
Breast Cancer Brain Metastases: Clonal Evolution in Clinical Context



Abstract
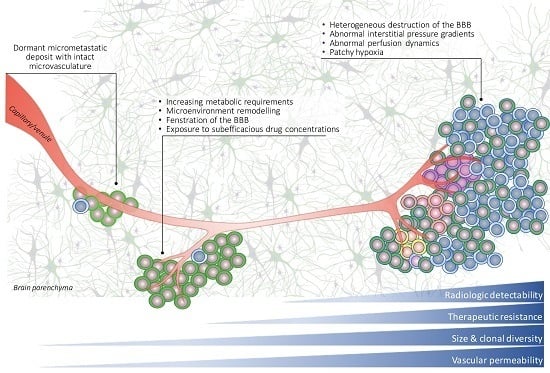
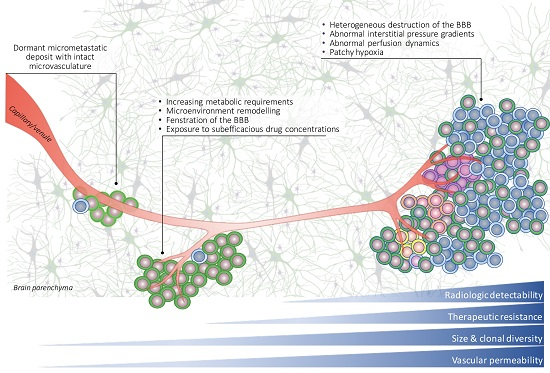
:
1. Clinico-Epidemiologic Profile of Brain Metastatic Breast Cancer
2. Breast Cancer Cell-Intrinsic Features Can Drive Breast Cancer Metastasis to the Brain
3. Extrinsic Factors That Drive Clonal Evolution in Brain Metastases
3.1. Microenvironment-Driven Selection Pressure
3.2. Therapy and Clonal Selection
4. The Molecular Portrait of Breast Cancer Brain Metastases
4.1. Analysis of ‘Brain-Seeking’ Clonal Cell Line Derivatives
4.2. Analysis of Human Clinical Samples
4.3. Subtractive Analysis of Breast Cancer-Brain Met Pairs
5. Factors Underlying the Recalcitrant Behaviour of Brain Metastases
5.1. Late Detection
5.2. Abnormal Vascular Perfusion and Hypoxia Leads to Inadequate Drug Uptake and Therapeutic Resistance
6. Future Directions and Final Comments
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Witzel, I.; Oliveira-Ferrer, L.; Pantel, K.; Muller, V.; Wikman, H. Breast cancer brain metastases: Biology and new clinical perspectives. Breast Cancer Res. 2016, 18, 8. [Google Scholar] [PubMed]
- Pelletier, E.M.; Shim, B.; Goodman, S.; Amonkar, M.M. Epidemiology and economic burden of brain metastases among patients with primary breast cancer: Results from a US claims data analysis. Breast Cancer Res. Treat. 2008, 108, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Sperduto, P.W.; Kased, N.; Roberge, D.; Xu, Z.; Shanley, R.; Luo, X.; Sneed, P.K.; Chao, S.T.; Weil, R.J.; Suh, J.; et al. Effect of tumor subtype on survival and the graded prognostic assessment for patients with breast cancer and brain metastases. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.; Bago-Horvath, Z.; de Vries, C.; Dubsky, P.; Pluschnig, U.; Rudas, M.; Rottenfusser, A.; Knauer, M.; Eiter, H.; Fitzal, F.; et al. Brain metastases free survival differs between breast cancer subtypes. Br. J. Cancer 2012, 106, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, C.A.; Flores, R.; Rojas, K.Y.; Castillo, M.; Dolores-Cerna, K.; Flores, C.; Belmar-Lopez, C.; Milla, E.; Gomez, H. Prognostic factors for patients with newly diagnosed brain metastasis from breast cancer. CNS Oncol. 2015, 4, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Boogerd, W.; Vos, V.W.; Hart, A.A.; Baris, G. Brain metastases in breast cancer; natural history, prognostic factors and outcome. J. Neurooncol. 1992, 15, 165–174. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Schur, S.; Fureder, L.M.; Gatterbauer, B.; Dieckmann, K.; Widhalm, G.; Hainfellner, J.; Zielinski, C.C.; Birner, P.; Bartsch, R.; et al. Descriptive statistical analysis of a real life cohort of 2419 patients with brain metastases of solid cancers. ESMO Open 2016, 1, 000024. [Google Scholar] [CrossRef] [PubMed]
- Sperduto, P.W.; Berkey, B.; Gaspar, L.E.; Mehta, M.; Curran, W. A new prognostic index and comparison to three other indices for patients with brain metastases: An analysis of 1960 patients in the RTOG database. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Sperduto, P.W.; Chao, S.T.; Sneed, P.K.; Luo, X.; Suh, J.; Roberge, D.; Bhatt, A.; Jensen, A.W.; Brown, P.D.; Shih, H.; et al. Diagnosis-specific prognostic factors, indexes, and treatment outcomes for patients with newly diagnosed brain metastases: A multi-institutional analysis of 4259 patients. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Bago-Horvath, Z.; Ilhan-Mutlu, A.; Magerle, M.; Dieckmann, K.; Marosi, C.; Birner, P.; Widhalm, G.; Steger, G.G.; Zielinski, C.C.; et al. Brain-only metastatic breast cancer is a distinct clinical entity characterised by favourable median overall survival time and a high rate of long-term survivors. Br. J. Cancer 2012, 107, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Nieder, C.; Oehlke, O.; Hintz, M.; Grosu, A.L. The challenge of durable brain control in patients with brain-only metastases from breast cancer. Springerplus 2015, 4, 585. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.A. Treatment of brain metastases: Chemotherapy. Curr. Oncol. Rep. 2012, 14, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Rosner, D.; Nemoto, T.; Lane, W.W. Chemotherapy induces regression of brain metastases in breast carcinoma. Cancer 1986, 58, 832–839. [Google Scholar] [CrossRef]
- Morikawa, A.; Peereboom, D.M.; Thorsheim, H.R.; Samala, R.; Balyan, R.; Murphy, C.G.; Lockman, P.R.; Simmons, A.; Weil, R.J.; Tabar, V.; et al. Capecitabine and lapatinib uptake in surgically resected brain metastases from metastatic breast cancer patients: A prospective study. Neuro Oncol. 2015, 17, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Ekenel, M.; Hormigo, A.M.; Peak, S.; Deangelis, L.M.; Abrey, L.E. Capecitabine therapy of central nervous system metastases from breast cancer. J. Neurooncol. 2007, 85, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Dieras, V.; Paul, D.; Lossignol, D.; Christodoulou, C.; Stemmler, H.J.; Roche, H.; Liu, M.C.; Greil, R.; Ciruelos, E.; et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin. Cancer Res. 2009, 15, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Eierman, W.; Greil, R.; Campone, M.; Kaufman, B.; Steplewski, K.; Lane, S.R.; Zembryki, D.; Rubin, S.D.; Winer, E.P. Randomized phase II study of lapatinib plus capecitabine or lapatinib plus topotecan for patients with HER2-positive breast cancer brain metastases. J. Neurooncol. 2011, 105, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Fulford, L.G.; Reis-Filho, J.S.; Ryder, K.; Jones, C.; Gillett, C.E.; Hanby, A.; Easton, D.; Lakhani, S.R. Basal-like grade III invasive ductal carcinoma of the breast: Patterns of metastasis and long-term survival. Breast Cancer Res. 2007, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Harrell, J.C.; Prat, A.; Parker, J.S.; Fan, C.; He, X.; Carey, L.; Anders, C.; Ewend, M.; Perou, C.M. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res. Treat. 2011, 132, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.S.; Kuo, S.H.; Huang, C.S. Recent advances in the management of primary breast cancers. J. Formos. Med. Assoc. 2004, 103, 579–598. [Google Scholar] [PubMed]
- Fisher, B.; Redmond, C.; Fisher, E.R.; Bauer, M.; Wolmark, N.; Wickerham, D.L.; Deutsch, M.; Montague, E.; Margolese, R.; Foster, R. Ten-year results of a randomized clinical trial comparing radical mastectomy and total mastectomy with or without radiation. N. Engl. J. Med. 1985, 312, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Fisher, B.; Jeong, J.H.; Anderson, S.; Bryant, J.; Fisher, E.R.; Wolmark, N. Twenty-five-year follow-up of a randomized trial comparing radical mastectomy, total mastectomy, and total mastectomy followed by irradiation. N. Engl. J. Med. 2002, 347, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Comen, E.; Norton, L. Self-seeding in cancer. Recent Results Cancer Res. 2012, 195, 13–23. [Google Scholar] [PubMed]
