
R-Flurbiprofen Traps Prostaglandins within Cells by Inhibition of Multidrug Resistance-Associated Protein-4

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
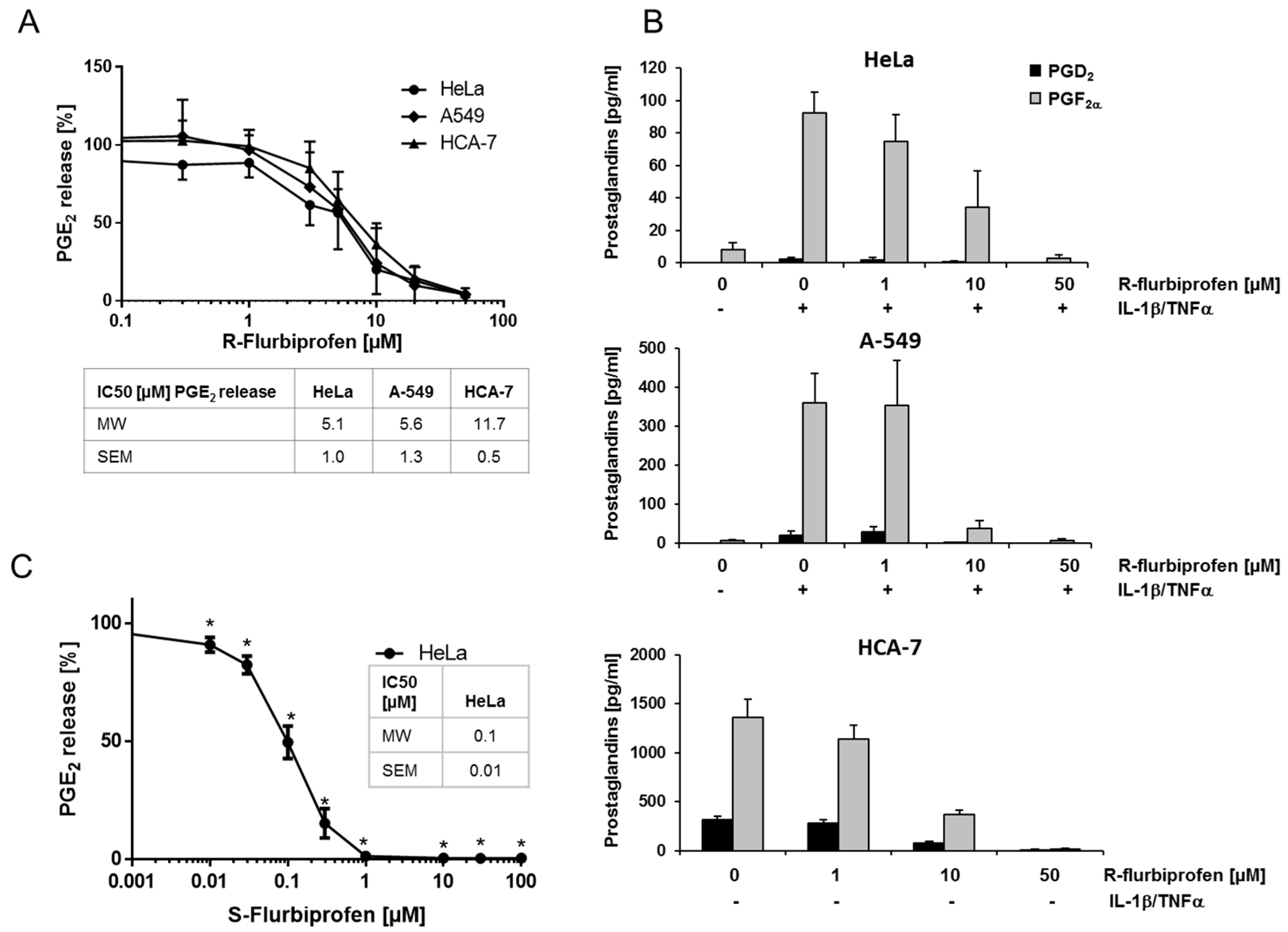
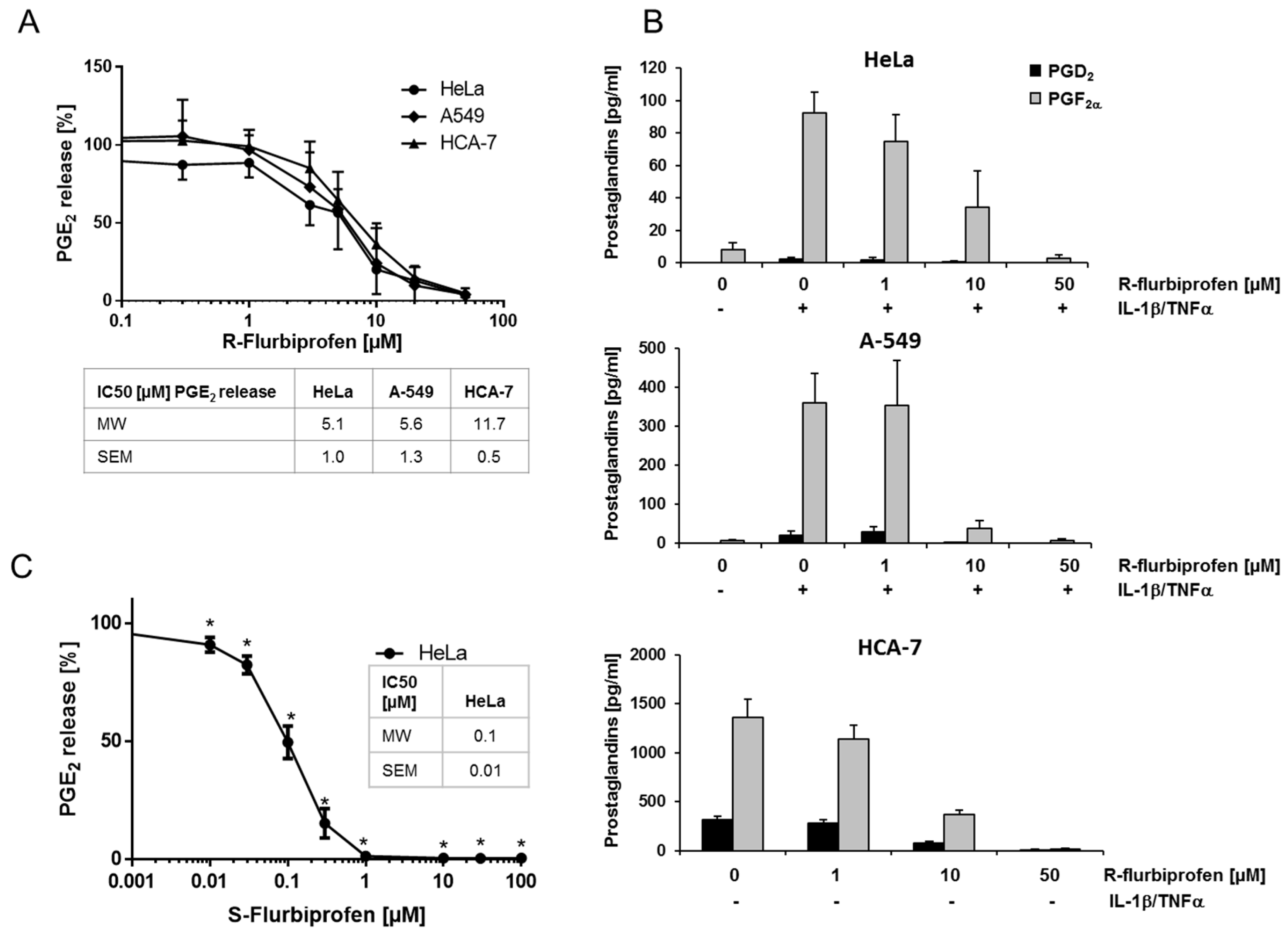
2.1. R-Flurbiprofen Reduces Prostaglandin Levels
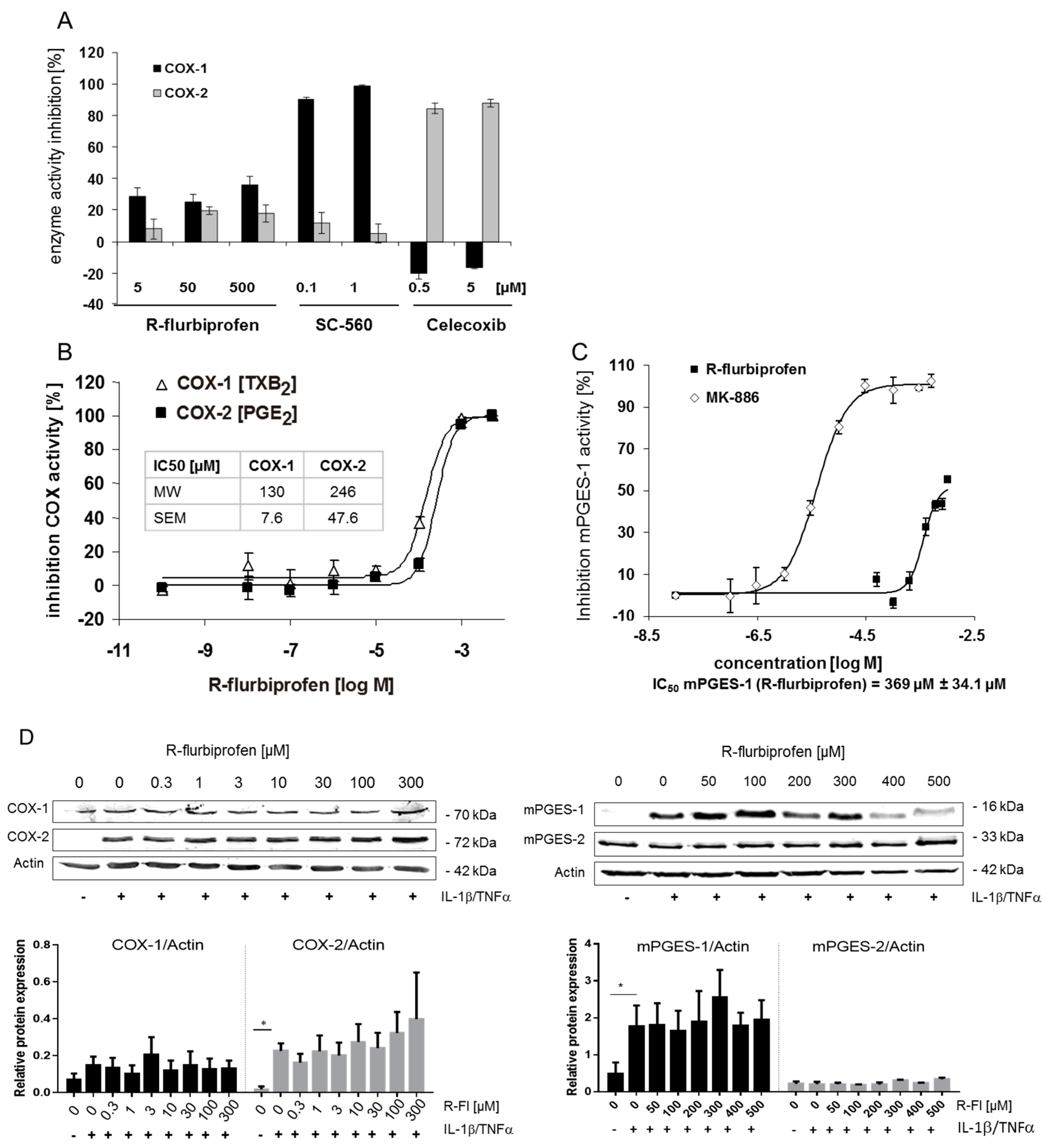
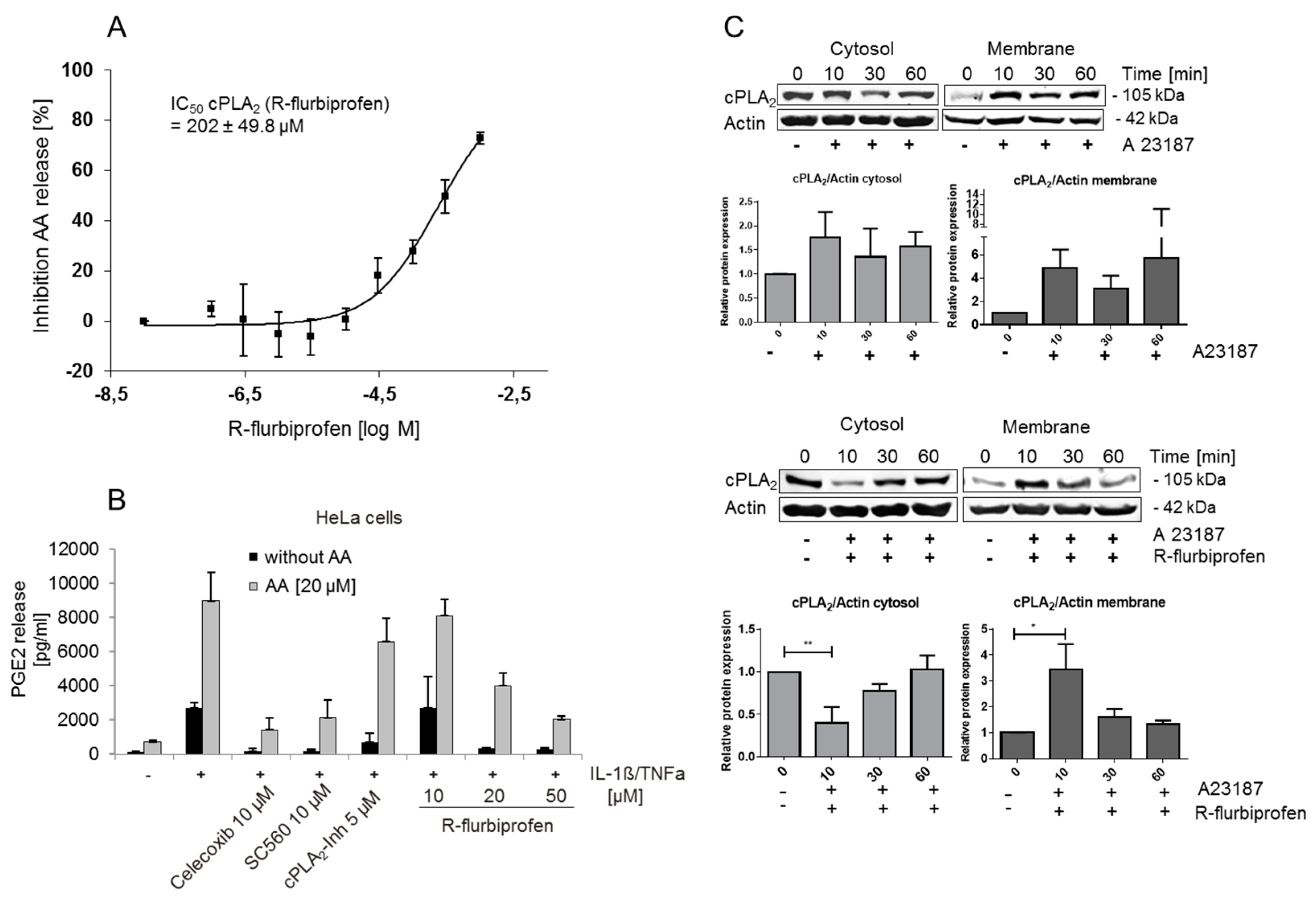
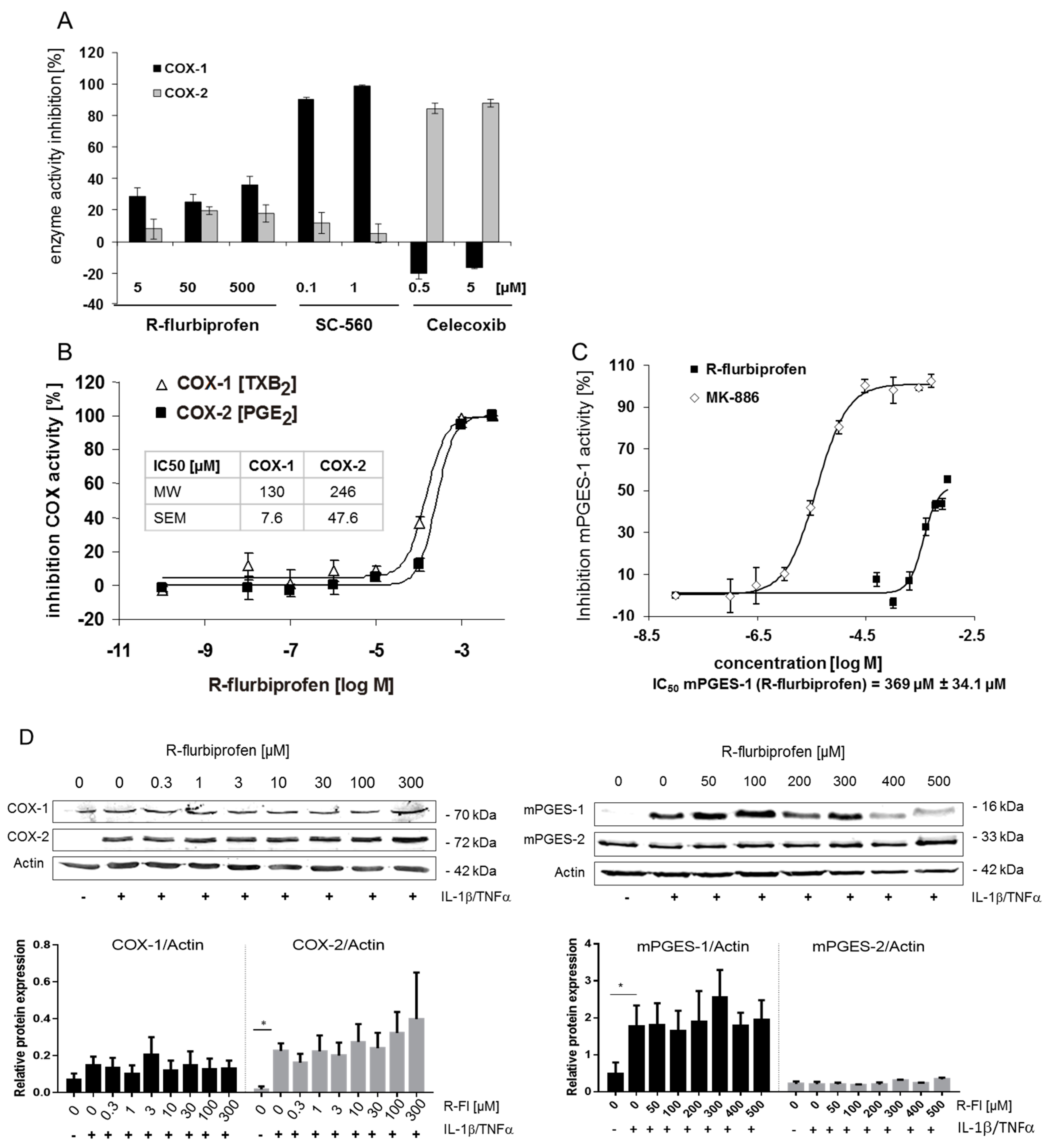
2.2. Inhibition of PG Release Is Not Mediated through COX-1/2- or mPGES-1
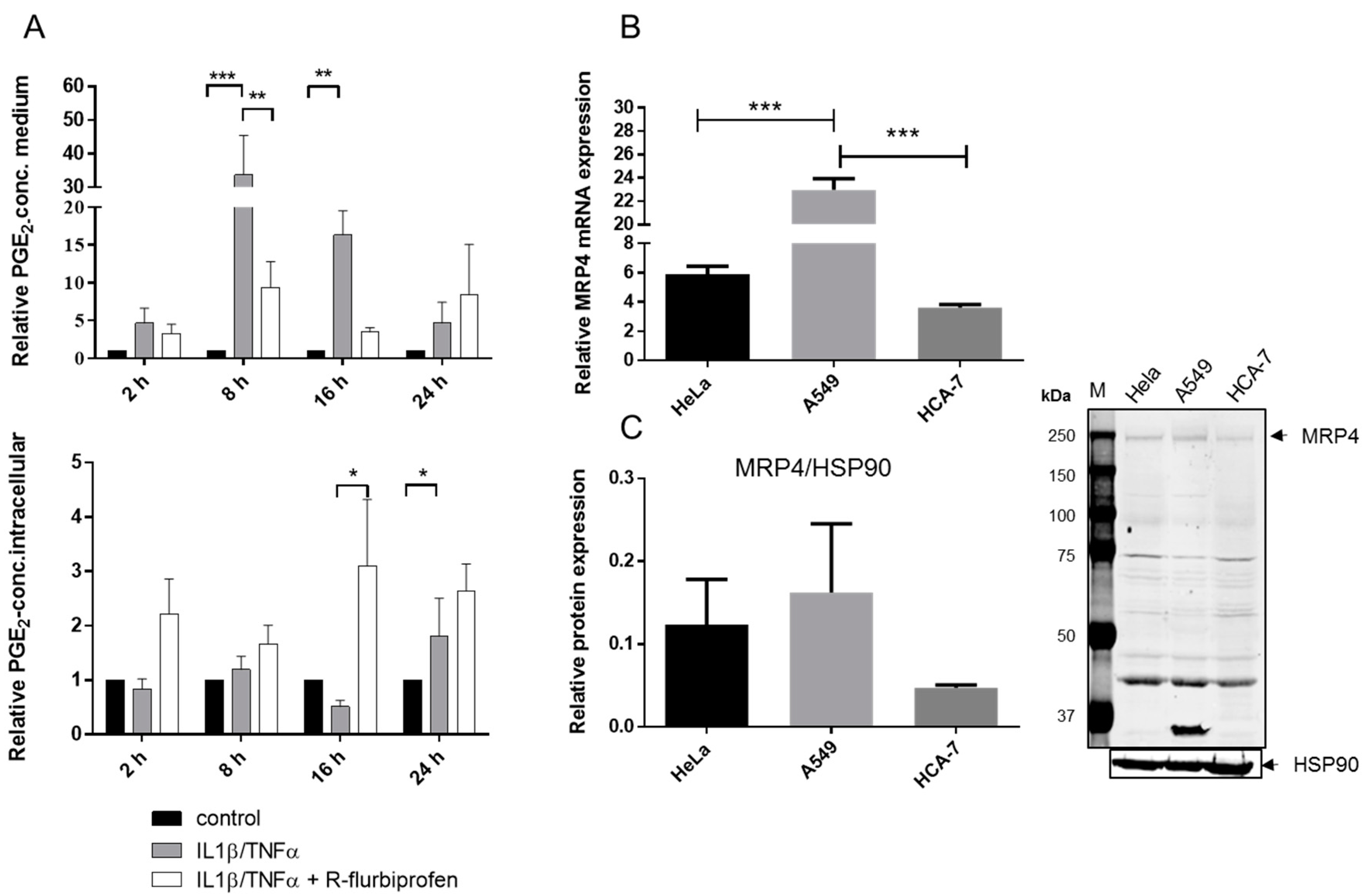
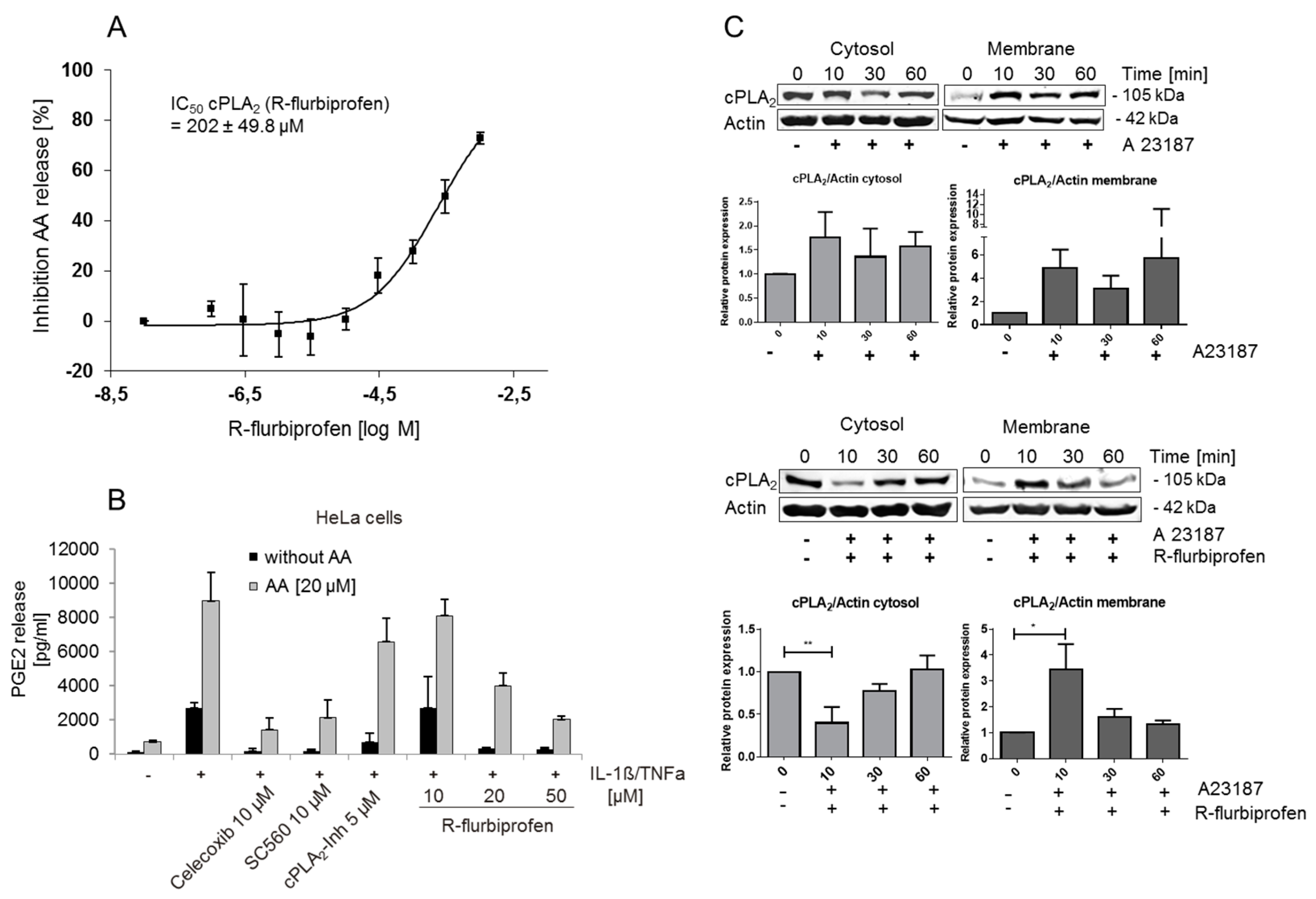
2.3. R-Flurbiprofen Targets Alternative Candidates in the PG Pathway
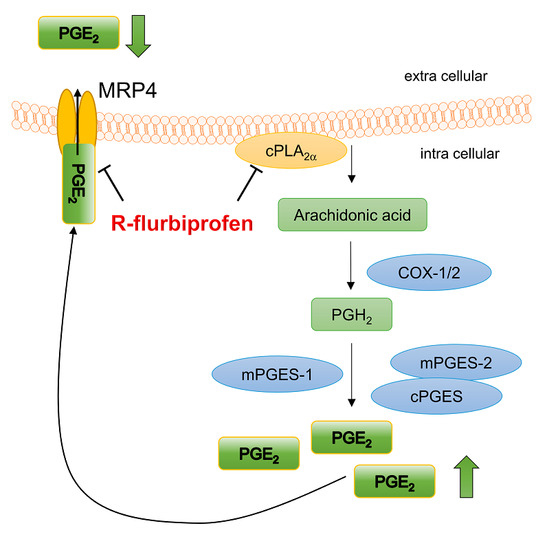
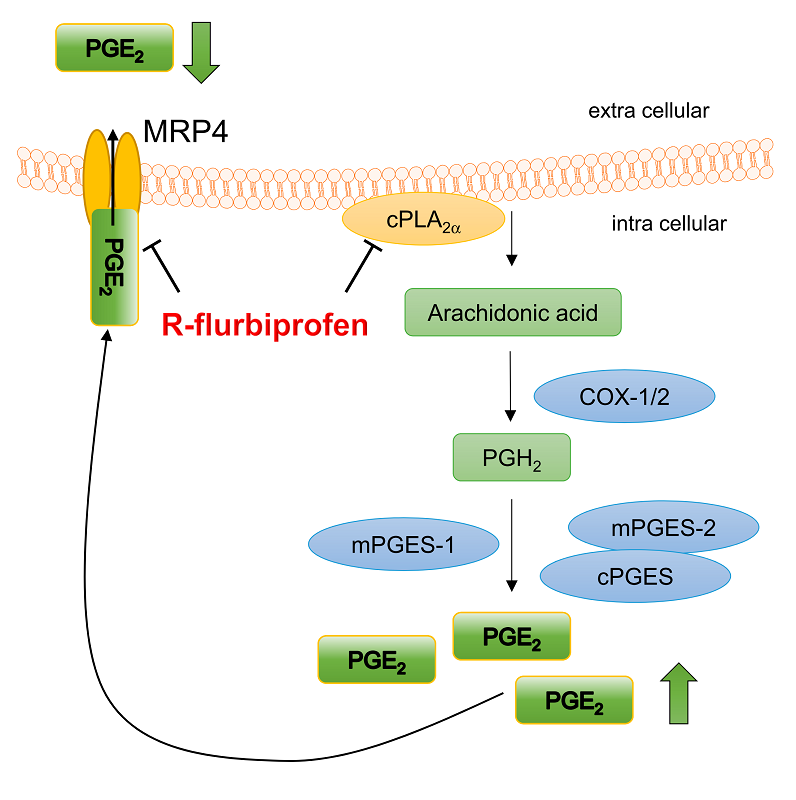
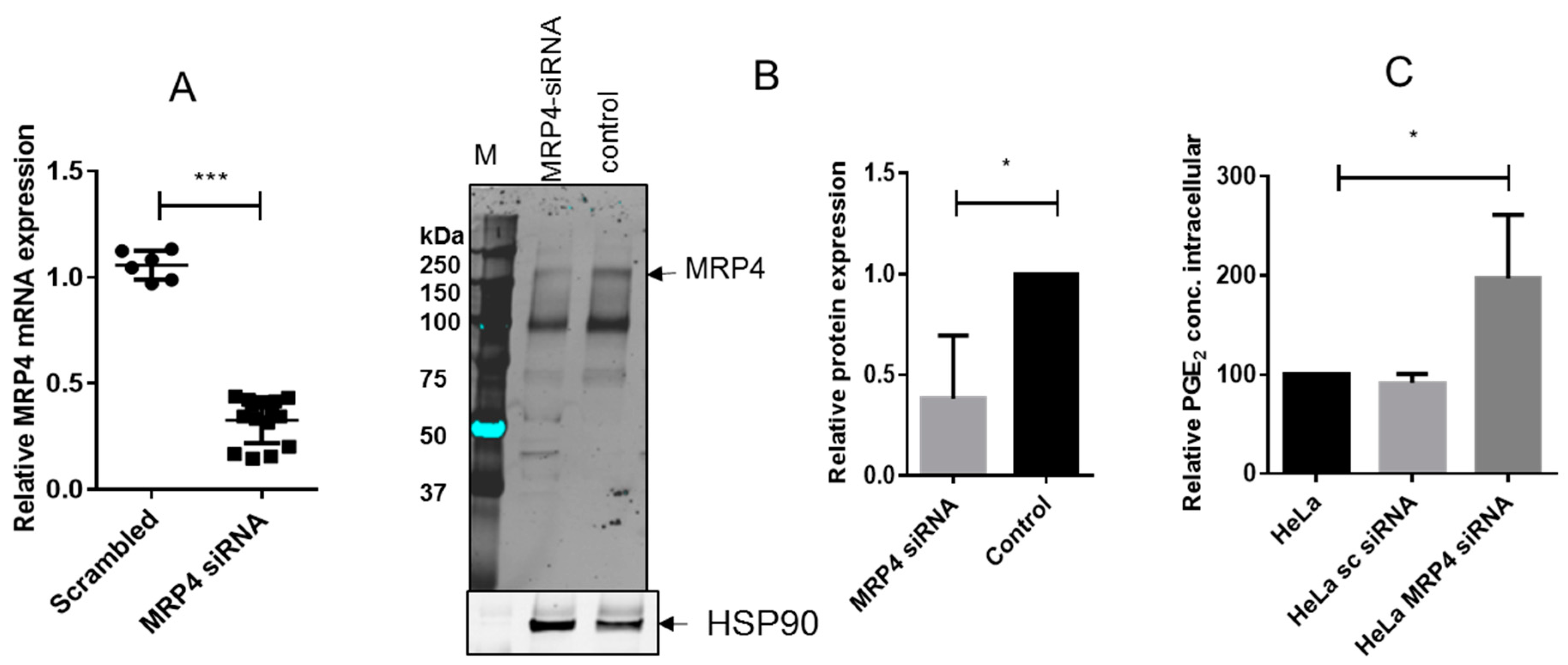
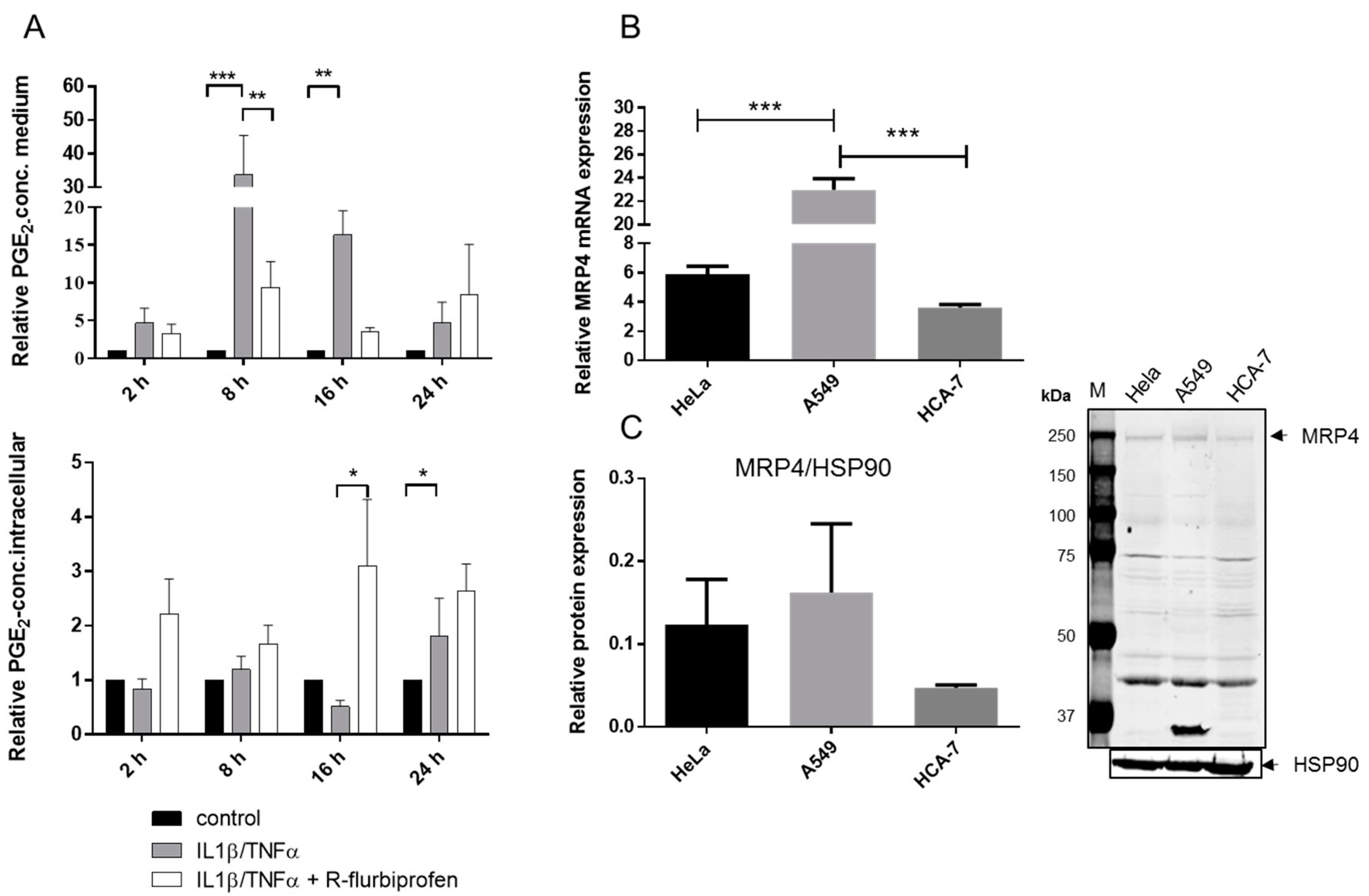
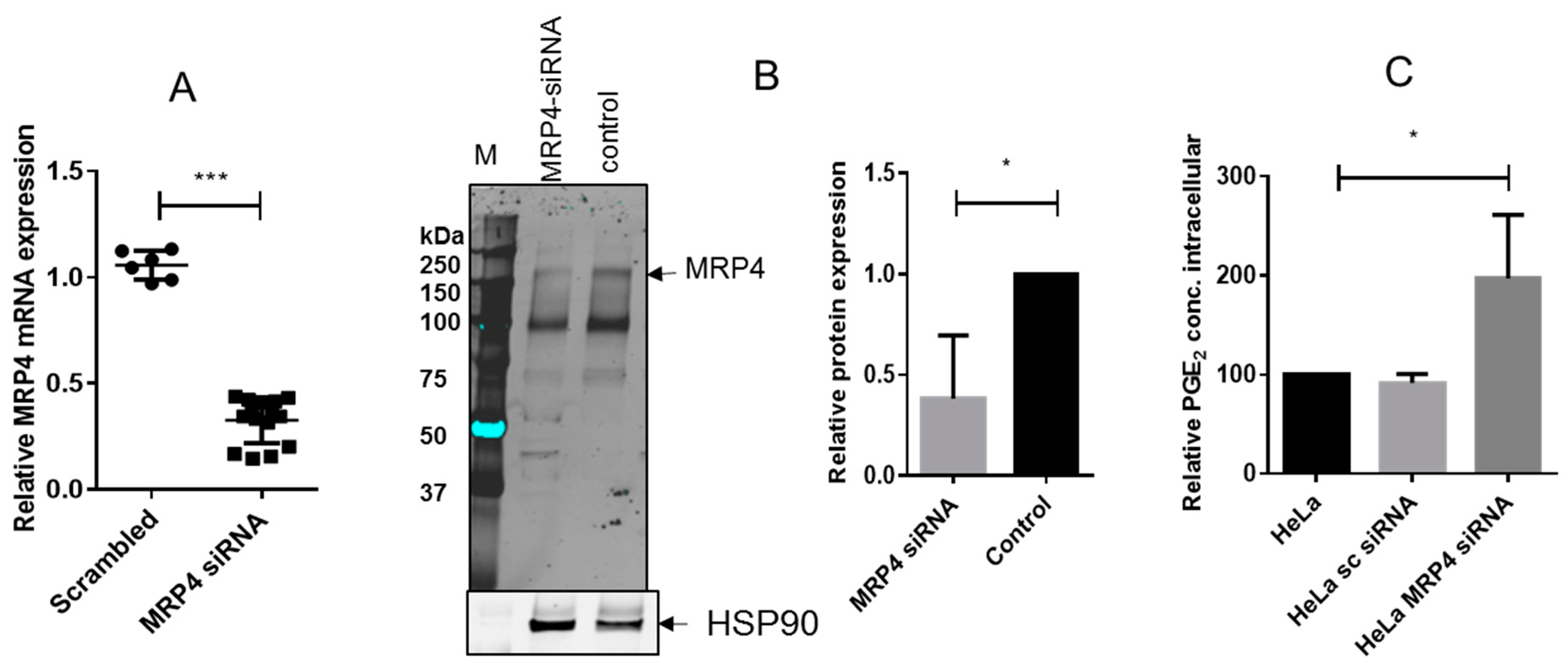
2.4. R-Flurbiprofen Traps PGs Inside of Cells
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. COX Inhibitor Screening Assay
4.3. Analysis of Prostaglandin Levels (PGE2, PGD2 and PGF2α)
4.4. Human Whole-Blood Assay In Vitro
4.5. mPGES-1 Activity Assay
4.6. Western Blot Analysis
4.7. cPLA2α Activity Assay
4.8. Silencing of MRP4
4.9. Quantitative Real-Time-PCR
4.10. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| cPLA2 | cytosolic phospholipase A2 |
| COX-1 | cyclooxygenase-1 |
| COX-2 | cyclooxygenase -2 |
| IL-1β | interleukin-1 beta |
| MRP4 | multidrug resistance protein 4 |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| PG | prostaglandin |
| PGES | PGE-synthases |
| PGT | prostaglandin transporter |
| 15-PGDH | 15-hydroxyprostaglandin dehydrogenase |
| TNFα | tumor necrosis factor alpha |
References
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, I.M.; Cathcart, B.J.; Kumar, E.B.; Dick, W.C.; Buchanan, W.W. Clinico-pharmacological studies and clinical evaluation of flurbiprofen. A new non-steroidal antirheumatic agent. Ann. Rheum. Dis. 1972, 31, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Grosch, S.; Tegeder, I.; Schilling, K.; Maier, T.J.; Niederberger, E.; Geisslinger, G. Activation of c-Jun-N-terminal-kinase is crucial for the induction of a cell cycle arrest in human colon carcinoma cells caused by flurbiprofen enantiomers. FASEB J. 2003, 17, 1316–1318. [Google Scholar] [CrossRef] [PubMed]
- Tegeder, I.; Niederberger, E.; Israr, E.; Guhring, H.; Brune, K.; Euchenhofer, C.; Grosch, S.; Geisslinger, G. Inhibition of NF-κB and AP-1 activation by R- and S-flurbiprofen. FASEB J. 2001, 15, 595–597. [Google Scholar] [PubMed]
