
Unconventional Pathways of Secretion Contribute to Inflammation

Abstract
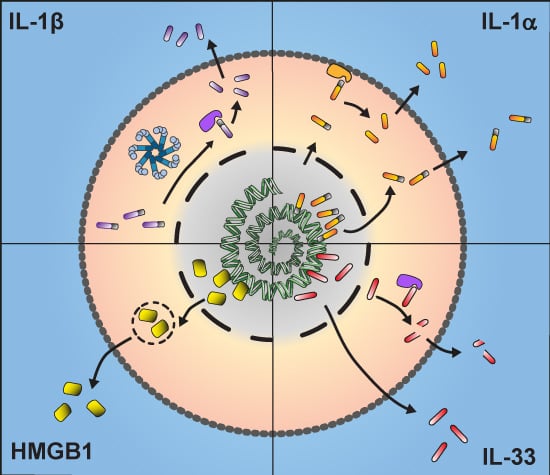
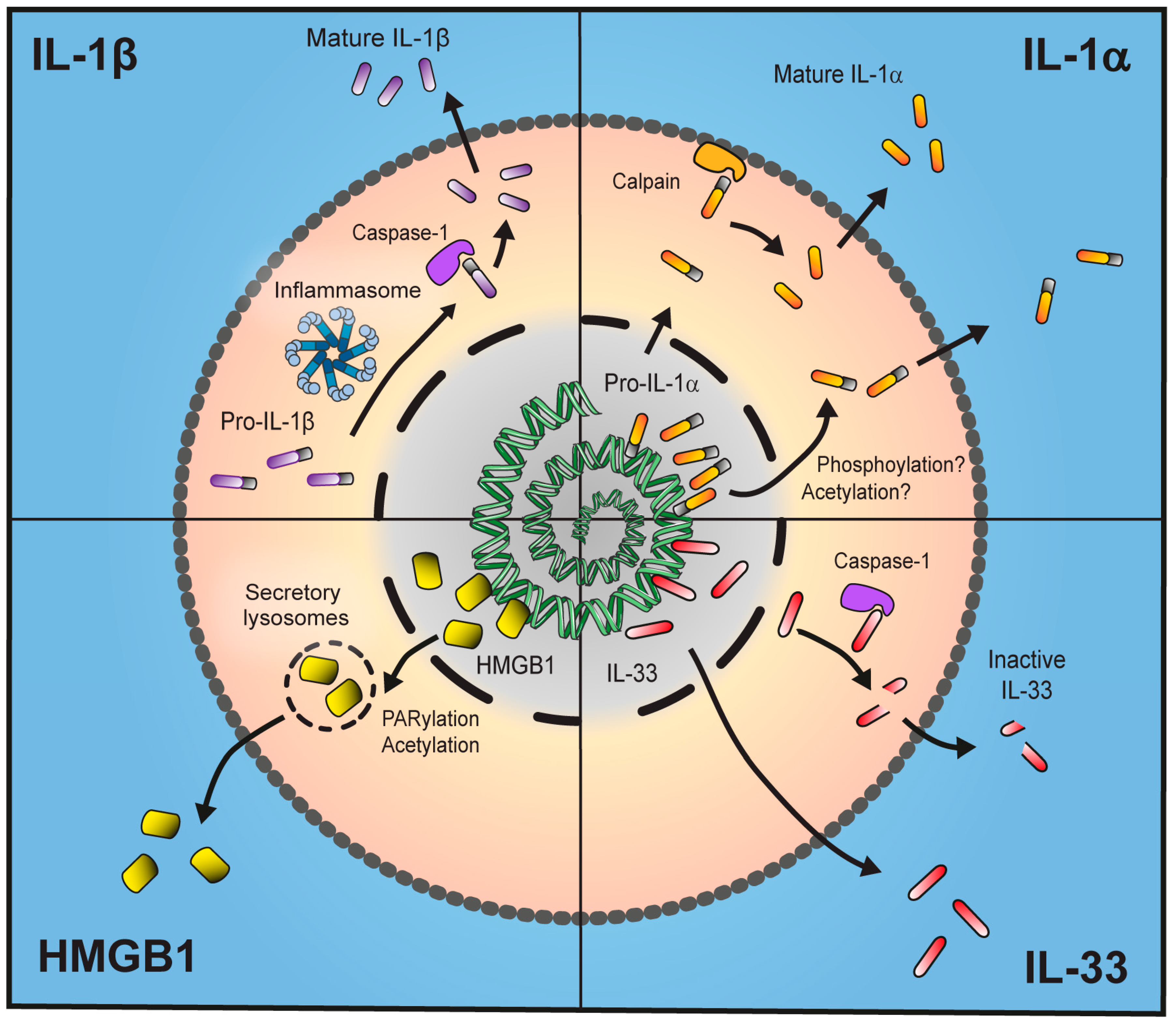
:
{kind=link}
{kind=link}
1. Introduction
1.1. Unconventional Protein Secretion
1.2. Inflammation
2. IL-1β
3. IL-1α
4. IL-33
5. HMGB1
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Lee, M.C.S.; Miller, E.A.; Goldberg, J.; Orci, L.; Schekman, R. Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 2004, 20, 87–123. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Engling, A.; Backhaus, R.; Stegmayer, C.; Zehe, C.; Seelenmeyer, C.; Kehlenbach, A.; Schwappach, B.; Wegehingel, S.; Nickel, W. Biosynthetic FGF-2 is targeted to non-lipid raft microdomains following translocation to the extracellular surface of CHO cells. J. Cell Sci. 2002, 115, 3619–3631. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.C. Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochim. Biophys. Acta. 1999, 1473, 172–185. [Google Scholar] [CrossRef]
- Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2009, 10, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Malhotra, V.; Nickel, W. Diversity in unconventional protein secretion. J. Cell Sci. 2012, 125, 5251–5255. [Google Scholar] [CrossRef] [PubMed]
- Bikfalvi, A.; Klein, S.; Pintucci, G.; Rifkin, D.B. Biological roles of fibroblast growth factor-2. Endocr. Rev. 1997, 18, 26–45. [Google Scholar] [CrossRef] [PubMed]
- Mignatti, P.; Morimoto, T.; Rifkin, D.B. Basic fibroblast growth factor, a protein devoid of secretory signal sequence, is released by cells via a pathway independent of the endoplasmic reticulum-Golgi complex. J. Cell. Physiol. 1992, 151, 81–93. [Google Scholar] [CrossRef] [PubMed]
- La Venuta, G.; Wegehingel, S.; Sehr, P.; Müller, H.-M.; Dimou, E.; Steringer, J.P.; Grotwinkel, M.; Hentze, N.; Mayer, M.P.; Will, D.W.; et al. Small molecule inhibitors targeting Tec kinase block unconventional secretion of fibroblast growth factor 2. J. Biol. Chem. 2016, 291, 17787–17803. [Google Scholar] [CrossRef] [PubMed]
- Steringer, J.P.; Müller, H.-M.; Nickel, W. Unconventional secretion of fibroblast growth factor 2—A novel type of protein translocation across membranes? J. Mol. Biol. 2015, 427, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N.; Barondes, S.H. Evidence for export of a muscle lectin from cytosol to extracellular matrix and for a novel secretory mechanism. J. Cell Biol. 1990, 110, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Seelenmeyer, C.; Wegehingel, S.; Tews, I.; Künzler, M.; Aebi, M.; Nickel, W. Cell surface counter receptors are essential components of the unconventional export machinery of galectin-1. J. Cell Biol. 2005, 171, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Seelenmeyer, C.; Stegmayer, C.; Nickel, W. Unconventional secretion of fibroblast growth factor 2 and galectin-1 does not require shedding of plasma membrane-derived vesicles. FEBS Lett. 2008, 582, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A.; Cozzolino, F.; Talio, M.; Sitia, R. A novel secretory pathway for interleukin-1β, a protein lacking a signal sequence. EMBO J. 1990, 9, 1503–1510. [Google Scholar] [PubMed]
- Luheshi, N.M.; Rothwell, N.J.; Brough, D. The dynamics and mechanisms of interleukin-1α and β nuclear import. Traffic 2009, 10, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Lopez-Castejon, G.; Brough, D. Caspase-1: Is IL-1 just the tip of the ICEberg? Cell Death Dis. 2012, 3, e338. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Perregaux, D.; Gabel, C.A. Interleukin-1β maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [PubMed]
- Ferrari, D.; Pizzirani, C.; Adinolfi, E.; Lemoli, R.M.; Curti, A.; Idzko, M.; Panther, E.; di Virgilio, F. The P2X7 receptor: A key player in IL-1 processing and release. J. Immunol. 2006, 176, 3877–3883. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A genome-wide CRISPR screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem. 2015. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Brough, D.; Rothwell, N.J. Caspase-1-dependent processing of pro-interleukin-1β is cytosolic and precedes cell death. J. Cell Sci. 2007, 120, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Martín-Sánchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.J.D.; Mortellaro, A.; et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.P.; Kearney, C.J.; Clancy, D.M.; Martin, S.J. Diverse activators of the NLRP3 inflammasome promote IL-1β secretion by triggering necrosis. Cell Rep. 2015, 11, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Viganò, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar] [CrossRef] [PubMed]
