Blocking of the Ubiquitin-Proteasome System Prevents Inflammation-Induced Bone Loss by Accelerating M-CSF Receptor c-Fms Degradation in Osteoclast Differentiation

Abstract
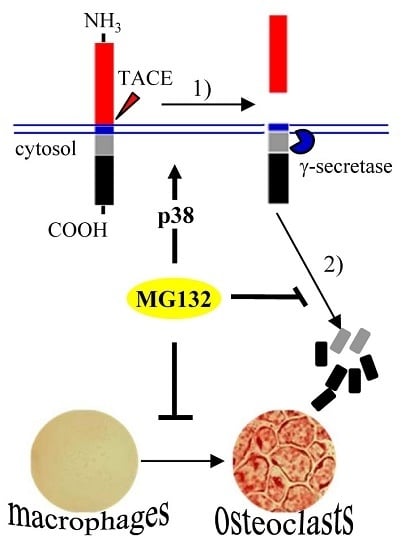
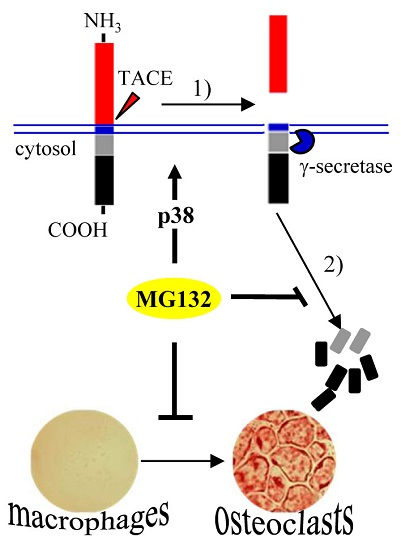{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
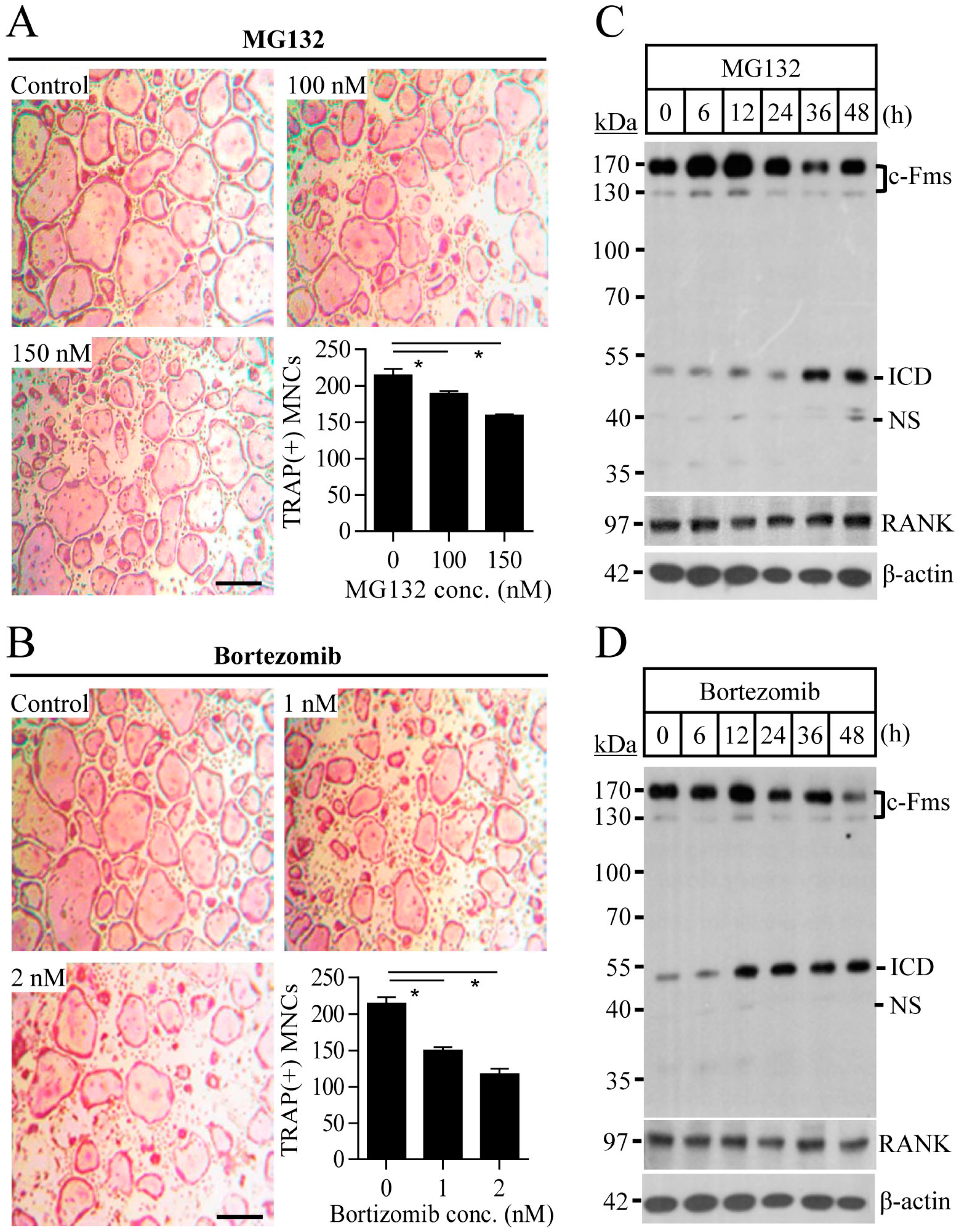
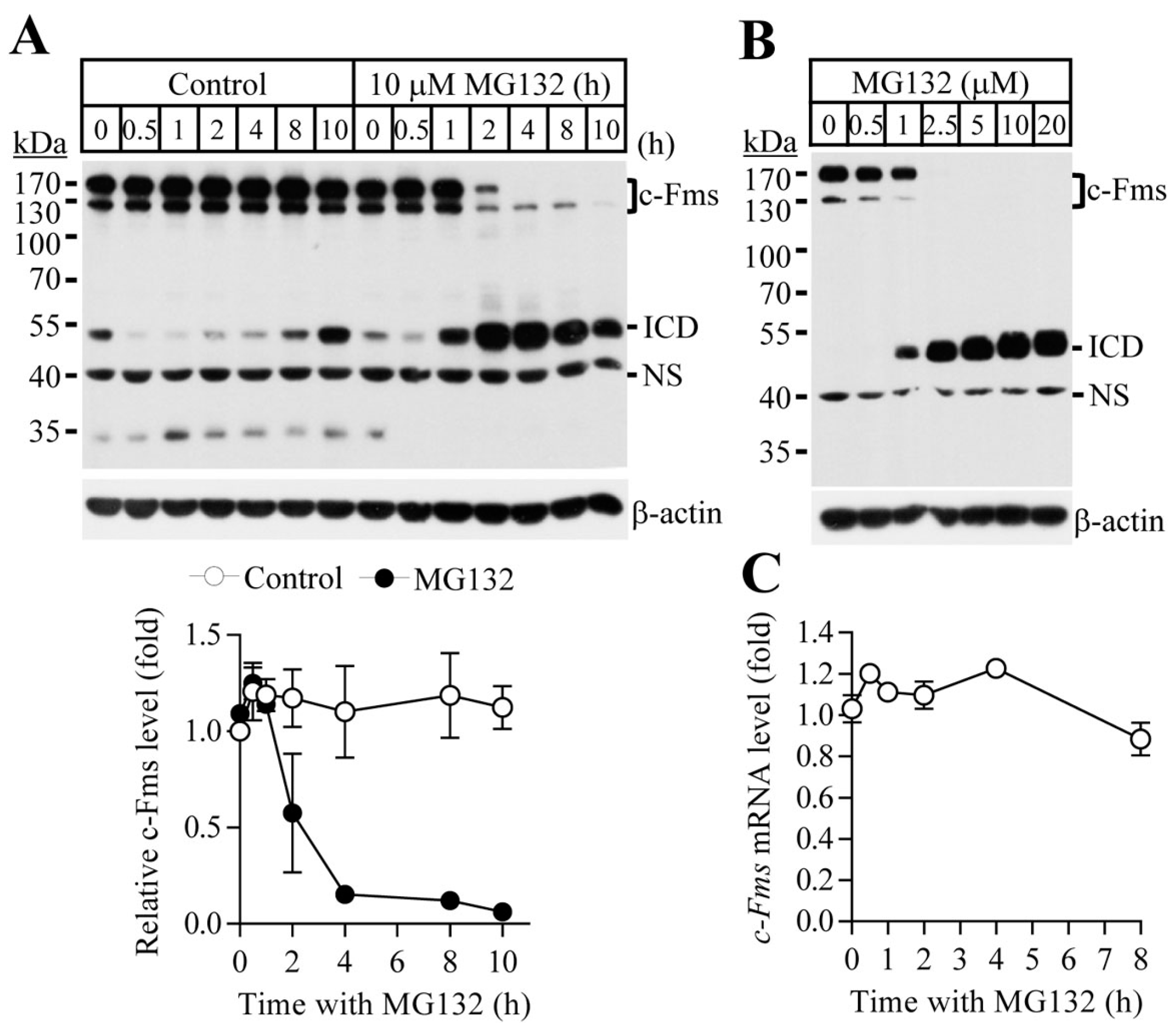
2.1. Proteasome Inhibition Suppresses Osteoclast Differentiation by Downregulating M-CSF Receptor c-Fms
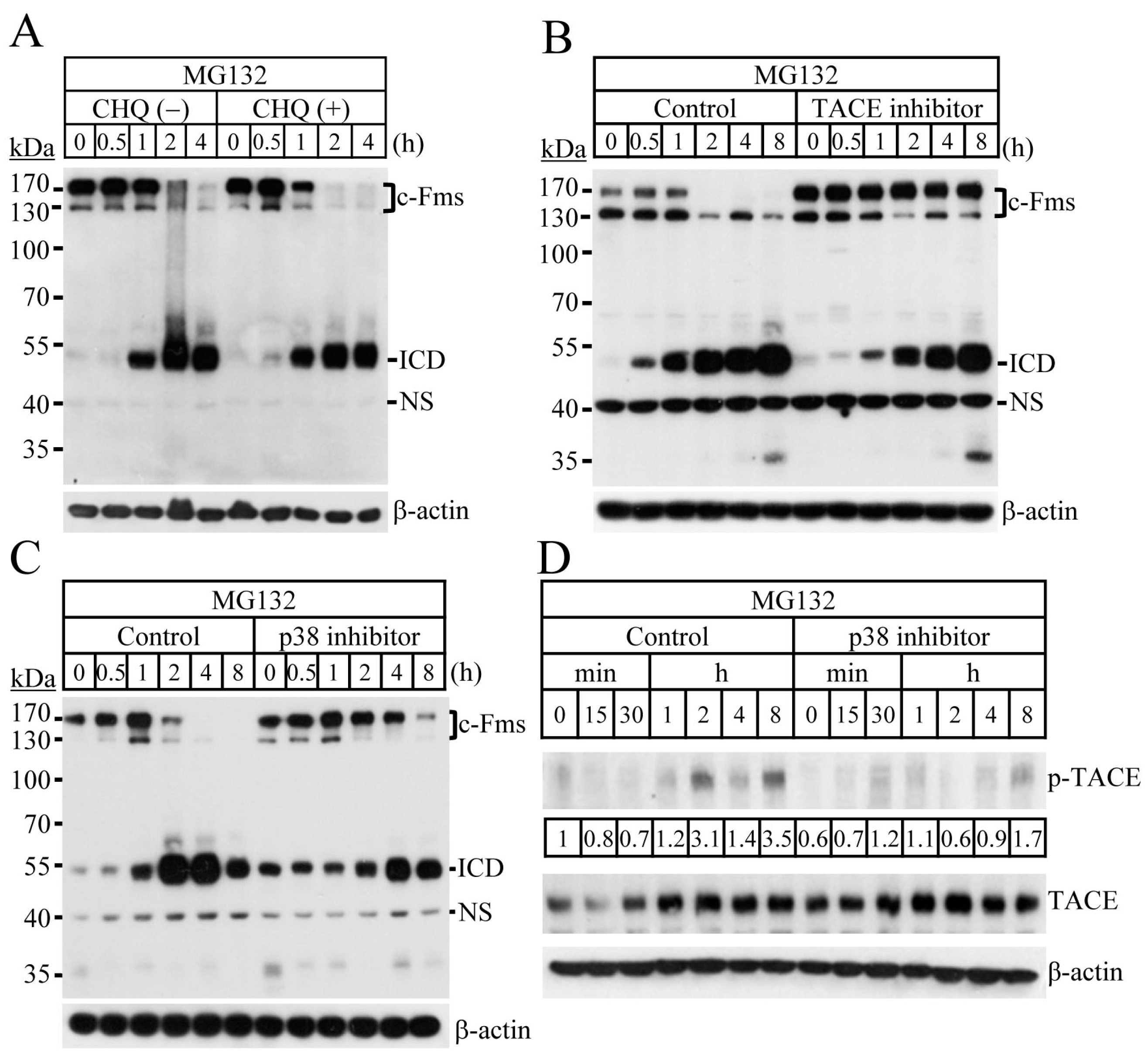
2.2. Blocking of the Proteasome System Induces c-Fms Degradation by Stimulating p38/TACE-Mediated RIPping
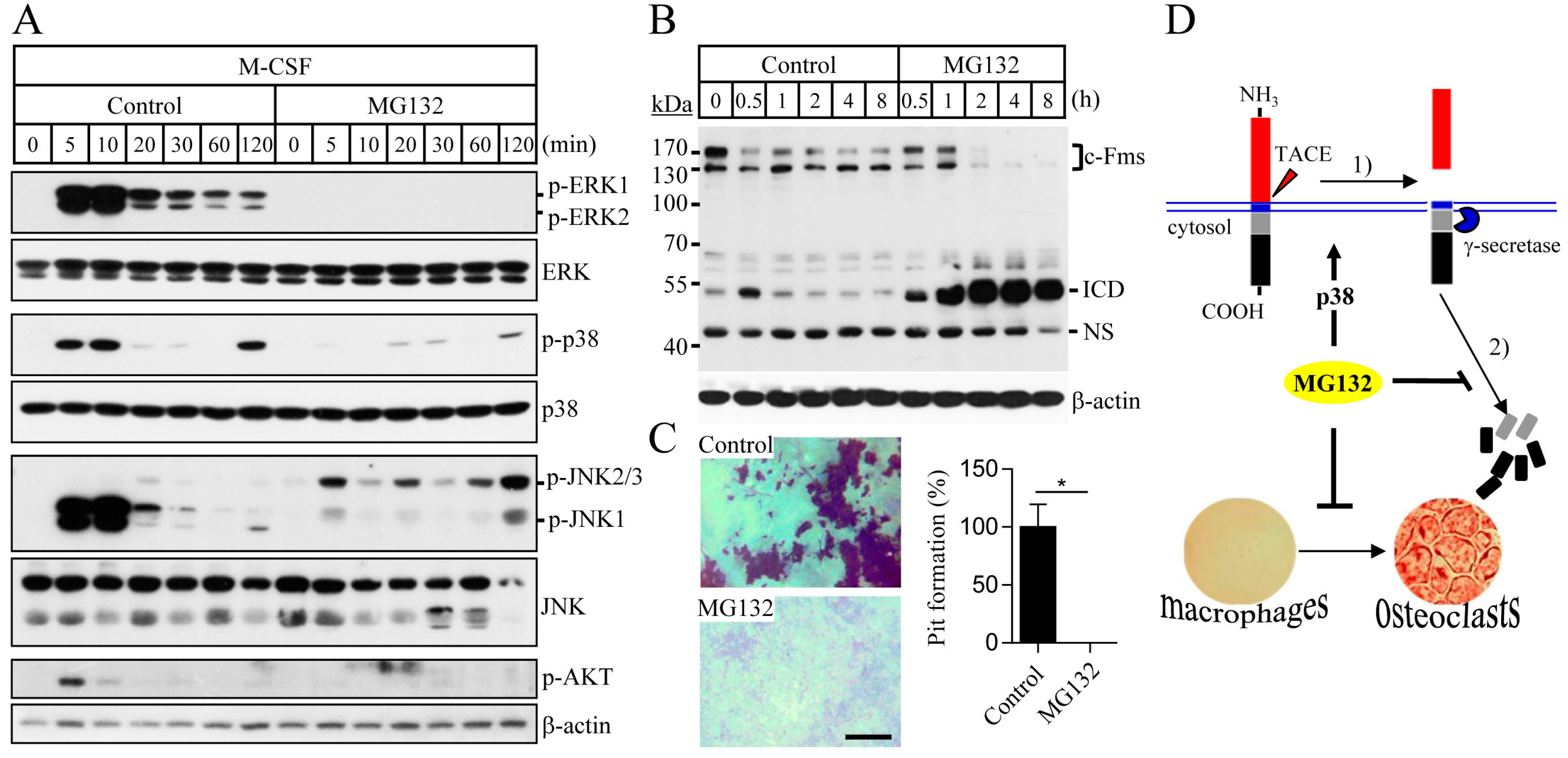
2.3. Proteasome Inhibition Suppresses M-CSF/c-Fms-Mediated Intrinsic Signalling and Bone Resorption Activity of Mature Osteoclasts
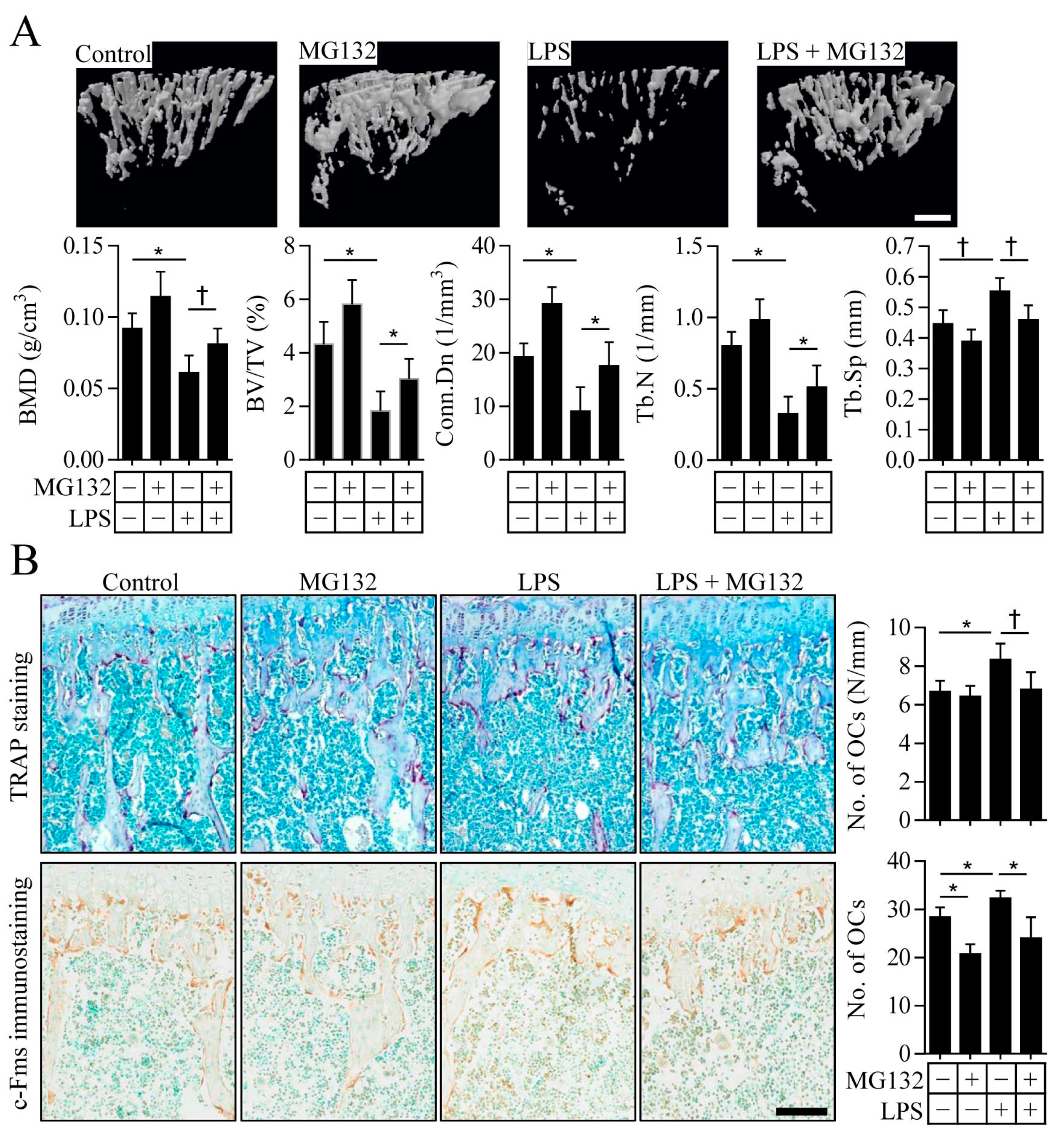
2.4. Proteasome Inhibition Prevents Inflammation-Induced Osteoporosis in Mice
3. Discussion
4. Materials and Methods
4.1. Osteoclast Progenitor Preparation and Osteoclast Differentiation
4.2. Pit Formation Assay
4.3. Immunoblot Analysis
4.4. Quantitative Real-Time PCR
4.5. Cell Proliferation and Apoptosis Assay
4.6. Mouse Model with Inflammation-Induced Bone Loss
4.7. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pixley, F.J.; Stanley, E.R. CSF-1 regulation of the wandering macrophage: Complexity in action. Trends Cell Biol. 2004, 14, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Mouchemore, K.A.; Pixley, F.J. CSF-1 signaling in macrophages: Pleiotrophy through phosphotyrosine-based signaling