- Geisslinger, G.; Schaible, H.G. New insights into the site and mode of antinociceptive action of flurbiprofen enantiomers. J. Clin. Pharmacol. 1996, 36, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, A.B.; Yaksh, T.L. Antinociception produced by spinal delivery of the S and R enantiomers of flurbiprofen in the formalin test. Eur. J. Pharmacol. 1994, 256, 205–209. [Google Scholar] [CrossRef]
- Shieh, W.R.; Chen, C.S. Purification and characterization of novel “2-arylpropionyl-CoA epimerases” from rat liver cytosol and mitochondria. J. Biol. Chem. 1993, 268, 3487–3493. [Google Scholar] [PubMed]
- Leaper, D.J.; French, B.; Bennett, A. Reduction by flurbiprofen of primary tumor growth and local metastasis formation in mice. Adv. Prostaglandin Thromboxane Res. 1980, 6, 591–593. [Google Scholar] [PubMed]
- Waddell, W.R.; Loughry, R.W. Sulindac for polyposis of the colon. J. Surg. Oncol. 1983, 24, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Grosch, S.; Schilling, K.; Janssen, A.; Maier, T.J.; Niederberger, E.; Geisslinger, G. Induction of apoptosis by R-flurbiprofen in human colon carcinoma cells: Involvement of p53. Biochem. Pharmacol. 2005, 69, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Schmassmann, A. Mechanisms of ulcer healing and effects of nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 43S–51S. [Google Scholar] [CrossRef]
- Rossat, J.; Maillard, M.; Nussberger, J.; Brunner, H.R.; Burnier, M. Renal effects of selective cyclooxygenase-2 inhibition in normotensive salt-depleted subjects. Clin. Pharmacol. Ther. 1999, 66, 76–84. [Google Scholar] [CrossRef]
- Bresalier, R.S.; Sandler, R.S.; Quan, H.; Bolognese, J.A.; Oxenius, B.; Horgan, K.; Lines, C.; Riddell, R.; Morton, D.; Lanas, A.; et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N. Engl. J. Med. 2005, 352, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Geisslinger, G.; Muth-Selbach, U.; Coste, O.; Vetter, G.; Schrodter, A.; Schaible, H.G.; Brune, K.; Tegeder, I. Inhibition of noxious stimulus-induced spinal prostaglandin E2 release by flurbiprofen enantiomers: A microdialysis study. J. Neurochem. 2000, 74, 2094–2100. [Google Scholar] [CrossRef] [PubMed]
- Peskar, B.M.; Kluge, S.; Peskar, B.A.; Soglowek, S.M.; Brune, K. Effects of pure enantiomers of flurbiprofen in comparison to racemic flurbiprofen on eicosanoid release from various rat organs ex vivo. Prostaglandins 1991, 42, 515–531. [Google Scholar] [CrossRef]