- Englezou, P.C.; Rothwell, S.W.; Ainscough, J.S.; Brough, D.; Landsiedel, R.; Verkhratsky, A.; Kimber, I.; Dearman, R.J. P2X7R activation drives distinct IL-1 responses in dendritic cells compared to macrophages. Cytokine 2015, 74, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, M.; Katsnelson, M.A.; Dubyak, G.R.; Pearlman, E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat. Commun. 2016, 7, 10555. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Zhang, Y.; Krantz, B.A.; Miao, E.A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 2016, 213, 2113–2128. [Google Scholar] [CrossRef] [PubMed]
- Andrei, C.; Dazzi, C.; Lotti, L.; Torrisi, M.R.; Chimini, G.; Rubartelli, A. The secretory route of the leaderless protein interleukin 1β involves exocytosis of endolysosome-related vesicles. Mol. Biol. Cell 1999, 10, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Franchi, L.; Nunez, G.; Dubyak, G.R. Nonclassical IL-1β secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J. Immunol. 2007, 179, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Ramachandra, L.; Mohr, S.; Franchi, L.; Harding, C.V.; Nunez, G.; Dubyak, G.R. P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase-1. J. Immunol. 2009, 182, 5052–5062. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.B.; Opitz, B.; van der Meer, J.H.M.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood 2009, 113, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Piccini, A.; Carta, S.; Tassi, S.; Lasiglié, D.; Fossati, G.; Rubartelli, A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1β and IL-18 secretion in an autocrine way. Proc. Natl. Acad. Sci. USA 2008, 105, 8067–8072. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1β secretion by targeting pro-IL-1β for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kenny, S.J.; Ge, L.; Xu, K.; Schekman, R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife 2015. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [PubMed]
- Shao, A.; Wu, H.; Hong, Y.; Tu, S.; Sun, X.; Wu, Q.; Zhao, Q.; Zhang, J.; Sheng, J. Hydrogen-rich saline attenuated subarachnoid hemorrhage-induced early brain injury in rats by suppressing inflammatory response: Possible involvement of NF-κB pathway and NLRP3 inflammasome. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Kuemmerle-Deschner, J.B. CAPS—Pathogenesis, presentation and treatment of an autoinflammatory disease. Semin. Immunopathol. 2015, 37, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Maltez, V.I.; Miao, E.A. Reassessing the evolutionary importance of inflammasomes. J. Immunol. 2016, 196, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.G.; Brough, D.; Freeman, S. Inhibiting the inflammasome: A chemical perspective. J. Med. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Perregaux, D.G.; McNiff, P.; Laliberte, R.; Hawryluk, N.; Peurano, H.; Stam, E.; Eggler, J.; Griffiths, R.; Dombroski, M.A.; Gabel, C.A. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J. Pharmacol. Exp. Ther. 2001, 299, 187–197. [Google Scholar] [PubMed]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hirano, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T.; et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.J.D.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [PubMed]
- Auron, P.E.; Webb, A.C.; Rosenwasser, L.J.; Mucci, S.F.; Rich, A.; Wolff, S.M.; Dinarello, C.A. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc. Natl. Acad. Sci. USA 1984, 81, 7907–7911. [Google Scholar] [CrossRef] [PubMed]