- Norton, L.; Massague, J. Is cancer a disease of self-seeding? Nat. Med. 2006, 12, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Li, Y.; Hameed, O.; Siegal, G.P.; Wei, S. Prognostic factors in patients with metastatic breast cancer at the time of diagnosis. Pathol. Res. Pract. 2014, 210, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Bartsch, R.; Preusser, M.; Ricken, G.; Steger, G.G.; Bago-Horvath, Z.; Rudas, M.; Streubel, B.; Dubsky, P.; Gnant, M.; et al. Co-overexpression of HER2/HER3 is a predictor of impaired survival in breast cancer patients. Breast 2014, 23, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Saunus, J.M.; Quinn, M.C.; Patch, A.M.; Pearson, J.V.; Bailey, P.J.; Nones, K.; McCart Reed, A.E.; Miller, D.; Wilson, P.J.; Al-Ejeh, F.; et al. Integrated genomic and transcriptomic analysis of human brain metastases identifies alterations of potential clinical significance. J. Pathol. 2015, 237, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Wikman, H.; Lamszus, K.; Detels, N.; Uslar, L.; Wrage, M.; Benner, C.; Hohensee, I.; Ylstra, B.; Eylmann, K.; Zapatka, M.; et al. Relevance of PTEN loss in brain metastasis formation in breast cancer patients. Breast Cancer Res. 2012, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Louie, E.; Chen, X.F.; Coomes, A.; Ji, K.; Tsirka, S.; Chen, E.I. Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene 2013, 32, 4064–4077. [Google Scholar] [CrossRef] [PubMed]
- Salhia, B.; Kiefer, J.; Ross, J.T.; Metapally, R.; Martinez, R.A.; Johnson, K.N.; di Perna, D.M.; Paquette, K.M.; Jung, S.; Nasser, S.; et al. Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS ONE 2014, 9, 85448. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.-P.; Goh, B.-C. Exosome-Mediated Metastasis: From Epithelial–Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Camacho, L.; Guerrero, P.; Marchetti, D. MicroRNA and protein profiling of brain metastasis competent cell-derived exosomes. PLoS ONE 2013, 8, 73790. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Ording, A.G.; Heide-Jorgensen, U.; Christiansen, C.F.; Norgaard, M.; Acquavella, J.; Sorensen, H.T. Site of metastasis and breast cancer mortality: A Danish nationwide registry-based cohort study. Clin. Exp. Metastasis 2016. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Ren, Z.; Hameed, O.; Chanda, D.; Morgan, C.J.; Siegal, G.P.; Wei, S. Breast cancer subtypes predispose the site of distant metastases. Am. J. Clin. Pathol. 2015, 143, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.G.; Lendeckel, U.; Bertram, I.; Bukowska, A.; Kanakis, D.; Dobrowolny, H.; Stauch, R.; Krell, D.; Mawrin, C.; Budinger, E.; et al. Localization of NRG1α and one of its receptors, ErbB-4 tyrosine kinase, in developing and adult human brain. Brain Res. Bull. 2006, 69, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Lok, J.; Sardi, S.P.; Guo, S.; Besancon, E.; Ha, D.M.; Rosell, A.; Kim, W.J.; Corfas, G.; Lo, E.H. Neuregulin-1 signaling in brain endothelial cells. J. Cereb. Blood Flow Metab. 2009, 29, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Pinkas-Kramarski, R.; Eilam, R.; Spiegler, O.; Lavi, S.; Liu, N.; Chang, D.; Wen, D.; Schwartz, M.; Yarden, Y. Brain neurons and glial cells express Neu differentiation factor/heregulin: A survival factor for astrocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 9387–9391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5, 180ra48. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Tobai, S.; Nakamura, K. Extravascular migration of tumor cells in the brain: An electron microscopic study. Invasion Metastasis 1982, 2, 40–50. [Google Scholar] [PubMed]
- Carbonell, W.S.; Ansorge, O.; Sibson, N.; Muschel, R. The vascular basement membrane as "soil" in brain metastasis. PLoS ONE 2009, 4, 5857. [Google Scholar] [CrossRef] [PubMed]
- Lorger, M.; Felding-Habermann, B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am. J. Pathol. 2010, 176, 2958–2971. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Avraham, H.K.; Jiang, S.; Avraham, S. Vascular endothelial growth factor modulates the transendothelial migration of MDA-MB-231 breast cancer cells through regulation of brain microvascular endothelial cell permeability. J. Biol. Chem. 2003, 278, 5277–5284. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Avraham, H.; Lee, S.H.; Avraham, S. Vascular endothelial growth factor modulates neutrophil transendothelial migration via up-regulation of interleukin-8 in human brain microvascular endothelial cells. J. Biol. Chem. 2002, 277, 10445–10451. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.A.; Hasan, N.; Mann, A.P.; Zheng, W.; Zhao, L.; Morris, L.; Zhu, W.; Zhao, Y.D.; Suh, K.S.; Dooley, W.C.; et al. Blocking the adhesion cascade at the premetastatic niche for prevention of breast cancer metastasis. Mol. Ther. 2015, 23, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Sevenich, L.; Bowman, R.L.; Mason, S.D.; Quail, D.F.; Rapaport, F.; Elie, B.T.; Brogi, E.; Brastianos, P.K.; Hahn, W.C.; Holsinger, L.J.; et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat. Cell Biol. 2014, 16, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Momeny, M.; Saunus, J.M.; Marturana, F.; McCart Reed, A.E.; Black, D.; Sala, G.; Iacobelli, S.; Holland, J.D.; Yu, D.; da Silva, L.; et al. Heregulin-HER3-HER2 signaling promotes matrix metalloproteinase-dependent blood-brain-barrier transendothelial migration of human breast cancer cell lines. Oncotarget 2015, 6, 3932–3946. [Google Scholar] [CrossRef] [PubMed]
- Neman, J.; Choy, C.; Kowolik, C.M.; Anderson, A.; Duenas, V.J.; Waliany, S.; Chen, B.T.; Chen, M.Y.; Jandial, R. Co-evolution of breast-to-brain metastasis and neural progenitor cells. Clin. Exp. Metastasis 2013, 30, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S.; Camphausen, K.A.; Smith, Q.R. Brain metastases as preventive and therapeutic targets. Nat. Rev. Cancer 2011, 11, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Baeten, K.M.; Akassoglou, K. Extracellular Matrix and Matrix Receptors in Blood-Brain Barrier Formation and Stroke. Dev. Neurobiol. 2011, 1013–1039. [Google Scholar] [CrossRef] [PubMed]