- Oelkers, R.; Neupert, W.; Williams, K.M.; Brune, K.; Geisslinger, G. Disposition and effects of flurbiprofen enantiomers in human serum and blister fluid. Br. J. Clin. Pharmacol. 1997, 43, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H.; Tarenflurbil Phase 3 Study Group. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: A randomized controlled trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.R.; Graff-Radford, N.; May, S.; Hendrix, S.; Cottrell, B.A.; Sagi, S.A.; Mather, G.; Laughlin, M.; Zavitz, K.H.; Swabb, E.; et al. Safety, tolerability, pharmacokinetics, and Abeta levels after short-term administration of R-flurbiprofen in healthy elderly individuals. Alzheimer Dis. Assoc. Disord. 2007, 21, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Brenneis, C.; Maier, T.J.; Schmidt, R.; Hofacker, A.; Zulauf, L.; Jakobsson, P.J.; Scholich, K.; Geisslinger, G. Inhibition of prostaglandin E2 synthesis by SC-560 is independent of cyclooxygenase 1 inhibition. FASEB J. 2006, 20, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Diez, E.; Louis-Flamberg, P.; Hall, R.H.; Mayer, R.J. Substrate specificities and properties of human phospholipases A2 in a mixed vesicle model. J. Biol. Chem. 1992, 267, 18342–18348. [Google Scholar] [PubMed]
- Leslie, C.C. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Wielinga, P.; Zelcer, N.; van der Heijden, I.; Kuil, A.; de Haas, M.; Wijnholds, J.; Borst, P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2003, 100, 9244–9249. [Google Scholar] [CrossRef] [PubMed]
- Schuster, V.L. Prostaglandin transport. Prostaglandins Other Lipid Mediat. 2002, 68–69, 633–647. [Google Scholar] [CrossRef]
- Chan, B.S.; Bao, Y.; Schuster, V.L. Role of conserved transmembrane cationic amino acids in the prostaglandin transporter PGT. Biochemistry 2002, 41, 9215–9221. [Google Scholar] [CrossRef] [PubMed]
- Kawase, A.; Yamamoto, T.; Egashira, S.; Iwaki, M. Stereoselective inhibition of methotrexate excretion by glucuronides of nonsteroidal anti-inflammatory drugs via multidrug resistance proteins 2 and 4. J. Pharmacol. Exp. Ther. 2016, 356, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.M. Stereoselective metabolism of anti-inflammatory 2-arylpropionates. Acta Pharm. Nord. 1990, 2, 197–216. [Google Scholar] [PubMed]