- McDowell, T.L.; Symons, J.A.; Duff, G.W. Human interleukin-1α gene expression is regulated by Sp1 and a transcriptional repressor. Cytokine 2005, 30, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Luheshi, N.M.; Kovács, K.J.; Lopez-Castejon, G.; Brough, D.; Denes, A. Interleukin-1α expression precedes IL-1β after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J. Neuroinflamm. 2011, 8, 186. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, A.D.; Brough, D.; Robinson, E.M.; Girard, S.; Rothwell, N.J.; Allan, S.M. Interleukin-1 receptor antagonist is beneficial after subarachnoid haemorrhage in rat by blocking haem-driven inflammatory pathology. Dis. Model. Mech. 2012, 5, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Singer, C.F.; Hudelist, G.; Gschwantler-Kaulich, D.; Fink-Retter, A.; Mueller, R.; Walter, I.; Czerwenka, K.; Kubista, E. Interleukin-1α protein secretion in breast cancer is associated with poor differentiation and estrogen receptor α negativity. Int. J. Gynecol. Cancer 2006, 16, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Freigang, S.; Ampenberger, F.; Weiss, A.; Kanneganti, T.-D.; Iwakura, Y.; Hersberger, M.; Kopf, M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 2013, 14, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.A.; Clark, R.R.; Bartling, T.R.; Trebak, M.; Melendez, J.A. Redox control of the senescence regulator interleukin-1α and the secretory phenotype. J. Biol. Chem. 2013, 288, 32149–32159. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Grando, S.A.; Li, Y.C. Regulation of IL-1 family cytokines IL-1α, IL-1 receptor antagonist, and IL-18 by 1,25-dihydroxyvitamin D3 in primary keratinocytes. J. Immunol. 2006, 176, 3780–3787. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Kaplanov, I.; Romzova, M.; Bernardis, L.; Braiman, A.; Voronov, E.; Apte, R.N. The transcription of the alarmin cytokine interleukin-1α is controlled by hypoxia inducible factors 1 and 2 α in hypoxic cells. Front. Immunol. 2012, 3, 290. [Google Scholar] [CrossRef] [PubMed]
- Hawn, T.R.; Ozinsky, A.; Underhill, D.M.; Buckner, F.S.; Akira, S.; Aderem, A. Leishmania major activates IL-1α expression in macrophages through a MyD88-dependent pathway. Microbes Infect. 2002, 4, 763–771. [Google Scholar] [CrossRef]
- Fettelschoss, A.; Kistowska, M.; LeibundGut-Landmann, S.; Beer, H.-D.; Johansen, P.; Senti, G.; Contassot, E.; Bachmann, M.F.; French, L.E.; Oxenius, A.; et al. Inflammasome activation and IL-1β target IL-1α for secretion as opposed to surface expression. Proc. Natl. Acad. Sci. USA 2011, 108, 18055–18060. [Google Scholar] [CrossRef] [PubMed]
- Enya, K.; Hayashi, H.; Takii, T.; Ohoka, N.; Kanata, S.; Okamoto, T.; Onozaki, K. The interaction with Sp1 and reduction in the activity of histone deacetylase 1 are critical for the constitutive gene expression of IL-1α in human melanoma cells. J. Leukoc. Biol. 2008, 83, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Alheim, K.; McDowell, T.L.; Symons, J.A.; Duff, G.W.; Bartfai, T. An AP-1 site is involved in the NGF induction of IL-1α in PC12 cells. Neurochem. Int. 1996, 29, 487–496. [Google Scholar] [CrossRef]
- Mori, N.; Prager, D. Transactivation of the interleukin-1α promoter by human T-cell leukemia virus type I and type II Tax proteins. Blood 1996, 87, 3410–3417. [Google Scholar] [PubMed]
- Chan, J.; Atianand, M.; Jiang, Z.; Carpenter, S.; Aiello, D.; Elling, R.; Fitzgerald, K.A.; Caffrey, D.R. Cutting edge: A natural antisense transcript, AS-IL1α, controls inducible transcription of the proinflammatory cytokine IL-1α. J. Immunol. 2015, 195, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Wessendorf, J.H.; Garfinkel, S.; Zhan, X.; Brown, S.; Maciag, T. Identification of a nuclear localization sequence within the structure of the human interleukin-1α precursor. J. Biol. Chem. 1993, 268, 22100–22104. [Google Scholar] [PubMed]
- Lange, A.; McLane, L.M.; Mills, R.E.; Devine, S.E.; Corbett, A.H. Expanding the definition of the classical bipartite nuclear localization signal. Traffic 2010, 11, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Werman, A.; Werman-Venkert, R.; White, R.; Lee, J.-K.; Werman, B.; Krelin, Y.; Voronov, E.; Dinarello, C.A.; Apte, R.N. The precursor form of IL-1α is an intracrine proinflammatory activator of transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Buryskova, M.; Pospisek, M.; Grothey, A.; Simmet, T.; Burysek, L. Intracellular interleukin-1α functionally interacts with histone acetyltransferase complexes. J. Biol. Chem. 2004, 279, 4017–4026. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Rider, P.; Carmi, Y.; Braiman, A.; Dotan, S.; White, M.R.; Voronov, E.; Martin, M.U.; Dinarello, C.A.; Apte, R.N. Differential release of chromatin-bound IL-1α discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 2574–2579. [Google Scholar] [CrossRef] [PubMed]