- Termini, J.; Neman, J.; Jandial, R. Role of the neural niche in brain metastatic cancer. Cancer Res. 2014, 74, 4011–4015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, D. Microenvironment determinants of brain metastasis. Cell Biosci. 2011, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Heyn, C.; Ronald, J.A.; Ramadan, S.S.; Snir, J.A.; Barry, A.M.; MacKenzie, L.T.; Mikulis, D.J.; Palmieri, D.; Bronder, J.L.; Steeg, P.S.; et al. In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain. Magn. Reson. Med. 2006, 56, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.N.; van Rossum, D.; Sieger, D.; Siam, L.; Klemm, F.; Bleckmann, A.; Bayerlova, M.; Farhat, K.; Scheffel, J.; Schulz, M.; et al. Carcinoma cells misuse the host tissue damage response to invade the brain. Glia 2013, 61, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Woditschka, S.; Evans, L.; Duchnowska, R.; Reed, L.T.; Palmieri, D.; Qian, Y.; Badve, S.; Sledge, G., Jr.; Gril, B.; Aladjem, M.I.; et al. DNA double-strand break repair genes and oxidative damage in brain metastasis of breast cancer. J. Natl. Cancer Inst. 2014, 106, 145. [Google Scholar] [CrossRef] [PubMed]
- Niikura, N.; Hayashi, N.; Masuda, N.; Takashima, S.; Nakamura, R.; Watanabe, K.; Kanbayashi, C.; Ishida, M.; Hozumi, Y.; Tsuneizumi, M.; et al. Treatment outcomes and prognostic factors for patients with brain metastases from breast cancer of each subtype: A multicenter retrospective analysis. Breast Cancer Res. Treat. 2014, 147, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Neman, J.; Termini, J.; Wilczynski, S.; Vaidehi, N.; Choy, C.; Kowolik, C.M.; Li, H.; Hambrecht, A.C.; Roberts, E.; Jandial, R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Sci. USA 2014, 111, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Gril, B.; Palmieri, D.; Qian, Y.; Anwar, T.; Liewehr, D.J.; Steinberg, S.M.; Andreu, Z.; Masana, D.; Fernandez, P.; Steeg, P.S.; et al. Pazopanib inhibits the activation of PDGFRbeta-expressing astrocytes in the brain metastatic microenvironment of breast cancer cells. Am. J. Pathol. 2013, 182, 2368–2379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Gritsenko, P.G.; Ilina, O.; Friedl, P. Interstitial guidance of cancer invasion. J. Pathol. 2012, 226, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Rajky, O.; Winkler, F.; Bartsch, R.; Furtner, J.; Hainfellner, J.A.; Goodman, S.L.; Weller, M.; Schittenhelm, J.; Preusser, M. Invasion patterns in brain metastases of solid cancers. Neuro Oncol. 2013, 15, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Yano, S.; Zhang, R.D.; Fujimaki, T.; Bucana, C.D. The seed and soil hypothesis: Vascularisation and brain metastases. Lancet Oncol. 2002, 3, 53–57. [Google Scholar] [CrossRef]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R., 3rd; Felding-Habermann, B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Ramkissoon, S.H.; Xie, S.; Goel, S.; Stover, D.G.; Guo, H.; Luu, V.; Marco, E.; Ramkissoon, L.A.; Kang, Y.J.; et al. Combination inhibition of PI3K and mTORC1 yields durable remissions in mice bearing orthotopic patient-derived xenografts of HER2-positive breast cancer brain metastases. Nat. Med. 2016, 22, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, L.; Simpson, P.T.; Smart, C.E.; Cocciardi, S.; Waddell, N.; Lane, A.; Morrison, B.J.; Vargas, A.; Healey, S.; Beesley, J.; et al. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res. 2010, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Lockman, P.R.; Mittapalli, R.K.; Taskar, K.S.; Rudraraju, V.; Gril, B.; Bohn, K.A.; Adkins, C.E.; Roberts, A.; Thorsheim, H.R.; Gaasch, J.A.; et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res. 2010, 16, 5664–5678. [Google Scholar] [CrossRef] [PubMed]
- Taskar, K.S.; Rudraraju, V.; Mittapalli, R.K.; Samala, R.; Thorsheim, H.R.; Lockman, J.; Gril, B.; Hua, E.; Palmieri, D.; Polli, J.W.; et al. Lapatinib Distribution in HER2 Overexpressing Experimental Brain Metastases of Breast Cancer. Pharm. Res. 2012, 29, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Dijkers, E.C.; Oude Munnink, T.H.; Kosterink, J.G.; Brouwers, A.H.; Jager, P.L.; de Jong, J.R.; van Dongen, G.A.; Schroder, C.P.; Lub-de Hooge, M.N.; de Vries, E.G. Biodistribution of 89Zr-trastuzumab and PET imaging of HER2-positive lesions in patients with metastatic breast cancer. Clin. Pharmacol. Ther. 2010, 87, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Kodack, D.P.; Askoxylakis, V.; Ferraro, G.B.; Fukumura, D.; Jain, R.K. Emerging Strategies for Treating Brain Metastases from Breast Cancer. Cancer Cell 2015, 27, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Laforest, R.; Lapi, S.E.; Oyama, R.; Bose, R.; Tabchy, A.; Marquez-Nostra, B.V.; Burkemper, J.; Wright, B.D.; Frye, J.; Frye, S.; et al. [89Zr]Trastuzumab: Evaluation of Radiation Dosimetry, Safety, and Optimal Imaging Parameters in Women with HER2-Positive Breast Cancer. Mol. Imaging Biol. 2016, 18, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, J.E.; Bading, J.R.; Colcher, D.M.; Conti, P.S.; Frankel, P.H.; Carroll, M.I.; Tong, S.; Poku, E.; Miles, J.K.; Shively, J.E.; et al. Functional imaging of HER2-positive metastatic breast cancer using (64)Cu-DOTA-trastuzumab PET. J. Nucl. Med. 2014, 55, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Kurihara, H.; Yonemori, K.; Tsuda, H.; Suzuki, J.; Kono, Y.; Honda, N.; Kodaira, M.; Yamamoto, H.; Yunokawa, M.; et al. 64Cu-DOTA-trastuzumab PET imaging in patients with HER2-positive breast cancer. J. Nucl. Med. 2013, 54, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Hatori, T.; Murata, N.; Zhang, Z.A.; Ishikawa, F.; Nonaka, H.; Iwabuchi, S.; Samejima, H. A double three-step theory of brain metastasis in mice: The role of the pia mater and matrix metalloproteinases. Neuropathol. Appl. Neurobiol. 2007, 33, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Chen, D.; Piccart, M.; Rugo, H.S.; Burris, H.A., 3rd; Pritchard, K.I.; Campone, M.; Noguchi, S.; Perez, A.T.; Deleu, I.; et al. Correlative Analysis of Genetic Alterations and Everolimus Benefit in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From BOLERO-2. J. Clin. Oncol. 2016, 34, 419–426. [Google Scholar] [PubMed]
- Garrett, J.T.; Olivares, M.G.; Rinehart, C.; Granja-Ingram, N.D.; Sanchez, V.; Chakrabarty, A.; Dave, B.; Cook, R.S.; Pao, W.; McKinely, E.; et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 5021–5026. [Google Scholar] [CrossRef]
- Sergina, N.V.; Rausch, M.; Wang, D.; Blair, J.; Hann, B.; Shokat, K.M.; Moasser, M.M. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007, 445, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Lyu, H.; Huang, J.; Liu, B. Targeting of erbB3 receptor to overcome resistance in cancer treatment. Mol. Cancer 2014, 13, 105. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.M.; Hutchinson, K.; Williams, M.M.; Stanford, J.C.; Balko, J.M.; Young, C.; Kuba, M.G.; Sanchez, V.; Williams, A.J.; Hicks, D.J.; et al. ErbB3 downregulation enhances luminal breast tumor response to antiestrogens. J. Clin. Investig. 2013, 123, 4329–4343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Chang, Y.; Rios, A.; An, Z. HER3/ErbB3, an emerging cancer therapeutic target. Acta Biochim. Biophys. Sin. 2016, 48, 39–48. [Google Scholar] [CrossRef] [PubMed]
- US National Institutes of Health. Available online: http://www.clinicaltrials.gov/ (accessed on 1 November 2016).