- Bishay, P.; Schmidt, H.; Marian, C.; Haussler, A.; Wijnvoord, N.; Ziebell, S.; Metzner, J.; Koch, M.; Myrczek, T.; Bechmann, I.; et al. R-flurbiprofen reduces neuropathic pain in rodents by restoring endogenous cannabinoids. PLoS ONE 2010, 5, E10628. [Google Scholar] [CrossRef] [PubMed]
- Geisslinger, G.; Ferreira, S.H.; Menzel, S.; Schlott, D.; Brune, K. Antinociceptive actions of R(−)-flurbiprofen—A non-cyclooxygenase inhibiting 2-arylpropionic acid—in rats. Life Sci. 1994, 54, L173–L177. [Google Scholar] [CrossRef]
- Lotsch, J.; Geisslinger, G.; Mohammadian, P.; Brune, K.; Kobal, G. Effects of flurbiprofen enantiomers on pain-related chemo-somatosensory evoked potentials in human subjects. Br. J. Clin. Pharmacol. 1995, 40, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.; de Bruin, N.; Bishay, P.; Mannich, J.; Haussler, A.; Altmann, C.; Ferreiros, N.; Lotsch, J.; Ultsch, A.; Parnham, M.J.; et al. R-flurbiprofen attenuates experimental autoimmune encephalomyelitis in mice. EMBO Mol. Med. 2014, 6, 1398–1422. [Google Scholar] [CrossRef] [PubMed]
- McCracken, J.D.; Wechter, W.J.; Liu, Y.; Chase, R.L.; Kantoci, D.; Murray, E.D., Jr.; Quiggle, D.D.; Mineyama, Y. Antiproliferative effects of the enantiomers of flurbiprofen. J. Clin. Pharmacol. 1996, 36, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Wechter, W.J.; Kantoci, D.; Murray, E.D., Jr.; Quiggle, D.D.; Leipold, D.D.; Gibson, K.M.; McCracken, J.D. R-flurbiprofen chemoprevention and treatment of intestinal adenomas in the APC(Min)/+ mouse model: Implications for prophylaxis and treatment of colon cancer. Cancer Res. 1997, 57, 4316–4324. [Google Scholar] [PubMed]
- Wechter, W.J.; Leipold, D.D.; Murray, E.D., Jr.; Quiggle, D.; McCracken, J.D.; Barrios, R.S.; Greenberg, N.M. E-7869 (R-flurbiprofen) inhibits progression of prostate cancer in the TRAMP mouse. Cancer Res. 2000, 60, 2203–2208. [Google Scholar] [PubMed]
- Wechter, W.J.; Murray, E.D., Jr.; Kantoci, D.; Quiggle, D.D.; Leipold, D.D.; Gibson, K.M.; McCracken, J.D. Treatment and survival study in the C57BL/6J-APC(Min)/+(Min) mouse with R-flurbiprofen. Life Sci. 2000, 66, 745–753. [Google Scholar] [CrossRef]