- Luheshi, N.M.; McColl, B.W.; Brough, D. Nuclear retention of IL-1α by necrotic cells: A mechanism to dampen sterile inflammation. Eur. J. Immunol. 2009, 39, 2973–2980. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Oppenheim, J.J.; Matsushima, K. Calcium-dependent binding of phosphorylated human pre interleukin 1α to phospholipids. J. Biochem. 1990, 107, 666–670. [Google Scholar] [PubMed]
- Sung, S.J.; Walters, J.A. Stimulation of interleukin-1α and interleukin-1β production in human monocytes by protein phosphatase 1 and 2A inhibitors. J. Biol. Chem. 1993, 268, 5802–5809. [Google Scholar] [PubMed]
- Cohen, I.; Idan, C.; Rider, P.; Peleg, R.; Vornov, E.; Elena, V.; Tomas, M.; Martin, T.; Tudor, C.; Cicerone, T.; et al. IL-1α is a DNA damage sensor linking genotoxic stress signaling to sterile inflammation and innate immunity. Sci. Rep. 2015, 5, 14756. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J. The interleukin-1 receptor/Toll-like receptor superfamily: 10 Years of progress. Immunol. Rev. 2008, 226, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Mosley, B.; Urdal, D.L.; Prickett, K.S.; Larsen, A.; Cosman, D.; Conlon, P.J.; Gillis, S.; Dower, S.K. The interleukin-1 receptor binds the human interleukin-1α precursor but not the interleukin-1β precursor. J. Biol. Chem. 1987, 262, 2941–2944. [Google Scholar] [PubMed]
- Kim, B.; Lee, Y.; Kim, E.; Kwak, A.; Ryoo, S.; Bae, S.H.; Azam, T.; Kim, S.; Dinarello, C.A. The Interleukin-1α precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front. Immunol. 2013, 4, 391. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Humphry, M.; Maguire, J.J.; Bennett, M.R.; Clarke, M.C.H. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1α, controlling necrosis-induced sterile inflammation. Immunity 2013, 38, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Tynan, G.A.; Logue, S.E.; Cullen, S.P.; Bots, M.; Lüthi, A.U.; Reeves, E.P.; McElvaney, N.G.; Medema, J.P.; Lavelle, E.C.; et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol. Cell 2011, 44, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Yazdi, A.S.; Thomas, C.J.; Masin, M.; Heinz, L.X.; Guarda, G.; Quadroni, M.; Drexler, S.K.; Tschopp, J. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 2012, 36, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Yamamoto, K.; Saido, T.; Kawasaki, H.; Oppenheim, J.J.; Matsushima, K. Identification of calcium-activated neutral protease as a processing enzyme of human interleukin-1α. Proc. Natl. Acad. Sci. USA 1990, 87, 5548–5552. [Google Scholar] [CrossRef] [PubMed]
- Carruth, L.M.; Demczuk, S.; Mizel, S.B. Involvement of a calpain-like protease in the processing of the murine interleukin-1α precursor. J. Biol. Chem. 1991, 266, 12162–12167. [Google Scholar] [PubMed]
- England, H.; Summersgill, H.R.; Edye, M.E.; Rothwell, N.J.; Brough, D. Release of interleukin-1α or interleukin-1β depends on mechanism of cell death. J. Biol. Chem. 2014, 289, 15942–15950. [Google Scholar] [CrossRef] [PubMed]
- Croall, D.E.; Ersfeld, K. The calpains: Modular designs and functional diversity. Genome Biol. 2007, 8, 218. [Google Scholar] [CrossRef] [PubMed]
- Rami, A.; Sims, J.; Botez, G.; Winckler, J. Spatial resolution of phospholipid scramblase 1 (PLSCR1), caspase-3 activation and DNA-fragmentation in the human hippocampus after cerebral ischemia. Neurochem. Int. 2003, 43, 79–87. [Google Scholar] [CrossRef]
- Leloup, L.; Shao, H.; Bae, Y.H.; Deasy, B.; Stolz, D.; Roy, P.; Wells, A. m-Calpain activation is regulated by its membrane localization and by its binding to phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2010, 285, 33549–33566. [Google Scholar] [CrossRef] [PubMed]