- Bos, P.D.; Zhang, X.H.F.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- McMullin, R.P.; Wittner, B.S.; Yang, C.; Denton-Schneider, B.R.; Hicks, D.; Singavarapu, R.; Moulis, S.; Lee, J.; Akbari, M.R.; Narod, S.A.; et al. A BRCA1 deficient-like signature is enriched in breast cancer brain metastases and predicts DNA damage-induced poly (ADP-ribose) polymerase inhibitor sensitivity. Breast Cancer Res. 2014, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Bollig-Fischer, A.; Michelhaugh, S.K.; Wijesinghe, P.; Dyson, G.; Kruger, A.; Palanisamy, N.; Choi, L.; Alosh, B.; Ali-Fehmi, R.; Mittal, S. Cytogenomic profiling of breast cancer brain metastases reveals potential for repurposing targeted therapeutics. Oncotarget 2015, 6, 14614–14624. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Park, K.; Lim, S.H.; Kim, H.S.; Yoo, K.H.; Jung, K.S.; Song, H.N.; Hong, M.; Do, I.G.; Ahn, T.; et al. Mutational profiling of brain metastasis from breast cancer: Matched pair analysis of targeted sequencing between brain metastasis and primary breast cancer. Oncotarget 2015, 6, 43731–43742. [Google Scholar] [PubMed]
- Varešlija, D.; Fagan, A.; Buckley, P.; Farrell, M.; Hill, A.; Young, L. Whole genome transcriptome analysis of sequential breast to brain metastasis uncovers new signalling pathways and druggable targets. Cancer Res. 2016, 76, P20503. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, K.; Lee, E.; Ahn, T.; Jung, H.H.; Lim, S.H.; Hong, M.; Do, I.G.; Cho, E.Y.; Kim, D.H.; et al. Gene Expression Profiling of Breast Cancer Brain Metastasis. Sci. Rep. 2016, 6, 28623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, W.C.; Zhang, L.; Zhang, C.; Lowery, F.J.; Ding, Z.; Guo, H.; Wang, H.; Huang, S.; Sahin, A.A.; et al. SRC family kinases as novel therapeutic targets to treat breast cancer brain metastases. Cancer Res. 2013, 73, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Bronder, J.L.; Herring, J.M.; Yoneda, T.; Weil, R.J.; Stark, A.M.; Kurek, R.; Vega-Valle, E.; Feigenbaum, L.; Halverson, D.; et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007, 67, 4190–4198. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, D.P.; Subramanian, P.; Deshpande, M.; Graves, C.; Gordon, I.; Qian, Y.; Snitkovsky, Y.; Liewehr, D.J.; Steinberg, S.M.; Paltan-Ortiz, J.D.; et al. Opposing effects of pigment epithelium-derived factor on breast cancer cell versus neuronal survival: Implication for brain metastasis and metastasis-induced brain damage. Cancer Res. 2012, 72, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Jacob, L.S.; Vanharanta, S.; Obenauf, A.C.; Pirun, M.; Viale, A.; Socci, N.D.; Massague, J. Metastatic Competence Can Emerge with Selection of Preexisting Oncogenic Alleles without a Need of New Mutations. Cancer Res. 2015, 75, 3713–3719. [Google Scholar] [CrossRef] [PubMed]
- Laimito, K.R.; Gamez-Pozo, A.; Sepulveda, J.; Manso, L.; Lopez-Vacas, R.; Pascual, T.; Fresno Vara, J.A.; Ciruelos, E. Characterisation of the triple negative breast cancer phenotype associated with the development of central nervous system metastases. Ecancermedicalscience 2016, 10, 632. [Google Scholar] [PubMed]
- Drolez, A.; Vandenhaute, E.; Delannoy, C.P.; Dewald, J.H.; Gosselet, F.; Cecchelli, R.; Julien, S.; Dehouck, M.P.; Delannoy, P.; Mysiorek, C. ST6GALNAC5 Expression Decreases the Interactions between Breast Cancer Cells and the Human Blood-Brain Barrier. Int. J. Mol. Sci. 2016, 17, 1309. [Google Scholar] [CrossRef] [PubMed]
- Vargo-Gogola, T.; Rosen, J.M. Modelling breast cancer: One size does not fit all. Nat. Rev. Cancer 2007, 7, 659–672. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Magerle, M.; Ilhan-Mutlu, A.; Dinhof, C.; Widhalm, G.; Dieckman, K.; Marosi, C.; Wohrer, A.; Hackl, M.; Zochbauer-Muller, S.; et al. Frequent overexpression of ErbB—Receptor family members in brain metastases of non-small cell lung cancer patients. Acta Pathol. Microbiol. Immunol. Scand. 2013, 121, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Behrens, C.; Feng, L.; Ozburn, N.; Tang, X.; Yin, G.; Komaki, R.; Varella-Garcia, M.; Hong, W.K.; Aldape, K.D.; et al. HER family receptor abnormalities in lung cancer brain metastases and corresponding primary tumors. Clin. Cancer Res. 2009, 15, 4829–4837. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Fitzgerald, D.; Shreeve, S.M.; Hua, E.; Bronder, J.L.; Weil, R.J.; Davis, S.; Stark, A.M.; Merino, M.J.; Kurek, R.; et al. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol. Cancer Res. 2009, 7, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Owonikoko, T.K.; Arbiser, J.; Zelnak, A.; Shu, H.K.; Shim, H.; Robin, A.M.; Kalkanis, S.N.; Whitsett, T.G.; Salhia, B.; Tran, N.L.; et al. Current approaches to the treatment of metastatic brain tumours. Nat. Rev. Clin. Oncol. 2014, 11, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.A.; Mietz, J.; Arteaga, C.L.; de Pinho, R.A.; Mohla, S. Brain metastasis: Opportunities in basic and translational research. Cancer Res. 2009, 69, 6015–6020. [Google Scholar] [CrossRef] [PubMed]
- Burvenich, I.J.; Lee, F.T.; Cartwright, G.A.; O’Keefe, G.J.; Makris, D.; Cao, D.; Gong, S.; Chueh, A.C.; Mariadason, J.M.; Brechbiel, M.W.; et al. Molecular imaging of death receptor 5 occupancy and saturation kinetics in vivo by humanized monoclonal antibody CS-1008. Clin. Cancer Res. 2013, 19, 5984–5993. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Baselga, J.; Miles, D.; Im, Y.H.; Quah, C.; Lee, L.F.; Cortes, J. Incidence of central nervous system metastases in patients with HER2-positive metastatic breast cancer treated with pertuzumab, trastuzumab, and docetaxel: Results from the randomized phase III study CLEOPATRA. Ann. Oncol. 2014, 25, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Amiri-Kordestani, L.; Palmieri, D.; Liewehr, D.J.; Steeg, P.S. CNS metastases in breast cancer: Old challenge, new frontiers. Clin. Cancer Res. 2013, 19, 6404–6418. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Winkler, F.; Collette, L.; Haller, S.; Marreaud, S.; Soffietti, R.; Klein, M.; Reijneveld, J.C.; Tonn, J.C.; Baumert, B.G.; et al. Trial design on prophylaxis and treatment of brain metastases: Lessons learned from the EORTC Brain Metastases Strategic Meeting 2012. Eur. J. Cancer 2012, 48, 3439–3447. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, J.S.; Park, E.S.; Lee, J.S.; Lin, Q.; Langley, R.R.; Maya, M.; He, J.; Kim, S.W.; Weihua, Z.; et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia 2011, 13, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Balasubramanian, K.; Lin, Q.; Kim, S.W.; Kim, S.J. The brain microenvironment and metastasis. Mol. Cells 2010, 30, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.A.; Ertugrul, H.; Firat, U.; Kucukoner, M.; Inal, A.; Urakci, Z.; Pekkolay, Z.; Isikdogan, A. Brain metastases in HER2-positive metastatic breast cancer patients who received chemotherapy with or without trastuzumab. Breast Cancer 2015, 22, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Nahas, G.; Bliss, S.A.; Sinha, G.; Ganta, T.; Greco, S.J.; Rameshwar, P. Is reduction of tumor burden sufficient for the 21st century? Cancer Lett. 2015, 356, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, L.; Cotte, F.E.; Mercier, F.; Vainchtock, A.; Vidal-Trecan, G.; Durand-Zaleski, I. Burden of breast cancer with brain metastasis: A French national hospital database analysis. J. Med. Econ. 2012, 15, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Koay, E.; Sulman, E.P. Management of brain metastasis: Past lessons, modern management, and future considerations. Curr. Oncol. Rep. 2012, 14, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Weathers, T.; Haney, L.G.; Timmerman, R.; Dickler, M.; Shen, J.; Sledge, G.W., Jr. Occult central nervous system involvement in patients with metastatic breast cancer: Prevalence, predictive factors and impact on overall survival. Ann. Oncol. 2003, 14, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Cummings, M.C.; Simpson, P.T.; Reid, L.E.; Jayanthan, J.; Skerman, J.; Song, S.; McCart Reed, A.E.; Kutasovic, J.R.; Morey, A.L.; Marquart, L.; et al. Metastatic progression of breast cancer: Insights from 50 years of autopsies. J. Pathol. 2014, 232, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T. Breast carcinoma: Pattern of metastasis at autopsy. J. Surg. Oncol. 1983, 23, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fouad, A.; Pickren, J.W.; Lane, W.W. Central nervous system metastasis from breast carcinoma: Autopsy study. Cancer 1983, 52, 2349–2354. [Google Scholar] [CrossRef]
- Murrell, D.H.; Foster, P.J.; Chambers, A.F. Brain metastases from breast cancer: Lessons from experimental magnetic resonance imaging studies and clinical implications. J. Mol. Med. 2014, 92, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Kalita-de Croft, P.; Al-Ejeh, F.; McCart Reed, A.E.; Saunus, J.M.; Lakhani, S.R. Omics Approaches in Breast Cancer Research and Clinical Practice. Adv. Anat. Pathol. 2016, 23, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Bellon, J.R.; Winer, E.P. CNS metastases in breast cancer. J. Clin. Oncol. 2004, 22, 3608–3617. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Monsky, W.L.; Mouta Carreira, C.; Tsuzuki, Y.; Gohongi, T.; Fukumura, D.; Jain, R.K. Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: Mammary fat pad versus cranial tumors. Clin. Cancer Res. 2002, 8, 1008–1013. [Google Scholar] [PubMed]
- Moeller, B.J.; Richardson, R.A.; Dewhirst, M.W. Hypoxia and radiotherapy: Opportunities for improved outcomes in cancer treatment. Cancer Metastasis Rev. 2007, 26, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Milojkovic Kerklaan, B.; van Tellingen, O.; Huitema, A.D.; Beijnen, J.H.; Boogerd, W.; Schellens, J.H.; Brandsma, D. Strategies to target drugs to gliomas and CNS metastases of solid tumors. J. Neurol. 2016, 263, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Tong, R.T.; Munn, L.L. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: Insights from a mathematical model. Cancer Res. 2007, 67, 2729–2735. [Google Scholar] [CrossRef] [PubMed]
- Kobus, T.; Zervantonakis, I.K.; Zhang, Y.; McDannold, N.J. Growth inhibition in a brain metastasis model by antibody delivery using focused ultrasound-mediated blood-brain barrier disruption. J. Control. Release 2016, 238, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Alkins, R.; Burgess, A.; Ganguly, M.; Francia, G.; Kerbel, R.; Wels, W.S.; Hynynen, K. Focused ultrasound delivers targeted immune cells to metastatic brain tumors. Cancer Res. 2013, 73, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, P.; Shalviri, A.; Henderson, J.T.; He, C.; Foltz, W.D.; Prasad, P.; Brodersen, P.M.; Chen, Y.; da Costa, R.; et al. A multifunctional polymeric nanotheranostic system delivers doxorubicin and imaging agents across the blood-brain barrier targeting brain metastases of breast cancer. ACS Nano 2014, 8, 9925–9940. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, R.K.; Liu, X.; Adkins, C.E.; Nounou, M.I.; Bohn, K.A.; Terrell, T.B.; Qhattal, H.S.; Geldenhuys, W.J.; Palmieri, D.; Steeg, P.S.; et al. Paclitaxel-hyaluronic nanoconjugates prolong overall survival in a preclinical brain metastases of breast cancer model. Mol. Cancer Ther. 2013, 12, 2389–2399. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.M.; Aidoudi-Ahmed, S.; Sharma, S.; Kotamraju, V.R.; Foster, P.J.; Sugahara, K.N.; Ruoslahti, E.; Rutt, B.K. Nanoparticles coated with the tumor-penetrating peptide iRGD reduce experimental breast cancer metastasis in the brain. J. Mol. Med. 2015, 93, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Patil, R.; Ljubimov, A.V.; Gangalum, P.R.; Ding, H.; Portilla-Arias, J.; Wagner, S.; Inoue, S.; Konda, B.; Rekechenetskiy, A.; Chesnokova, A.; et al. MRI virtual biopsy and treatment of brain metastatic tumors with targeted nanobioconjugates: Nanoclinic in the brain. ACS Nano 2015, 9, 5594–5608. [Google Scholar] [CrossRef] [PubMed]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Preusser, M. The inflammatory microenvironment in brain metastases: Potential treatment target? Chin. Clin. Oncol. 2015, 4, 21. [Google Scholar] [PubMed]
- Beham, H.S. Hercules Slaying the Hydra. Available online: https://de.wikipedia.org/wiki/Datei:Hercules_slaying_the_Hydra.jpg (accessed on 1 November 2016).