- Eriksen, J.L.; Sagi, S.A.; Smith, T.E.; Weggen, S.; Das, P.; McLendon, D.C.; Ozols, V.V.; Jessing, K.W.; Zavitz, K.H.; Koo, E.H.; et al. NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Abeta 42 in vivo. J. Clin. Investig. 2003, 112, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Zemskova, M.; Wechter, W.; Bashkirova, S.; Chen, C.S.; Reiter, R.; Lilly, M.B. Gene expression profiling in R-flurbiprofen-treated prostate cancer: R-Flurbiprofen regulates prostate stem cell antigen through activation of AKT kinase. Biochem. Pharmacol. 2006, 72, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Quann, E.J.; Khwaja, F.; Zavitz, K.H.; Djakiew, D. The aryl propionic acid R-flurbiprofen selectively induces p75NTR-dependent decreased survival of prostate tumor cells. Cancer Res. 2007, 67, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Duggan, K.C.; Hermanson, D.J.; Musee, J.; Prusakiewicz, J.J.; Scheib, J.L.; Carter, B.D.; Banerjee, S.; Oates, J.A.; Marnett, L.J. (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat. Chem. Biol. 2011, 7, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, D.J.; Gamble-George, J.C.; Marnett, L.J.; Patel, S. Substrate-selective COX-2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation. Trends Pharmacol. Sci. 2014, 35, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Hu, M.; Pearlman, A.; Rohan, L.C. Expression, regulation, and function of drug transporters in cervicovaginal tissues of a mouse model used for microbicide testing. Biochem. Pharmacol. 2016, 116, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Renaud, H.J.; Klaassen, C.D.; Cui, J.Y. Age-specific regulation of drug-processing genes in mouse liver by ligands of xenobiotic-sensing transcription factors. Drug Metab. Dispos. 2016, 44, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Honjo, H.; Uwai, Y.; Aoki, Y.; Iwamoto, K. Stereoselective inhibitory effect of flurbiprofen, ibuprofen and naproxen on human organic anion transporters hOAT1 and hOAT3. Biopharm. Drug Dispos. 2011, 32, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Holla, V.R.; Backlund, M.G.; Yang, P.; Newman, R.A.; DuBois, R.N. Regulation of prostaglandin transporters in colorectal neoplasia. Cancer Prev. Res. 2008, 1, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gu, J.; Xu, L.; Qu, C.; Zhang, Y.; Zhang, W. RNAi-mediated silencing of ATP-binding cassette C4 protein inhibits cell growth in MGC80–3 gastric cancer cell lines. Cell. Mol. Biol. 2014, 60, 1–5. [Google Scholar] [PubMed]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; DuBois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adeno carcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Al-Kharusi, M.R.; Smartt, H.J.; Greenhough, A.; Collard, T.J.; Emery, E.D.; Williams, A.C.; Paraskeva, C. LGR5 promotes survival in human colorectal adenoma cells and is upregulated by PGE2: Implications for targeting adenoma stem cells with NSAIDs. Carcinogenesis 2013, 34, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Houchen, C.W.; Sturmoski, M.A.; Anant, S.; Breyer, R.M.; Stenson, W.F. Prosurvival and antiapoptotic effects of PGE2 in radiation injury are mediated by EP2 receptor in intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G490–G498. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Montrose, D.C.; Clark, P.; Nambiar, P.R.; Belinsky, G.S.; Claffey, K.P.; Xu, D.; Rosenberg, D.W. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008, 68, 3251–3259. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Hu, B.; Tang, L.; Zheng, S.; Sun, Y.; Sheng, Z.; Yao, Y.; Lin, F. The overexpression of MRP4 is related to multidrug resistance in osteosarcoma cells. J. Cancer Res. Ther. 2015, 11, 18–23. [Google Scholar] [PubMed]
- Arber, N.; Kuwada, S.; Leshno, M.; Sjodahl, R.; Hultcrantz, R.; Rex, D. Sporadic adenomatous polyp regression with exisulind is effective but toxic: A randomised, double blind, placebo controlled, dose-response study. Gut 2006, 55, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Hakozaki, M.; Tajino, T.; Konno, S.; Kikuchi, S.; Yamada, H.; Yanagisawa, M.; Nishida, J.; Nagasawa, H.; Tsuchiya, T.; Ogose, A.; et al. Overexpression of cyclooxygenase-2 in malignant peripheral nerve sheath tumor and selective cyclooxygenase-2 inhibitor-induced apoptosis by activating caspases in human malignant peripheral nerve sheath tumor cells. PLoS ONE 2014, 9, E88035. [Google Scholar] [CrossRef] [PubMed]
- Jerussi, T.P.; Caubet, J.F.; McCray, J.E.; Handley, D.A. Clinical endoscopic evaluation of the gastroduodenal tolerance to (R)-ketoprofen, (R)-flurbiprofen, racemic ketoprofen, and paracetamol: A randomized, single-blind, placebo-controlled trial. J. Clin. Pharmacol. 1998, 38, 19S–24S. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, G.K.; Black, S.E.; Hendrix, S.B.; Zavitz, K.H.; Swabb, E.A.; Laughlin, M.A.; Tarenflurbil Phase II Study Investigators. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer's disease: A randomised phase II trial. Lancet Neurol. 2008, 7, 483–493. [Google Scholar] [CrossRef]