- Välimäki, E.; Cypryk, W.; Virkanen, J.; Nurmi, K.; Turunen, P.M.; Eklund, K.K.; Åkerman, K.E.; Nyman, T.A.; Matikainen, S. Calpain activity is essential for ATP-driven unconventional vesicle-mediated protein secretion and inflammasome activation in human macrophages. J. Immunol. 2016, 197, 3315–3325. [Google Scholar] [CrossRef] [PubMed]
- Casson, C.N.; Copenhaver, A.M.; Zwack, E.E.; Nguyen, H.T.; Strowig, T.; Javdan, B.; Bradley, W.P.; Fung, T.C.; Flavell, R.A.; Brodsky, I.E.; et al. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog. 2013, 9, e1003400. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-J.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 2007, 13, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Millis, A.J.; Baglioni, C. Expression of interleukin 1-inducible genes and production of interleukin 1 by aging human fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 4683–4687. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J. Cell death and inflammation: The case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J. 2016, 283, 2599–2615. [Google Scholar] [CrossRef] [PubMed]
- Baekkevold, E.S.; Roussigné, M.; Yamanaka, T.; Johansen, F.-E.; Jahnsen, F.L.; Amalric, F.; Brandtzaeg, P.; Erard, M.; Haraldsen, G.; Girard, J.-P. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am. J. Pathol. 2003, 163, 69–79. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Wu, Y.; Zhou, Y.; Zeng, L.; Huang, T. Anti-IL-33 antibody treatment inhibits airway inflammation in a murine model of allergic asthma. Biochem. Biophys. Res. Commun. 2009, 386, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Préfontaine, D.; Lajoie-Kadoch, S.; Foley, S.; Audusseau, S.; Olivenstein, R.; Halayko, A.J.; Lemière, C.; Martin, J.G.; Hamid, Q. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J. Immunol. 2009, 183, 5094–5103. [Google Scholar] [CrossRef] [PubMed]
- Kearley, J.; Silver, J.S.; Sanden, C.; Liu, Z.; Berlin, A.A.; White, N.; Mori, M.; Pham, T.-H.; Ward, C.K.; Criner, G.J.; et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 2015, 42, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.; Talabot-Ayer, D.; Lamacchia, C.; Toy, D.; Seemayer, C.A.; Viatte, S.; Finckh, A.; Smith, D.E.; Gabay, C. Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum. 2009, 60, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jiang, H.-R.; Kewin, P.; Li, Y.; Mu, R.; Fraser, A.R.; Pitman, N.; Kurowska-Stolarska, M.; McKenzie, A.N.J.; McInnes, I.B.; et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. USA 2008, 105, 10913–10918. [Google Scholar] [CrossRef] [PubMed]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.-P. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel IL-33-LacZ gene trap reporter strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef] [PubMed]
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel “alarmin”? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [PubMed]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.-A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.-P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Roussel, L.; Erard, M.; Cayrol, C.; Girard, J.-P. Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Rep. 2008, 9, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Mohs, A.; Thomas, M.; Klare, J.; Ross, R.; Schmitz, M.L.; Martin, M.U. The dual function cytokine IL-33 interacts with the transcription factor NF-κB to dampen NF-κB-stimulated gene transcription. J. Immunol. 2011, 187, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Bessa, J.; Meyer, C.A.; de Vera Mudry, M.C.; Schlicht, S.; Smith, S.H.; Iglesias, A.; Cote-Sierra, J. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J. Autoimmun. 2014, 55, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, A.A.; Oldham, E.R.; Murphy, E.E.; Schmitz, J.; Pflanz, S.; Kastelein, R.A. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J. Immunol. 2007, 179, 2551–2555. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Edwards, J.E. Type 1/Type 2 immunity in infectious diseases. Clin. Infect. Dis. 2001, 32, 76–102. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.-P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Oboki, K.; Kajiwara, N.; Morii, E.; Aozasa, K.; Flavell, R.A.; Okumura, K.; Saito, H.; Nakae, S. Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. J. Immunol. 2009, 183, 7890–7897. [Google Scholar] [CrossRef] [PubMed]