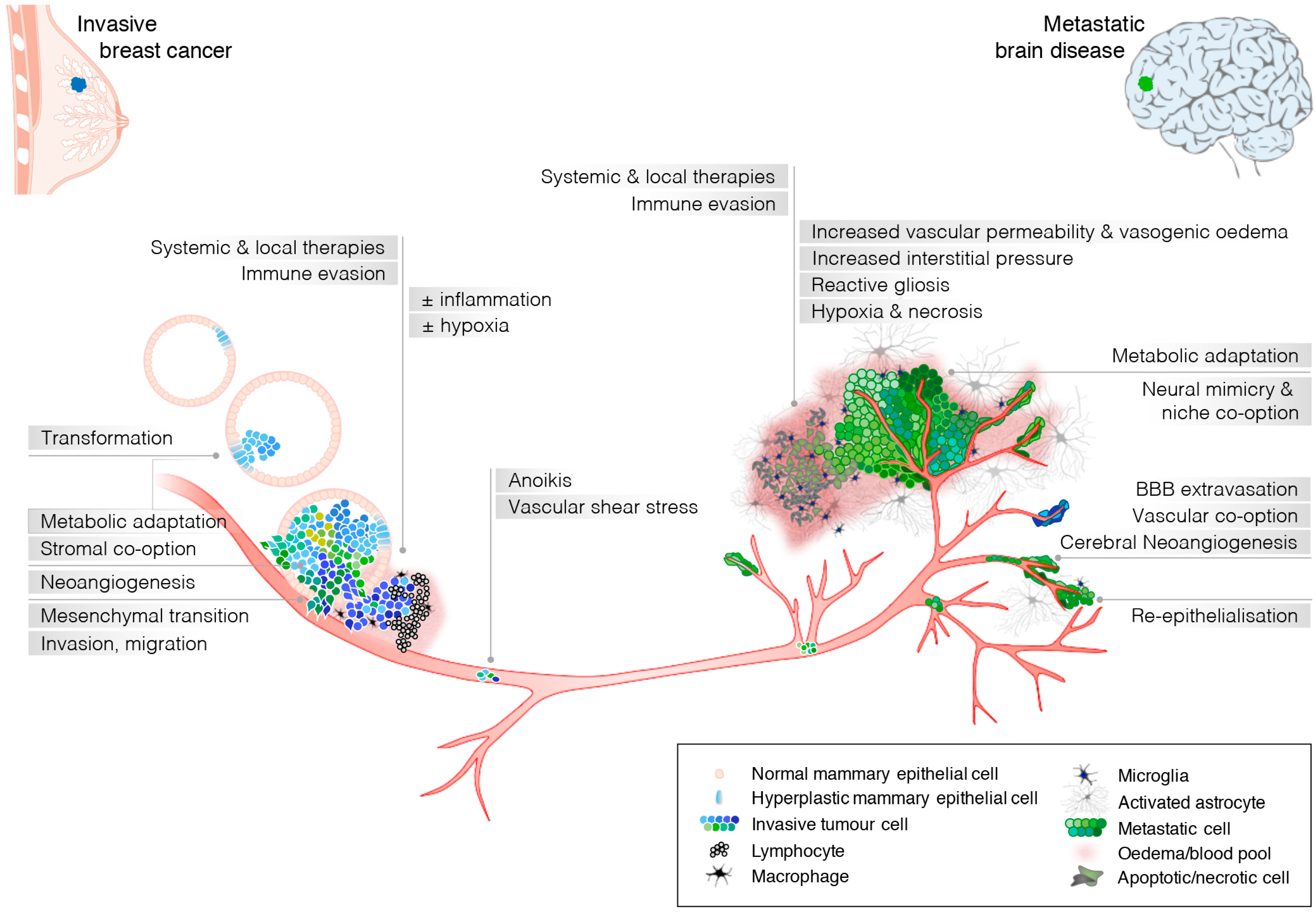


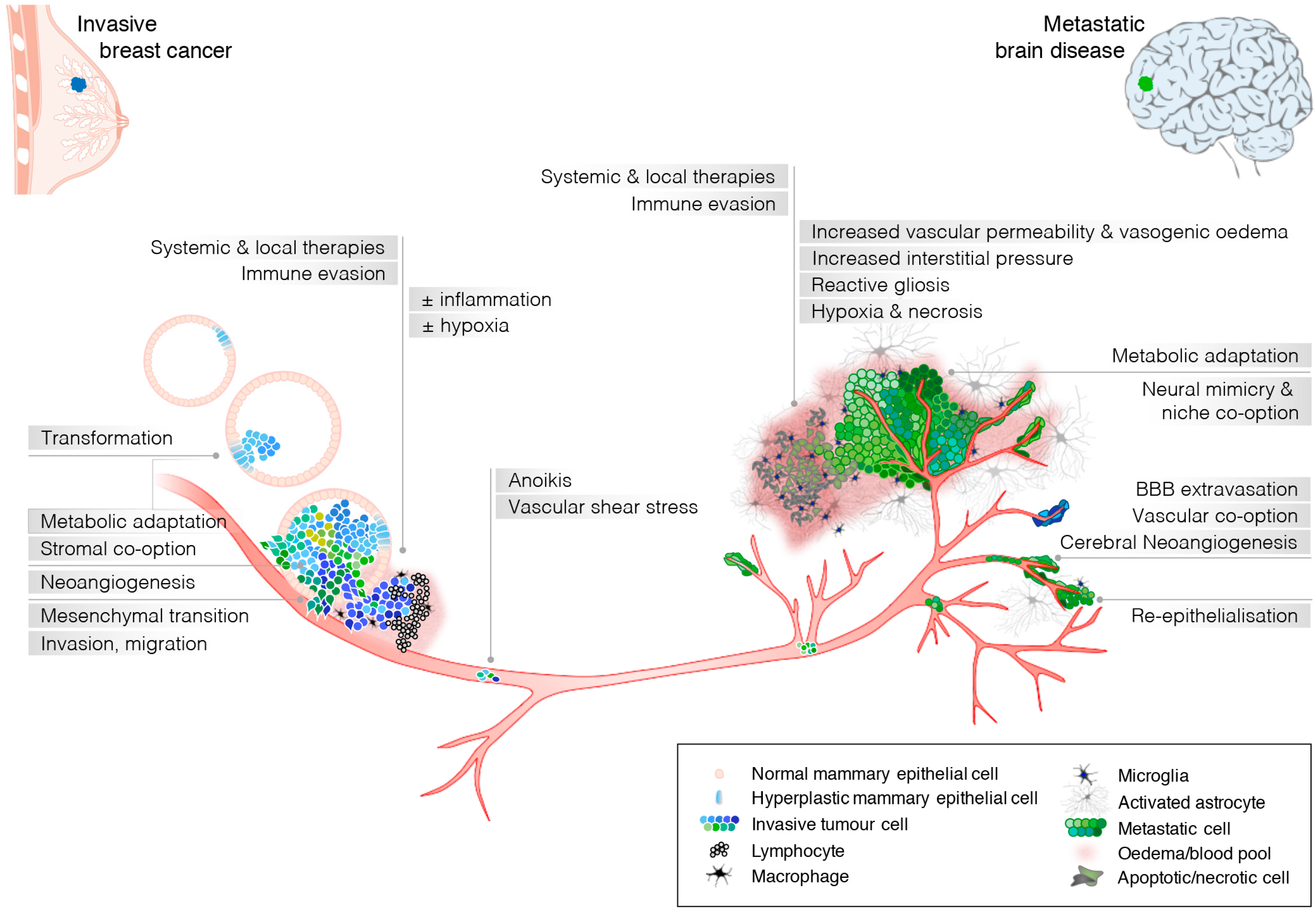
{kind=link}
{kind=link}
{kind=link}
| NCT-ID | Subtype | Phase | Experimental Arm(s) | Comparator Arm | Approach | Primary Endpoints |
|---|---|---|---|---|---|---|
| 02429570 | All | 0 | Meclofenamate | NA | GAP junction modulator | ORR, PFS, safety |
| 01621906 | All | 0 | WBRT + Sorafenib + [18F]FLT PET at baseline | WBRT + [18F]FLT PET at baseline | XRT + VEGFR | RR (radiographic) |
| 01386580 | All | 1/2 | Glutathione pegylated liposomal doxorubicin | Glutathione-pegylated liposomal dox + Trastuz | Carrier (CTx + HER2) | MTD, safety |
| 01132664 | HER2+ | 1/2 | Buparlisib + Trastuz | Buparlisib + Trastuz + Capecitabine | VEGFR + HER2 + CTx | MTD, RR, PFS, safety |
| 02154529 | HER2+ | 1/2 | Tesevatinib + Trastuz | Tesevatinib dose escalation + Trastuz | Broad-spec RTKi | MTD, PFS, RR, safety |
| 01921335 | HER2+ | 1 | ARRY-380 + Trastuz | ARRY-380 dose escalation + Trastuz | HER2 | MTD, RR and PFS |
| 01332929 | All | 1 | Bevacizumab + WRBT | Bevacizumab dose escalation + WRBT | XRT + VEGFR | MTD, RR, PFS |
| 02598427 | HER2+ | 1 | Intrathecal Pertuzumab + Trastuz | Pertuzumab dose escalation + Trastuz | HER2 (CSF delivery) | MTD, safety |
| 02650752 | HER2+ | 1 | Lapatinib + Capecitabine | Lapatinib dose escalation + Capecitabine | CTx + HER2 | MTD, RR, PFS |
| 01276210 | All | 1 | Sorafenib tosylate + SRS | Sorafenib tosylate dose escalation + SRS | VEGFR + Raf kinase | MTD, RR, PFS |
| 00981890 | All | 1 | Sunitinib + SRS | NA | XRT + VEGFR | Safety, MTD |
| 00649207 | All | 1 | Veliparib + WBRT | Veliparib dose escalation + WBRT | PARPi | MTD, safety |
| 01724606 | All | 1 | Sorafenib + WBRT | Sorafenib dose escalation + WBRT | XRT + VEGFR | MTD, safety |
| 02308020 | All | 2 | Abemaciclib | NA | CDK4/6i | RR, PFS, safety |
| 02768337 | All | 2 | Afatinib + 4 Gy XRT | Afatinib | XRT + HER2 | Drug uptake |
| 01441596 | HER2+ | 2 | Afatinib + vinorelbine | Afatinib | CTx + HER2 | PFS |
| 02048059 | All | 2 | ANG1005 (formerly GRN1005) | NA | Carrier (CTx) | RR, PFS, OS |
| 01898130 | All | 2 | Bevacizumab | NA | VEGFR + HER2 | RR, PFS, safety |
| 02000882 | All | 2 | Buparlisib + Capecitabine (+Trastuz if HER2+) | NA | CTx + panPI3Ki | RR |
| 01934894 | HER2+ | 2 | Cabazitaxel + Lapatinib | Cabazitaxel + Lapatinib (different doses) | CTx + HER2 | RR, MTD, safety |
| 02260531 | All | 2 | Cabozantinib + Trastuz | Cabozantinib | c-met + VEGFR | RR, PFS, safety |
| 02669914 | All | 2 | Durvalumab (MEDI4736) | NA | PDL1i | RR, PFS, safety |
| 01305941 | HER2+ | 2 | Everolimus + Vinorelbine + Trastuz | NA | CTx + HER2 | RR, PFS, safety |
| 01480583 | HER2+ | 2 | GRN1005 + Trastuz | GRN1005 alone | Carrier (CTx + HER2) | RR, PFS, safety |
| 01494662 | HER2+ | 2 | Neratinib (HKI-272) | Neratinib (HKI-272) + Capecitabine | CTx + HER2 | RR, PFS, safety |
| 01173497 | TNBC | 2 | Iniparib + Irinotecan | NA | CTx + PARPi | Efficacy, RR |
| 01783756 | HER2+ | 2 | Lapatinib + Everolimus + Capecitabine | NA | CTx + HER2 + mTORi | RR, PFS, safety |
| 01622868 | HER2+ | 2 | Lapatinib + WBRT or SRS | WBRT or SRS | XRT + HER2 | RR, PFS, safety |
| 01218529 | All | 2 | Lapatinib + WRBT | NA | XRT + HER2 | RR |
| 02614794 | HER2+ | 2 | ONT-380 + Capecitabine + Trastuz | Placebo + Capecitabine + Trastuz | CTx + HER2 | PFS, RR, safety |