- Uchida, Y.; Zhang, Z.; Tachikawa, M.; Terasaki, T. Quantitative targeted absolute proteomics of rat blood-cerebrospinal fluid barrier transporters: Comparison with a human specimen. J. Neurochem. 2015, 134, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Chapy, H.; Saubamea, B.; Tournier, N.; Bourasset, F.; Behar-Cohen, F.; Decleves, X.; Scherrmann, J.M.; Cisternino, S. Blood-brain and retinal barriers show dissimilar ABC transporter impacts and concealed effect of P-glycoprotein on a novel verapamil influx carrier. Br. J. Pharmacol. 2016, 173, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Belleville-Rolland, T.; Sassi, Y.; Decouture, B.; Dreano, E.; Hulot, J.S.; Gaussem, P.; Bachelot-Loza, C. MRP4 (ABCC4) as a potential pharmacologic target for cardiovascular disease. Pharmacol. Res. 2016, 107, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Sassi, Y.; Lipskaia, L.; Vandecasteele, G.; Nikolaev, V.O.; Hatem, S.N.; Cohen Aubart, F.; Russel, F.G.; Mougenot, N.; Vrignaud, C.; Lechat, P.; et al. Multidrug resistance-associated protein 4 regulates cAMP-dependent signaling pathways and controls human and rat SMC proliferation. J. Clin. Investig. 2008, 118, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, S.; Weigert, A.; Mannich, J.; Eberle, M.; Birod, K.; Haussler, A.; Ferreiros, N.; Schreiber, Y.; Kunkel, H.; Grez, M.; et al. PGE2/EP4 signaling in peripheral immune cells promotes development of experimental autoimmune encephalomyelitis. Biochem. Pharmacol. 2014, 87, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Linke, B.; Schreiber, Y.; Zhang, D.D.; Pierre, S.; Coste, O.; Henke, M.; Suo, J.; Fuchs, J.; Angioni, C.; Ferreiros-Bouzas, N.; et al. Analysis of sphingolipid and prostaglandin synthesis during zymosan-induced inflammation. Prostaglandins Other Lipid Mediat. 2012, 99, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Suo, J.; Ulshofer, T.; Jordan, H.; de Bruin, N.; Scholich, K.; Geisslinger, G.; Ferreiros, N. Nano-LC-MS/MS for the quantitation of prostanoids in immune cells. Anal. Bioanal. Chem. 2014, 406, 7103–7116. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.J.; Tausch, L.; Hoernig, M.; Coste, O.; Schmidt, R.; Angioni, C.; Metzner, J.; Groesch, S.; Pergola, C.; Steinhilber, D.; et al. Celecoxib inhibits 5-lipoxygenase. Biochem. Pharmacol. 2008, 76, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Thoren, S.; Jakobsson, P.J. Coordinate up- and down-regulation of glutathione-dependent prostaglandin E synthase and cyclooxygenase-2 in A549 cells. Inhibition by NS-398 and leukotriene C4. Eur. J. Biochem. 