- Andronicos, N.M.; McNally, J.; Kotze, A.C.; Hunt, P.W.; Ingham, A. Trichostrongylus colubriformis larvae induce necrosis and release of IL-33 from intestinal epithelial cells in vitro: implications for gastrointestinal nematode vaccine design. Int. J. Parasitol. 2012, 42, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sun, Y.; Lai, L.; Wu, H.; Xiao, Y.; Ming, B.; Gao, M.; Zou, H.; Xiong, P.; Xu, Y.; et al. Interleukin-33 is released in spinal cord and suppresses experimental autoimmune encephalomyelitis in mice. Neuroscience 2015, 308, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Kouzaki, H.; Iijima, K.; Kobayashi, T.; O’Grady, S.M.; Kita, H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J. Immunol. 2011, 186, 4375–4387. [Google Scholar] [CrossRef] [PubMed]
- Bustin, M.; Reeves, R. High-mobility-group chromosomal proteins: Architectural components that facilitate chromatin function. Prog. Nucleic Acid Res. Mol. Biol. 1996, 54, 35–100. [Google Scholar] [PubMed]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E.; Agresti, A.; et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wong, B.; Yen, Y.-M.; Oettinger, M.A.; Kwon, J.; Johnson, R.C. Determinants of HMGB proteins required to promote RAG1/2-Recombination signal sequence complex assembly and catalysis during V(D)J recombination. Mol. Cell. Biol. 2005, 25, 4413–4425. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, P.; Oñate, S.A.; Christensen, K.; Edwards, D.P. Nuclear accessory factors enhance the binding of progesterone receptor to specific target DNA. J. Steroid Biochem. Mol. Biol. 1994, 48, 1–13. [Google Scholar] [CrossRef]
- Calogero, S.; Grassi, F.; Aguzzi, A.; Voigtländer, T.; Ferrier, P.; Ferrari, S.; Bianchi, M.E. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 1999, 22, 276–280. [Google Scholar] [PubMed]
- Baker, C.; Isenberg, I.; Goodwin, G.H.; Johns, E.W. Physical studies of the nonhistone chromosomal proteins HMG-U and HMG-2. Biochemistry 1976, 15, 1645–1649. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Wang, H.; Yuan, R.; Li, H.; Ochani, M.; Ochani, K.; Rosas-Ballina, M.; Czura, C.J.; Huston, J.M.; Miller, E.; et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J. Exp. Med. 2006, 203, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Arcaroli, J.; Carmody, A.; Wang, H.; Tracey, K.J. HMG-1 as a mediator of acute lung inflammation. J. Immunol. 2000, 165, 2950–2954. [Google Scholar] [CrossRef] [PubMed]
- Kokkola, R.; Li, J.; Sundberg, E.; Aveberger, A.-C.; Palmblad, K.; Yang, H.; Tracey, K.J.; Andersson, U.; Harris, H.E. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 2003, 48, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Takahashi, H.K.; Liu, K.; Wake, H.; Liu, R.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Mori, S.; et al. Anti-high mobility group box-1 monoclonal antibody protects the blood–brain barrier from ischemia-induced disruption in rats. Stroke 2011, 42, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Mori, S.; Takahashi, H.K.; Tomono, Y.; Wake, H.; Kanke, T.; Sato, Y.; Hiraga, N.; Adachi, N.; Yoshino, T.; et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007, 21, 3904–3916. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Harada, T.; Mollen, K.P.; Prince, J.M.; Levy, R.M.; Englert, J.A.; Gallowitsch-Puerta, M.; Yang, L.; Yang, H.; Tracey, K.J.; et al. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol. Med. 2006, 12, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Rouhiainen, A.; Kuja-Panula, J.; Wilkman, E.; Pakkanen, J.; Stenfors, J.; Tuominen, R.K.; Lepäntalo, M.; Carpén, O.; Parkkinen, J.; Rauvala, H. Regulation of monocyte migration by amphoterin (HMGB1). Blood 2004, 104, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.-J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Arcaroli, J.; Yum, H.-K.; Yang, H.; Wang, H.; Yang, K.-Y.; Choe, K.-H.; Strassheim, D.; Pitts, T.M.; Tracey, K.J.; et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am. J. Physiol. Cell Physiol. 2003, 284, C870–C879. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Jiang, Y.; Wang, J.; Shi, X.; Liu, Q.; Liu, Z.; Li, Y.; Scott, M.J.; Xiao, G.; Li, S.; et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014, 21, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Merenmies, J.; Pihlaskari, R.; Laitinen, J.; Wartiovaara, J.; Rauvala, H. 30-kDa heparin-binding protein of brain (amphoterin) involved in neurite outgrowth. Amino acid sequence and localization in the filopodia of the advancing plasma membrane. J. Biol. Chem. 1991, 266, 16722–16729. [Google Scholar] [PubMed]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.; Pelovsky, P.; Ugrinova, I.; Takahashi, M.; Pasheva, E. The DNA binding and bending activities of truncated tail-less HMGB1 protein are differentially affected by Lys-2 and Lys-81 residues and their acetylation. Int. J. Biol. Sci. 2011, 7, 691–699. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhang, B.; Wei, X.; Wang, Z.; Fan, B.; Du, P.; Zhang, Y.; Jian, W.; Chen, L.; Wang, L.; et al. HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J. Cell. Mol. Med. 2013, 17, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Evankovich, J.; Cho, S.W.; Zhang, R.; Cardinal, J.; Dhupar, R.; Zhang, L.; Klune, J.R.; Zlotnicki, J.; Billiar, T.; Tsung, A. High mobility group box-1 release from hepatocytes during Ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J. Biol. Chem. 2010, 285, 39888–39897. [Google Scholar] [CrossRef] [PubMed]
- Rabadi, M.M.; Xavier, S.; Vasko, R.; Kaur, K.; Goligorksy, M.S.; Ratliff, B.B. High-mobility group box-1 is a novel deacetylation target of Sirtuin1. Kidney Int. 2015, 87, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Ditsworth, D.; Zong, W.-X.; Thompson, C.B. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J. Biol. Chem. 2007, 282, 17845–17854. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, L.; Chen, L.; Yuan, W.; Dong, L.; Zhang, Y.; Wu, H.; Wang, C. PARP-1 mediates LPS-induced HMGB1 release by macrophages through regulation of HMGB1 acetylation. J. Immunol. 2014, 193, 6114–6123. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.H.; Shin, J.-S. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J. Immunol. 2006, 177, 7889–7897. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Kazama, H.; Ricci, J.-E.; Herndon, J.M.; Hoppe, G.; Green, D.R.; Ferguson, T.A. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008, 29, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Towne, J.E.; Renshaw, B.R.; Douangpanya, J.; Lipsky, B.P.; Shen, M.; Gabel, C.A.; Sims, J.E. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 2011, 286, 42594–42602. [Google Scholar] [CrossRef] [PubMed]
- Bulau, A.-M.; Nold, M.F.; Li, S.; Nold-Petry, C.A.; Fink, M.; Mansell, A.; Schwerd, T.; Hong, J.; Rubartelli, A.; Dinarello, C.A.; et al. Role of caspase-1 in nuclear translocation of IL-37, release of the cytokine, and IL-37 inhibition of innate immune responses. Proc. Natl. Acad. Sci. USA 2014, 111, 2650–2655. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Ho, A.S.; Haley-Vicente, D.; Zhang, J.; Bernal-Fussell, J.; Pace, A.M.; Hansen, D.; Schweighofer, K.; Mize, N.K.; Ford, J.E. Cloning and characterization of IL-1HY2, a novel interleukin 1 family member. J. Biol. Chem. 2001, 276, 20597–20602. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G.; Reed, D.A.; Dinarello, C.A.; Trinchieri, G.; Gerosa, F.; Dinarello, C.; Car, B.; Orange, J.; Sacco, S.; Cohen, J.; et al. IL-12-induced IFN-γ is dependent on caspase-1 processing of the IL-18 precursor. J. Clin. Investig. 1999, 104, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Mehul, B.; Hughes, R.C. Plasma membrane targetting, vesicular budding and release of galectin 3 from the cytoplasm of mammalian cells during secretion. J. Cell Sci. 1997, 110, 1169–1178. [Google Scholar] [PubMed]
- Liu, F.T.; Hsu, D.K.; Zuberi, R.I.; Kuwabara, I.; Chi, E.Y.; Henderson, W.R. Expression and function of galectin-3, a β-galactoside-binding lectin, in human monocytes and macrophages. Am. J. Pathol. 1995, 147, 1016–1028. [Google Scholar] [PubMed]