| 02774681 | All | 2 | Palbociclib (+Trastuz if HER2+) | NA | CDK4/6i | RR (radiographic), PFS, safety |
| 02312622 | All | 2 | Pegylated irinotecan (NKTR 102) | NA | Carrier (CTx) | Disease control rate, PFS |
| 02536339 | HER2+ | 2 | Pertuzumab + Trastuz | NA | HER2 | RR, PFS, OS, safety |
| 01924351 | HER2+ | 2 | SRS + HER-2 directed therapy | NA | XRT + HER2 | Relapse rate |
| 02571530 | HER2+ | 2 | Intra-arterial cerebral infusion of Trastuz | May consider dose escalation | HER2 | MTD, OS, PFS |
| 00303992 | HER2+ | 2 | Trastuz + Irinotecan | NA | CTx + HER2 | RR, disease progression |
| 02185352 | All | 2 | WBRT + Bevacizumab, Etoposide, Cisplatin | WBRT alone | XRT + VEGFR | RR, PFS |
| 00820222 | HER2+ | 3 | Lapatinib + Capecitabine | Trastuzumab + capecitabine | CTx + HER2 | PFS, RR |
| 00073528 | ER/HER2+ | 3 | Lapatinib + Letrozole | Placebo + Letrozole | CTx (aromatase-i) + HER2 | RR, PFS, safety |
| Study | BCBM Only? | Matched Pairs? | Cohort Size | FF or FFPE | GEX | CNA | Mutation Analysis | Exome | WGS | Targeted or Discovery | Key Findings |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bos 2009 [89] | Yes | No | 1 * | F | Array | No | No | No | No | D | COX2, HBEGF (EGFR ligand), ST6GALNAC5 (a 2,6-sialyltransferase) over-expressed, mediating BC cell passage through the BBB, with ST6GALNAC5 expression enhancing BC cell adhesion to brain endothelial cells |
| da Silva 2010 [73] | No | Some | 78 | FFPE | DASL (512 genes) | No | OncoCarta | No | No | T/D | Over-expression of ≥1 HER, esp HER3 (relative to matched primary tumours); Somatic mutations in EGFR, HRAS, KRAS, NRAS, PIK3CA; Increased activation of MAPK pathway in BM vs. primary tumours |
| Ding 2010 [90] | Yes | Yes | 1 | FF | No | SNP | No | No | Yes | D | Matched peripheral blood, primary tumour, BM and PdX; BM: 2 private mutations, a large deletion, 20 enriched mutations (PdX similar); 2 overlapping large deletions (CTNNA1) in all 3 tumour samples; Variation frequencies indicate metastases arise from a minority of cells in the BC |
| Wikman 2012 [30] | Yes | Some | 25 | FF | in silico | aCGH/AI | GSS | No | No | T/D | 9 loci with significant differences, incl. EGFR amp (7p11.2) & 10q22.3-qter loss; AI at PTEN more frequent in BM (52%) and brain relapsing BC (59%) compared with BC without relapse (18%; p = 0.003) or relapse other than brain (12%; p = 0.006); Loss of PTEN was especially frequent in HER2-negative BM (64%); PTEN mRNA was suppressed in BM compared with primary tumours; PTEN mutations were frequently found in BM |
| McMullin 2014 [91] | Yes | No | 19 | FF | Array | No | GSS | No | No | T/D | BRCA1 deficient-like GEX signature in HER2+ BCBM in absence of BRCA1 mutations; Values significantly higher in HER2-/ER- vs. HER2+/ER+ and HER2-/ER+ tumours |
| Salhia 2014 [32] | Yes | No | 35 | FF | Array | aCGH ^ | No | No | No | D | Frequent large gains 1q, 5p, 8q, 11q, 20q; broad-level deletions (8p, 17p, 21p, Xq); ATAD2, BRAF, DERL1, DNMTRB and NEK2A frequently amplified & overexpressed; ATM, CRYAB and HSPB2 commonly deleted & down-regulated Enrichment in cell cycle and G2/M pathways (incl. AURKA, AURKB & FOXM1; Defects in cell migration and adhesion due to hypermethylation + suppression of PENK, EDN3 and ITGAM; Hypomethylation + induction of KRT8 likely affects adhesion and permeability |
