
Molecular Pathogenesis of NASH



Abstract
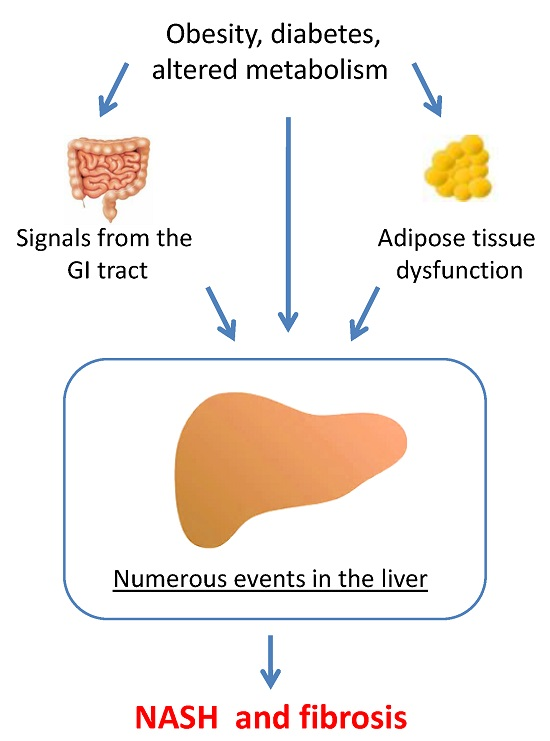
:
1. Introduction
2. Genetic Factors
3. Epigenetics
4. Dietary Factors
5. Mitochondrial Dysfunction and Apoptosis
6. Necroptosis
7. Endoplasmic Reticulum Stress
8. Hypoxia
9. Inflammation
10. Hedgehog
11. Nuclear Receptors
12. Pattern Recognition Receptors and the Inflammasomes
13. Adipokines
14. Microbiota
15. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Satapathy, S.K.; Sanyal, A.J. Epidemiology and natural history of nonalcoholic fatty liver disease. Semin. Liver Dis. 2015, 35, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; Network, N.C.R. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Charlotte, F.; Fedchuk, L.; Bedossa, P.; Lebray, P.; Poynard, T.; Ratziu, V. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol. 2013, 59, 550–556. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. Long-term mortality in nonalcoholic fatty liver disease: Is liver histology of any prognostic significance? Hepatology 2010, 51, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Lotersztajn, S. Pathophysiology of NASH: Perspectives for a targeted treatment. Curr. Pharm. Des. 2013, 19, 5250–5269. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Schork, N.; Chen, C.H.; Bettencourt, R.; Bhatt, A.; Ang, B.; Nguyen, P.; Hernandez, C.; Richards, L.; Salotti, J.; et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology 2015, 149, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic factors that affect risk of alcoholic and nonalcoholic fatty liver disease. Gastroenterology 2016, 150, 1728–1744. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Butler, J.L.; Palmer, C.D.; Voight, B.F.; Consortium, G.; Consortium, M.I.; Nash, C.R.N.; Hirschhorn, J.N. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 2010, 52, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Pirazzi, C.; Adiels, M.; Burza, M.A.; Mancina, R.M.; Levin, M.; Stahlman, M.; Taskinen, M.R.; Orho-Melander, M.; Perman, J.; Pujia, A.; et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (RS738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 2012, 57, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A.; et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chang, B.; Li, L.; Chan, L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010, 52, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Lai, K.M.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. PNPLA3 I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Mondul, A.; Mancina, R.M.; Merlo, A.; Dongiovanni, P.; Rametta, R.; Montalcini, T.; Valenti, L.; Albanes, D.; Romeo, S. PNPLA3 I148M variant influences circulating retinol in adults with nonalcoholic fatty liver disease or obesity. J. Nutr. 2015, 145, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Kovarova, M.; Konigsrainer, I.; Konigsrainer, A.; Machicao, F.; Haring, H.U.; Schleicher, E.; Peter, A. The genetic variant I148M in PNPLA3 is associated with increased hepatic retinyl-palmitate storage in humans. J. Clin. Endocrinol. Metab. 2015, 100, E1568–E1574. [Google Scholar] [CrossRef] [PubMed]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.C.; Anty, R.; et al. TM6SF2 RS58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; van’t Hooft, F. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L.; Rametta, R.; Daly, A.K.; Nobili, V.; Mozzi, E.; Leathart, J.B.; Pietrobattista, A.; Burt, A.D.; Maggioni, M.; et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010, 59, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Zhang, C.K.; Zhao, H.; Pakstis, A.J.; Kim, G.; Kursawe, R.; Dykas, D.J.; Bale, A.E.; Giannini, C.; Pierpont, B.; et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 2012, 55, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Tonjes, A.; Scholz, M.; Loeffler, M.; Stumvoll, M. Association of Pro12Ala polymorphism in peroxisome proliferator-activated receptor γ with pre-diabetic phenotypes: Meta-analysis of 57 studies on nondiabetic individuals. Diabetes Care 2006, 29, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Bhattacharya, R.; Lindor, K.D.; Chalasani, N.; Raaka, S.; Heathcote, E.J.; Miskovsky, E.; Shaffer, E.; Rulyak, S.J.; Kowdley, K.V. HFE C282Y mutations are associated with advanced hepatic fibrosis in caucasians with nonalcoholic steatohepatitis. Hepatology 2007, 46, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Manzini, P.; D’Antico, S.; Vanni, E.; Longo, F.; Leone, N.; Massarenti, P.; Piga, A.; Marchesini, G.; Rizzetto, M. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology 2004, 39, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010, 138, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Raszeja-Wyszomirska, J.; Kurzawski, G.; Lawniczak, M.; Miezynska-Kurtycz, J.; Lubinski, J. Nonalcoholic fatty liver disease and HFE gene mutations: A polish study. World J. Gastroenterol. 2010, 16, 2531–2536. [Google Scholar] [CrossRef] [PubMed]
- Al-Serri, A.; Anstee, Q.M.; Valenti, L.; Nobili, V.; Leathart, J.B.; Dongiovanni, P.; Patch, J.; Fracanzani, A.; Fargion, S.; Day, C.P.; et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012, 56, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Boren, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016, 150, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fan, J.G.; Qiao, L. Potential epigenetic mechanism in non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2015, 16, 5161–5179. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Zhang, H.W.; Zhou, J.Y.; Liu, Y.; Yang, Y.; Chen, X.L.; Zhu, C.H.; Zheng, R.D.; Ling, W.H.; Zhu, H.L. Betaine attenuates hepatic steatosis by reducing methylation of the MTTP promoter and elevating genomic methylation in mice fed a high-fat diet. J. Nutr. Biochem. 2014, 25, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.P.; Han, T.; Muskhelishvili, L.; Fuscoe, J.C.; Ross, S.A.; Beland, F.A.; Pogribny, I.P. Coupling global methylation and gene expression profiles reveal key pathophysiological events in liver injury induced by a methyl-deficient diet. Mol. Nutr. Food Res. 2011, 55, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Kalhan, S.C.; Edmison, J.; Marczewski, S.; Dasarathy, S.; Gruca, L.L.; Bennett, C.; Duenas, C.; Lopez, R. Methionine and protein metabolism in non-alcoholic steatohepatitis: Evidence for lower rate of transmethylation of methionine. Clin. Sci. 2011, 121, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, A.M.; Bruce German, J.; Esfandiari, F.; Halsted, C.H. Quantitative lipid metabolomic changes in alcoholic micropigs with fatty liver disease. Alcohol. Clin. Exp. Res. 2009, 33, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Campion, J.; Milagro, F.I.; Martinez, J.A. Transcriptomic and epigenetic changes in early liver steatosis associated to obesity: Effect of dietary methyl donor supplementation. Mol. Genet. Metab. 2013, 110, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueno, A.L.; Fernandez Gianotti, T.; Castano, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1α promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar] [PubMed]
- Pruis, M.G.; Lendvai, A.; Bloks, V.W.; Zwier, M.V.; Baller, J.F.; de Bruin, A.; Groen, A.K.; Plosch, T. Maternal western diet primes non-alcoholic fatty liver disease in adult mouse offspring. Acta Physiol. 2014, 210, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Broseus, J.; Hergalant, S.; Donnart, A.; Chevalier, C.; Bolanos-Jimenez, F.; Gueant, J.L.; Houlgatte, R. Identification of master genes involved in liver key functions through transcriptomics and epigenomics of methyl donor deficiency in rat: Relevance to nonalcoholic liver disease. Mol. Nutr. Food Res. 2015, 59, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, J.; Burgueno, A.L.; Rosselli, M.S.; Gianotti, T.F.; Lago, N.R.; Pirola, C.J.; Sookoian, S. High fat diet-induced liver steatosis promotes an increase in liver mitochondrial biogenesis in response to hypoxia. J. Cell. Mol. Med. 2011, 15, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Gianotti, T.F.; Burgueno, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castano, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Aagaard-Tillery, K.M.; Grove, K.; Bishop, J.; Ke, X.; Fu, Q.; McKnight, R.; Lane, R.H. Developmental origins of disease and determinants of chromatin structure: Maternal diet modifies the primate fetal epigenome. J. Mol. Endocrinol. 