pathways. Crit. Rev. Clin. Lab. Sci. 2012, 49, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Rettenmier, C.W.; Sacca, R.; Roussel, M.F.; Look, A.T.; Stanley, E.R. The c-Fms proto-oncogene product is related to the receptor for the mononuclear phagocyte growth factor, CSF-1. Cell 1985, 41, 665–676. [Google Scholar] [CrossRef]
- Coussens, L.; Van Beveren, C.; Smith, D.; Chen, E.; Mitchell, R.L.; Isacke, C.M.; Verma, I.M.; Ullrich, A. Structural alteration of viral homologue of receptor proto-oncogene Fms at carboxyl terminus. Nature 1986, 320, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, K.; Tapley, P.; Haystead, C.; Rohrschneider, L. The role of kinase activity and the kinase insert region in ligand-induced internalization and degradation of the c-Fms protein. EMBO J. 1991, 10, 877–883. [Google Scholar] [PubMed]
- Glenn, G.; van der Geer, P. CSF-1 and TPA stimulate independent pathways leading to lysosomal degradation or regulated intramembrane proteolysis of the CSF-1 receptor. FEBS Lett. 2007, 581, 5377–5381. [Google Scholar] [CrossRef] [PubMed]
- Glenn, G.; van der Geer, P. Toll-like receptors stimulate regulated intramembrane proteolysis of the CSF-1 receptor through erk activation. FEBS Lett. 2008, 582, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Vahidi, A.; Glenn, G.; van der Geer, P. Identification and mutagenesis of the TACE and γ-secretase cleavage sites in the colony-stimulating factor 1 receptor. Biochem. Biophys. Res. Commun. 