| Bollig-Fischer 2015 [92] | Yes | No | 10 | FF & FFPE | No | aCGH | No | No | No | T/D | Stem cell pluripotency pathway enrichment; Recurring amplification of SOX2, PIK3CA, NTRK1, GNAS, CTNNB1, & FGFR1 |
| Brastianos 2015 [93] | No | Yes | 86 | FF & FFPE | No | No | No | Yes | No | D | 86 trios: matched BM, primary tumours, & normal tissue 53% cases had potentially clinically informative alterations in BM; Individual BM deposits genetically homogenous; Distal extracranial and regional node metastases highly divergent from BM; Alterations associated with PI3K/AKT/mTOR, CDK, & HER2/EGFRi sensitivity in BM |
| Lee 2015 [94] | Yes | Some | 42 | FFPE | No | No | Ion AmpliSeq Cancer | No | No | T | Frequent somatic mutations (e.g., TP53 59.5%, MLH1 14.3%, PIK3CA 14.3%, KIT 7.1%); No significant differences in mutation profiles between BCBM and BC; TP53 mutation frequency higher in BCBM than in primary BC (59.5% vs. 38.9%) |
| Saunus 2015 [29] | No | No | 36 | FF | RNASeq | SNP | No | Yes | No | D | Novel candidate genes: significantly mutated DSC2, ST7, PIK3R1 and SMC5; DNA repair, HER signalling, axon guidance & protein kinase-A signalling pathways; Potentially actionable genomic alterations in 31/36 BMs (86%); Altered patient management (+trastuz) in a case of HER2 status conversion; ERBB2 expression correlated with ERBB3 (p < 0.0001); HER3 & HER4 frequently activated in a cohort of 167 BM (7 primary cancer types); HER3 ligands NRG1/2 barely detectable by RNAseq, with NRG1 (8p12) genomic loss in 63.6% BCBM, suggesting a microenvironmental source of ligand; Mutational signature analysis facilitated identification of primary type for two CUP |
| Vareslija 2015 [95] | Yes | Yes | 7 | U | RNASeq | No | No | No | No | D | ER-specific metastatic pathways; Common pathways altered incl. ECM, adhesion & neuronal differentiation; ANTRX1, THBS2, FAP, VCAN & TIMP2 (invasion/migration/extravasation; EMT/stemness signalling driven by ANTRX1; WNT-driven RUNX prominent in cells acquiring migration ability |
| Lee 2016 [96] | Yes | Some | 41 | FFPE | Nanostring (252 genes) | No | No | No | No | T | 22/252 genes differentially expressed between BC and BCBM; CXCL12, MMP2, MMP11, VCAM1 & MME higher in BC, SOX2 & OLIG2 higher in BM; PAM50 molecular subtype conversion observed in 8/17 pairs (47.1%) |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saunus, J.M.; McCart Reed, A.E.; Lim, Z.L.; Lakhani, S.R. Breast Cancer Brain Metastases: Clonal Evolution in Clinical Context. Int. J. Mol. Sci. 2017, 18, 152. https://doi.org/10.3390/ijms18010152
Saunus JM, McCart Reed AE, Lim ZL, Lakhani SR. Breast Cancer Brain Metastases: Clonal Evolution in Clinical Context. International Journal of Molecular Sciences. 2017; 18(1):152. https://doi.org/10.3390/ijms18010152
Chicago/Turabian StyleSaunus, Jodi M., Amy E. McCart Reed, Zhun Leong Lim, and Sunil R. Lakhani. 2017. "Breast Cancer Brain Metastases: Clonal Evolution in Clinical Context" International Journal of Molecular Sciences 18, no. 1: 152. https://doi.org/10.3390/ijms18010152
APA StyleSaunus, J. M., McCart Reed, A. E., Lim, Z. L., & Lakhani, S. R. (2017). Breast Cancer Brain Metastases: Clonal Evolution in Clinical Context. International Journal of Molecular Sciences, 18(1), 152. https://doi.org/10.3390/ijms18010152