2008, 41, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Investig. 2010, 120, 4316–4331. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.J.; Kim, J.; Hoang, M.H.; Lee, S.J. Hepatic lipid accumulation alters global histone H3 Lysine 9 and 4 trimethylation in the peroxisome proliferator-activated receptor α network. PLoS ONE 2012, 7, e44345. [Google Scholar] [CrossRef]
- Li, J.; Huang, J.; Li, J.S.; Chen, H.; Huang, K.; Zheng, L. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J. Hepatol. 2012, 56, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Colak, Y.; Yesil, A.; Mutlu, H.H.; Caklili, O.T.; Ulasoglu, C.; Senates, E.; Takir, M.; Kostek, O.; Yilmaz, Y.; Yilmaz Enc, F.; et al. A potential treatment of non-alcoholic fatty liver disease with SIRT1 activators. J. Gastrointest. Liver Dis. 2014, 23, 311–319. [Google Scholar]
- Colak, Y.; Ozturk, O.; Senates, E.; Tuncer, I.; Yorulmaz, E.; Adali, G.; Doganay, L.; Enc, F.Y. SIRT1 as a potential therapeutic target for treatment of nonalcoholic fatty liver disease. Med. Sci. Monit. 2011, 17, HY5–HY9. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.A.; Chen, A.; Burdine, M.S.; Choudhury, M.; Harris, R.A.; Lane, R.H.; Friedman, J.E.; Grove, K.L.; Tackett, A.J.; Aagaard, K.M. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB J. 2012, 26, 5106–5114. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stancakova, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Rottiers, V.; Naar, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D.; Mitchell, G.M. MicroRNAs in insulin resistance and obesity. Exp. Diabetes Res. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.M.; Simao, A.L.; Rodrigues, C.M.; Castro, R.E. Revisiting the metabolic syndrome and paving the way for microRNAs in non-alcoholic fatty liver disease. FEBS J. 2014, 281, 2503–2524. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.E.; Ferreira, D.M.; Afonso, M.B.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Francl, J.M.; Boehme, S.; Chiang, J.Y. Regulation of cholesterol and bile acid homeostasis by the cholesterol 7α-hydroxylase/steroid response element-binding protein 2/microRNA-33a axis in mice. Hepatology 2013, 58, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xiong, Y.; Sheng, Q.; Zhao, S.; Wattacheril, J.; Flynn, C.R. A micro-RNA expression signature for human NAFLD progression. J. Gastroenterol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Soronen, J.; Yki-Jarvinen, H.; Zhou, Y.; Sadevirta, S.; Sarin, A.P.; Leivonen, M.; Sevastianova, K.; Perttila, J.; Laurila, P.P.; Sigruener, A.; et al. Novel hepatic microRNAs upregulated in human nonalcoholic fatty liver disease. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, G.; Qin, Y.; Zhang, C.; Tang, H.; Yin, Y.; Xiang, X.; Li, Y.; Zhao, J.; Mulholland, M.; et al. Ghrelin promotes hepatic lipogenesis by activation of mtor-PPARγ signaling pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 13163–13168. [Google Scholar] [CrossRef] [PubMed]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Chan, R.S.; Wong, G.L.; Cheung, B.H.; Chu, W.C.; Yeung, D.K.; Chim, A.M.; Lai, J.W.; Li, L.S.; Sea, M.M.; et al. Community-based lifestyle modification programme for non-alcoholic fatty liver disease: A randomized controlled trial. J. Hepatol. 2013, 59, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast food diet mouse: Novel small animal model of nash with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G825–G834. [Google Scholar] [CrossRef] [PubMed]