2000, 267, 6428–6434. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.J.; Janssen, A.; Schmidt, R.; Geisslinger, G.; Grosch, S. Targeting the β-catenin/APC pathway: A novel mechanism to explain the cyclooxygenase-2-independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. FASEB J. 2005, 19, 1353–1355. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Lopez, J.J.; Pergola, C.; Feisst, C.; Pawelczik, S.; Jakobsson, P.J.; Sorg, B.L.; Glaubitz, C.; Steinhilber, D.; Werz, O. Hyperforin induces Ca2+-independent arachidonic acid release in human platelets by facilitating cytosolic phospholipase A(2) activation through select phospholipid interactions. Biochim. Biophys. Acta 2010, 1801, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P. An reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 1993, 15, 532–537. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wobst, I.; Ebert, L.; Birod, K.; Wegner, M.-S.; Hoffmann, M.; Thomas, D.; Angioni, C.; Parnham, M.J.; Steinhilber, D.; Tegeder, I.; et al. R-Flurbiprofen Traps Prostaglandins within Cells by Inhibition of Multidrug Resistance-Associated Protein-4. Int. J. Mol. Sci. 2017, 18, 68. https://doi.org/10.3390/ijms18010068
Wobst I, Ebert L, Birod K, Wegner M-S, Hoffmann M, Thomas D, Angioni C, Parnham MJ, Steinhilber D, Tegeder I, et al. R-Flurbiprofen Traps Prostaglandins within Cells by Inhibition of Multidrug Resistance-Associated Protein-4. International Journal of Molecular Sciences. 2017; 18(1):68. https://doi.org/10.3390/ijms18010068
Chicago/Turabian StyleWobst, Ivonne, Lisa Ebert, Kerstin Birod, Marthe-Susanna Wegner, Marika Hoffmann, Dominique Thomas, Carlo Angioni, Michael J. Parnham, Dieter Steinhilber, Irmgard Tegeder, and et al. 2017. "R-Flurbiprofen Traps Prostaglandins within Cells by Inhibition of Multidrug Resistance-Associated Protein-4" International Journal of Molecular Sciences 18, no. 1: 68. https://doi.org/10.3390/ijms18010068
APA StyleWobst, I., Ebert, L., Birod, K., Wegner, M.-S., Hoffmann, M., Thomas, D., Angioni, C., Parnham, M. J., Steinhilber, D., Tegeder, I., Geisslinger, G., & Grösch, S. (2017). R-Flurbiprofen Traps Prostaglandins within Cells by Inhibition of Multidrug Resistance-Associated Protein-4. International Journal of Molecular Sciences, 18(1), 68. https://doi.org/10.3390/ijms18010068