2014, 450, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.; Welsh, J.B.; Lazar, C.S.; Wiley, H.S.; Gill, G.N.; Rosenfeld, M.G. Ligand-induced transformation by a noninternalizing epidermal growth factor receptor. Science 1990, 247, 962–964. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, K.; van der Geer, P. Phorbol 12-myristate 13-acetate-induced release of the colony-stimulating factor 1 receptor cytoplasmic domain into the cytosol involves two separate cleavage events. Mol. Cell. Biol. 2004, 24, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Scholl, S.M.; Bascou, C.H.; Mosseri, V.; Olivares, R.; Magdelenat, H.; Dorval, T.; Palangie, T.; Validire, P.; Pouillart, P.; Stanley, E.R. Circulating levels of colony-stimulating factor 1 as a prognostic indicator in 82 patients with epithelial ovarian cancer. Br. J. Cancer 1994, 69, 342–346. [Google Scholar] [CrossRef] [PubMed]
- McDermott, R.S.; Deneux, L.; Mosseri, V.; Vedrenne, J.; Clough, K.; Fourquet, A.; Rodriguez, J.; Cosset, J.M.; Sastre, X.; Beuzeboc, P.; et al. Circulating macrophage colony stimulating factor as a marker of tumour progression. Eur. Cytokine Netw. 2002, 13, 121–127. [Google Scholar] [PubMed]
- Webster, J.A.; Beck, A.H.; Sharma, M.; Espinosa, I.; Weigelt, B.; Schreuder, M.; Montgomery, K.D.; Jensen, K.C.; van de Rijn, M.; West, R. Variations in stromal signatures in breast and colorectal cancer metastases. J. Pathol. 2010, 222, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Richardsen, E.; Uglehus, R.D.; Due, J.; Busch, C.; Busund, L.T. The prognostic impact of m-CSF, CSF-1 receptor, CD68 and CD3 in prostatic carcinoma. Histopathology 2008, 53, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Richardsen, E.; Sorbye, S.W.; Crowe, J.P.; Yang, J.L.; Busund, L.T. Expression of m-CSF and CSF-1R is correlated with histological grade in soft tissue tumors. Anticancer Res. 2009, 29, 3861–3866. [Google Scholar] [PubMed]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Paniagua, R.T.; Chang, A.; Mariano, M.M.; Stein, E.A.; Wang, Q.; Lindstrom, T.M.; Sharpe, O.; Roscow, C.; Ho, P.P.; Lee, D.M.; et al. c-FMS-mediated differentiation and priming of monocyte lineage cells play a central role in autoimmune arthritis. Arthritis Res. Ther. 2010, 12, R32. [Google Scholar] [CrossRef] [PubMed]
- Murayama, T.; Yokode, M.; Kataoka, H.; Imabayashi, T.; Yoshida, H.; Sano, H.; Nishikawa, S.; Nishikawa, S.; Kita, T. Intraperitoneal administration of anti-c-fms monoclonal antibody prevents initial events of atherogenesis but does not reduce the size of advanced lesions in apolipoprotein e-deficient mice. Circulation 1999, 99, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Garrett, I.R.; Chen, D.; Gutierrez, G.; Zhao, M.; Escobedo, A.; Rossini, G.; Harris, S.E.; Gallwitz, W.; Kim, K.B.; Hu, S.; et al. Selective inhibitors of the osteoblast proteasome stimulate bone formation in vivo and in vitro. J. Clin. Investig. 2003, 111, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Qiao, M.; Oyajobi, B.O.; Mundy, G.R.; Chen, D. E3 ubiquitin ligase Smurf1 mediates core-binding factor alpha1/Runx2 degradation and plays a specific role in osteoblast differentiation. J. Biol. Chem. 2003, 278, 27939–27944. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Morandi, F.; Tagliaferri, S.; Lazzaretti, M.; Bonomini, S.; Crugnola, M.; Mancini, C.; Martella, E.; Ferrari, L.; Tabilio, A.; et al. The proteasome inhibitor bortezomib affects osteoblast differentiation in vitro and in vivo in multiple myeloma patients. Blood 2007, 110, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Zavrski, I.; Krebbel, H.; Wildemann, B.; Heider, U.; Kaiser, M.; Possinger, K.; Sezer, O. Proteasome inhibitors abrogate osteoclast differentiation and osteoclast function. Biochem. Biophys. Res. Commun. 2005, 333, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Kang, J.H.; Kim, H.J.; Kim, H.J.; Kim, H.H.; Kim, J.Y.; Lee, Y. Bortezomib inhibits osteoclastogenesis and porphyromonas gingivalis lipopolysaccharide-induced alveolar bone resorption. J. Dent. Res. 2015, 94, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Boissy, P.; Andersen, T.L.; Lund, T.; Kupisiewicz, K.; Plesner, T.; Delaisse, J.M. Pulse treatment with the proteasome inhibitor bortezomib inhibits osteoclast resorptive activity in clinically relevant conditions. Leuk. Res. 2008, 32, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Peng, Z. MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia Pac. J. Clin. Oncol. 2013, 9, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.P. m-CSF, c-FMS, and signaling in osteoclasts and their precursors. Ann. N. Y. Acad. Sci. 2006, 1068, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Sezer, O.; Croucher, P.; Dimopoulos, M.A. Myeloma bone disease and proteasome inhibition therapies. Blood 2007, 110, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Zavrski, I.; Kleeberg, L.; Kaiser, M.; Fleissner, C.; Heider, U.; Sterz, J.; Jakob, C.; Sezer, O. Proteasome as an emerging therapeutic target in cancer. Curr. Pharm. Des. 2007, 13, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Rizzoli, V.; Roodman, G.D. Multiple myeloma bone disease: Pathophysiology of osteoblast inhibition. Blood 2006, 108, 3992–3996. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A. Multiple myeloma: Review of 869 cases. Mayo Clin. Proc. 1975, 50, 29–40. [Google Scholar] [PubMed]
- Galson, D.L.; Silbermann, R.; Roodman, G.D. Mechanisms of multiple myeloma bone disease. Bonekey Rep. 2012, 1, 135. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Rahemtulla, A.; Dimopoulos, M.A. Current treatment options for myeloma. Expert Opin. Pharmacother. 2005, 6, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Heath, D.J.; Rahemtulla, A.; Zervas, K.; Chantry, A.; Anagnostopoulos, A.; Pouli, A.; Katodritou, E.; Verrou, E.; Vervessou, E.C.; et al. Bortezomib reduces serum dickkopf-1 and receptor activator of nuclear factor-κb ligand concentrations and normalises indices of bone remodelling in patients with relapsed multiple myeloma. Br. J. Haematol. 2006, 135, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Hongming, H.; Jian, H. Bortezomib inhibits maturation and function of osteoclasts from pbmcs of patients with multiple myeloma by downregulating traf6. Leuk. Res. 2009, 33, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.F.; Courtneidge, S.A. The adams