- Garbow, J.R.; Doherty, J.M.; Schugar, R.C.; Travers, S.; Weber, M.L.; Wentz, A.E.; Ezenwajiaku, N.; Cotter, D.G.; Brunt, E.M.; Crawford, P.A. Hepatic steatosis, inflammation, and ER stress in mice maintained long term on a very low-carbohydrate ketogenic diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G956–G967. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Miao, J.; Chanda, D.; Wang, Y.; Zhao, E.; Haas, M.E.; Hirschey, M.; Vaitheesvaran, B.; Farese, R.V., Jr.; Kurland, I.J.; et al. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 2012, 15, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.I.; Szendroedi, J.; Chmelik, M.; Krssak, M.; Moser, E.; Roden, M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011, 34, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.; Pan, X.; Malladi, P.; Wainwright, M.S.; Whitington, P.F. Mitochondrial reactive oxygen species signal hepatocyte steatosis by regulating the phosphatidylinositol 3-kinase cell survival pathway. J. Biol. Chem. 2007, 282, 21327–21336. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Munetsuna, E.; Yamada, H.; Ando, Y.; Yamazaki, M.; Taromaru, N.; Nagura, A.; Ishikawa, H.; Suzuki, K.; Teradaira, R.; et al. High fructose consumption induces DNA methylation at PPARα and CPT1A promoter regions in the rat liver. Biochem. Biophys. Res. Commun. 2015, 468, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Munetsuna, E.; Yamada, H.; Ando, Y.; Mizuno, G.; Murase, Y.; Kondo, K.; Ishikawa, H.; Teradaira, R.; Suzuki, K.; et al. Fructose consumption induces hypomethylation of hepatic mitochondrial DNA in rats. Life Sci. 2016, 149, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Handa, P.; Morgan-Stevenson, V.; Maliken, B.D.; Nelson, J.E.; Washington, S.; Westerman, M.; Yeh, M.M.; Kowdley, K.V. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G117–G127. [Google Scholar] [CrossRef] [PubMed]
- Tallino, S.; Duffy, M.; Ralle, M.; Cortes, M.P.; Latorre, M.; Burkhead, J.L. Nutrigenomics analysis reveals that copper deficiency and dietary sucrose up-regulate inflammation, fibrosis and lipogenic pathways in a mature rat model of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2015, 26, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of nash: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Unalp-Arida, A.; Wilson, L.A.; Chalasani, N.; Network, N.C.R. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.; Sanyal, A.J. Abnormalities of lipid metabolism in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab. 2012, 23, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, B.; Summers, S.A. Ceramides—Lipotoxic inducers of metabolic disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- McClain, C.J.; Barve, S.; Deaciuc, I. Good fat/bad fat. Hepatology 2007, 45, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J. Reply: To PMID 24818764. Gastroenterology 2015, 148, 262–263. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, A.; Milani, S.; Vizzutti, F.; Delogu, W.; Navari, N.; Novo, E.; Maggiora, M.; Maurino, V.; Laffi, G.; Parola, M.; et al. N-3 polyunsaturated fatty acids worsen inflammation and fibrosis in experimental nonalcoholic steatohepatitis. Liver Int. 2014, 34, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Caballero, F.; Fernandez, A.; De Lacy, A.M.; Fernandez-Checa, J.C.; Caballeria, J.; Garcia-Ruiz, C. Enhanced free cholesterol, SREBP-2 and star expression in human nash. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and FAS-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Savard, C.; Tartaglione, E.V.; Kuver, R.; Haigh, W.G.; Farrell, G.C.; Subramanian, S.; Chait, A.; Yeh, M.M.; Quinn, L.S.; Ioannou, G.N. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013, 57, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Van Rooyen, D.M.; Larter, C.Z.; Haigh, W.G.; Yeh, M.M.; Ioannou, G.; Kuver, R.; Lee, S.P.; Teoh, N.C.; Farrell, G.C. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 2011, 141, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Wouters, K.; van Gorp, P.J.; Bieghs, V.; Gijbels, M.J.; Duimel, H.; Lutjohann, D.; Kerksiek, A.; van Kruchten, R.; Maeda, N.; Staels, B.; et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 2008, 48, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Min, H.K.; Kapoor, A.; Fuchs, M.; Mirshahi, F.; Zhou, H.; Maher, J.; Kellum, J.; Warnick, R.; Contos, M.J.; Sanyal, A.J. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012, 15, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Simonen, P.; Kotronen, A.; Hallikainen, M.; Sevastianova, K.; Makkonen, J.; Hakkarainen, A.; Lundbom, N.; Miettinen, T.A.; Gylling, H.; Yki-Jarvinen, H. Cholesterol synthesis is increased and absorption decreased in non-alcoholic fatty liver disease independent of obesity. J. Hepatol. 2011, 54, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Cortes, V.A.; Busso, D.; Maiz, A.; Arteaga, A.; Nervi, F.; Rigotti, A. Physiological and pathological implications of cholesterol. Front. Biosci. 2014, 19, 416–428. [Google Scholar] [CrossRef]
- Walenbergh, S.M.; Koek, G.H.; Bieghs, V.; Shiri-Sverdlov, R. Non-alcoholic steatohepatitis: The role of oxidized low-density lipoproteins. J. Hepatol. 2013, 58, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Walenbergh, S.M.; Hofker, M.H.; Shiri-Sverdlov, R. Lysosomal cholesterol accumulation: Driver on the road to inflammation during atherosclerosis and non-alcoholic steatohepatitis. Obes. Rev. 2014, 15, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American association for the study of liver diseases, American college of gastroenterology, and the American gastroenterological association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Spolding, B.; Connor, T.; Wittmer, C.; Abreu, L.L.; Kaspi, A.; Ziemann, M.; Kaur, G.; Cooper, A.; Morrison, S.; Lee, S.; et al. Rapid development of non-alcoholic steatohepatitis in psammomys obesus (Israeli Sand Rat). PLoS ONE 2014, 9, e92656. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Mitarotonda, D.; Tamborra, R.; Blonda, M.; Iannelli, G.; Petrella, A.; Sanginario, V.; Iuliano, L.; Vendemiale, G.; Serviddio, G. Oxysterols induce mitochondrial impairment and hepatocellular toxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2014, 75, S16–S17. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Cuffe, H.; Marshall, S.M.; McDaniel, A.L.; Ha, J.H.; Kavanagh, K.; Hong, C.; Tontonoz, P.; Temel, R.E.; Parks, J.S. Dietary cholesterol promotes adipocyte hypertrophy and adipose tissue inflammation in visceral, but not in subcutaneous, fat in monkeys. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Vendemiale, G.; Altomare, E. Mitochondrial dysfunction in nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Tessari, P.; Coracina, A.; Cosma, A.; Tiengo, A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Florian, M.; Chen, Q.; Yan, J.; Petrov, I.; Coughlan, M.C.; Laziyan, M.; Caldwell, D.; Lalande, M.; Patry, D.; et al. Exposure to a northern contaminant mixture (NCM) alters hepatic energy and lipid metabolism exacerbating hepatic steatosis in obese JCR rats. PLoS ONE 2014, 9, e106832. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.; Allouche, M.; Esposti, D.D.; Fanelli, E.; Boursier, C.; Henry, C.; Chopineau, J.; Calamita, G.; Kroemer, G.; Lemoine, A.; et al. Glycogen synthase kinase 3-mediated voltage-dependent anion channel phosphorylation controls outer mitochondrial membrane permeability during lipid accumulation. Hepatology 2013, 57, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Le, B.H.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Penke, M.; Larsen, P.S.; Schuster, S.; Dall, M.; Jensen, B.A.; Gorski, T.; Meusel, A.; Richter, S.; Vienberg, S.G.; Treebak, J.T.; et al. Hepatic nad salvage pathway is enhanced in mice on a high-fat diet. Mol. Cell. Endocrinol. 2015, 412, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, B.; Kruk, J. Occurrence, biosynthesis and function of isoprenoid quinones. Biochim. Biophys. Acta 2010, 1797, 1587–1605. [Google Scholar] [CrossRef] [PubMed]
