple Sclerosis. International Journal of Molecular Sciences, 18(10), 2070. https://doi.org/10.3390/ijms18102070