family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev. 2003, 17, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Garbers, C.; Rose-John, S. ADAM17: A molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011, 32, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 2012, 5, ra34. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Derynck, R. Direct activation of tace-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 2010, 37, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Killock, D.J.; Ivetic, A. The cytoplasmic domains of tnfalpha-converting enzyme (TACE/ADAM17) and l-selectin are regulated differently by p38 MAPK and PKC to promote ectodomain shedding. Biochem. J. 2010, 428, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; MacDonald, K.P. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 2012, 119, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Kinne, R.W.; Brauer, R.; Stuhlmuller, B.; Palombo-Kinne, E.; Burmester, G.R. Macrophages in rheumatoid arthritis. Arthritis Res. 2000, 2, 189–202. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mahida, Y.R. The key role of macrophages in the immunopathogenesis of inflammatory bowel disease. Inflamm. Bowel Dis. 2000, 6, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar] [CrossRef] [PubMed]
- Staines, K.A.; Prideaux, M.; Allen, S.; Buttle, D.J.; Pitsillides, A.A.; Farquharson, C. E11/podoplanin protein stabilization through inhibition of the proteasome promotes osteocyte differentiation in murine in vitro models. J. Cell. Physiol. 2016, 231, 1392–1404. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Matsunaga, A.; Ono, T.; Hayashi, M.; Takayanagi, H.; Moriyama, K.; Nakashima, T. Osteocyte regulation of orthodontic force-mediated tooth movement via RANKL expression. Sci. Rep. 2017, 7, 8753. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.; Kim, M.Y.; Ahn, H.; Kim, H.-S.; Shin, H.-I.; Jeong, D. Blocking of the Ubiquitin-Proteasome System Prevents Inflammation-Induced Bone Loss by Accelerating M-CSF Receptor c-Fms Degradation in Osteoclast Differentiation. Int. J. Mol. Sci. 2017, 18, 2054. https://doi.org/10.3390/ijms18102054
Lee K, Kim MY, Ahn H, Kim H-S, Shin H-I, Jeong D. Blocking of the Ubiquitin-Proteasome System Prevents Inflammation-Induced Bone Loss by Accelerating M-CSF Receptor c-Fms Degradation in Osteoclast Differentiation. International Journal of Molecular Sciences. 2017; 18(10):2054. https://doi.org/10.3390/ijms18102054
Chicago/Turabian StyleLee, Kyunghee, Mi Yeong Kim, Heejin Ahn, Han-Sung Kim, Hong-In Shin, and Daewon Jeong. 2017. "Blocking of the Ubiquitin-Proteasome System Prevents Inflammation-Induced Bone Loss by Accelerating M-CSF Receptor c-Fms Degradation in Osteoclast Differentiation" International Journal of Molecular Sciences 18, no. 10: 2054. https://doi.org/10.3390/ijms18102054
APA StyleLee, K., Kim, M. Y., Ahn, H., Kim, H.-S., Shin, H.-I., & Jeong, D. (2017). Blocking of the Ubiquitin-Proteasome System Prevents Inflammation-Induced Bone Loss by Accelerating M-CSF Receptor c-Fms Degradation in Osteoclast Differentiation. International Journal of Molecular Sciences, 18(10), 2054. https://doi.org/10.3390/ijms18102054