- Van Stijn, C.M.W.; van den Broek, M.; van de Weerd, R.; Visser, M.; Taşdelen, I.; Tefsen, B.; van Die, I. Regulation of expression and secretion of galectin-3 in human monocyte-derived dendritic cells. Mol. Immunol. 2009, 46, 3292–3299. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Ouellet, N.; Pelletier, I.; Simard, M.; Rancourt, A.; Bergeron, M.G. Role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia. J. Immunol. 2002, 168, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Colnot, C.; Ripoche, M.A.; Milon, G.; Montagutelli, X.; Crocker, P.R.; Poirier, F. Maintenance of granulocyte numbers during acute peritonitis is defective in galectin-3-null mutant mice. Immunology 1998, 94, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.C.; Frigeri, L.G.; Liu, F.T. An endogenous lectin, galectin-3 (epsilon BP/Mac-2), potentiates IL-1 production by human monocytes. Immunol. Lett. 1994, 42, 113–116. [Google Scholar] [CrossRef]
- Sano, H.; Hsu, D.K.; Yu, L.; Apgar, J.R.; Kuwabara, I.; Yamanaka, T.; Hirashima, M.; Liu, F.T. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J. Immunol. 2000, 165, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A.B. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol. 2011, 32, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of interferon-γ inducing factor mediated by interleukin-1β converting enzyme. Science 1997, 275, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, S.; Uehara, A.; Nochi, T.; Yamaguchi, T.; Ueda, H.; Sugiyama, A.; Hanzawa, K.; Kumagai, K.; Okamura, H.; Takada, H. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J. Immunol. 2001, 167, 6568–6575. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kayagaki, N.; Kuida, K.; Nakano, H.; Hayashi, N.; Takeda, K.; Matsui, K.; Kashiwamura, S.; Hada, T.; Akira, S.; et al. Caspase-1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity 1999, 11, 359–367. [Google Scholar] [CrossRef]
- Novick, D.; Kim, S.H.; Fantuzzi, G.; Reznikov, L.L.; Dinarello, C.A.; Rubinstein, M. Interleukin-18 binding protein: A novel modulator of the Th1 cytokine response. Immunity 1999, 10, 127–136. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 binding protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef] [PubMed]
- Wegehingel, S.; Zehe, C.; Nickel, W. Rerouting of fibroblast growth factor 2 to the classical secretory pathway results in post-translational modifications that block binding to heparan sulfate proteoglycans. FEBS Lett. 2008, 582, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Pecceu, F.; Dousset, P.; Shire, D.; Cavrois, E.; Marchese, E.; Ferrara, P.; Kaghad, M.; Dumont, X.; Lupker, J. Human interleukin 1β fused to the human growth hormone signal peptide is N-glycosylated and secreted by Chinese hamster ovary cells. Gene 1991, 97, 253–258. [Google Scholar] [CrossRef]
- Fleer, R.; Chen, X.J.; Amellal, N.; Yeh, P.; Fournier, A.; Guinet, F.; Gault, N.; Faucher, D.; Folliard, F.; Fukuhara, H. High-level secretion of correctly processed recombinant human interleukin-1β in Kluyveromyces lactis. Gene 1991, 107, 285–295. [Google Scholar] [CrossRef]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nuñez, G.; He, Y.; Yin, X.-M.; O’Riordan, M.X.D. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and Caspase-2-driven mitochondrial damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef] [PubMed]
- McCrossan, M.; Windsor, M.; Ponnambalam, S.; Armstrong, J.; Wileman, T. The trans Golgi network is lost from cells infected with African swine fever virus. J. Virol. 2001, 75, 11755–11765. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.-Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daniels, M.J.D.; Brough, D. Unconventional Pathways of Secretion Contribute to Inflammation. Int. J. Mol. Sci. 2017, 18, 102. https://doi.org/10.3390/ijms18010102
Daniels MJD, Brough D. Unconventional Pathways of Secretion Contribute to Inflammation. International Journal of Molecular Sciences. 2017; 18(1):102. https://doi.org/10.3390/ijms18010102
Chicago/Turabian StyleDaniels, Michael J. D., and David Brough. 2017. "Unconventional Pathways of Secretion Contribute to Inflammation" International Journal of Molecular Sciences 18, no. 1: 102. https://doi.org/10.3390/ijms18010102
APA StyleDaniels, M. J. D., & Brough, D. (2017). Unconventional Pathways of Secretion Contribute to Inflammation. International Journal of Molecular Sciences, 18(1), 102. https://doi.org/10.3390/ijms18010102