- Laredj, L.N.; Licitra, F.; Puccio, H.M. The molecular genetics of coenzyme Q biosynthesis in health and disease. Biochimie 2014, 100, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—Biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Huertas, J.R.; Battino, M.; Lenaz, G.; Mataix, F.J. Changes in mitochondrial and microsomal rat liver coenzyme Q9 and Q10 content induced by dietary fat and endogenous lipid peroxidation. FEBS Lett. 1991, 287, 89–92. [Google Scholar] [CrossRef]
- Bravo, E.; Palleschi, S.; Rossi, B.; Napolitano, M.; Tiano, L.; D’Amore, E.; Botham, K.M. Coenzyme Q metabolism is disturbed in high fat diet-induced non-alcoholic fatty liver disease in rats. Int. J. Mol. Sci. 2012, 13, 1644–1657. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-α expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.A.; Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Yoo, S.H.; Jang, S.; Gonzalez, F.J.; Song, B.J. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J. Hepatol. 2012, 57, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Malhi, H.; Mott, J.L.; Gores, G.J. Apoptosis and necrosis in the liver. Compr. Physiol. 2013, 3, 977–1010. [Google Scholar] [PubMed]
- Hirsova, P.; Gores, G.J. Death receptor-mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.M.; Cook, W.D.; Okamoto, T.; Murphy, J.; Lawlor, K.E.; Vince, J.E.; Vaux, D.L. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Vucur, M.; Reisinger, F.; Cardenas, D.V.; Roderburg, C.; Koppe, C.; Kreggenwinkel, K.; Schneider, A.T.; Bartneck, M.; Neumann, U.P.; et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 2014, 6, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Afonso, M.B.; Rodrigues, P.M.; Carvalho, T.; Caridade, M.; Borralho, P.; Cortez-Pinto, H.; Castro, R.E.; Rodrigues, C.M. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. 2015, 129, 721–739. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Zhou, Y.; Lee, J.; Lu, A.; Sun, C.; Chung, J.; Ueki, K.; Ozcan, U. The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 2010, 16, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Sanyal, A.J. Endoplasmic reticulum stress and the unfolded protein response. Clin. Liver Dis. 2009, 13, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Padilla, A.; Descorbeth, M.; Almeyda, A.L.; Payne, K.; de Leon, M. Hyperglycemia magnifies Schwann cell dysfunction and cell death triggered by PA-induced lipotoxicity. Brain Res. 2011, 1370, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E275–E281. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Arias-Loste, M.T.; Fabrega, E.; Lopez-Hoyos, M.; Crespo, J. The crosstalk between hypoxia and innate immunity in the development of obesity-related nonalcoholic fatty liver disease. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Qu, A.; Taylor, M.; Xue, X.; Matsubara, T.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible transcription factor 2α promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology 2011, 54, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Gao, Z.; Yin, J.; He, Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1118–E1128. [Google Scholar] [CrossRef] [PubMed]
- Hodson, L. Adipose tissue oxygenation: Effects on metabolic function. Adipocyte 2014, 3, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Hodson, L.; Humphreys, S.M.; Karpe, F.; Frayn, K.N. Metabolic signatures of human adipose tissue hypoxia in obesity. Diabetes 2013, 62, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [PubMed]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Kuhlicke, J.; Frick, J.S.; Morote-Garcia, J.C.; Rosenberger, P.; Eltzschig, H.K. Hypoxia inducible factor (HIF)-1 coordinates induction of toll-like receptors TLR2 and TLR6 during hypoxia. PLoS ONE 2007, 2, e1364. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Choi, Y.J.; Joung, S.M.; Lee, B.H.; Jung, Y.S.; Lee, J.Y. Hypoxic stress up-regulates the expression of toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology 2010, 129, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Minville, C.; Tordjman, J.; Levy, P.; Bouillot, J.L.; Basdevant, A.; Bedossa, P.; Clement, K.; Pepin, J.L. Chronic intermittent hypoxia is a major trigger for non-alcoholic fatty liver disease in morbid obese. J. Hepatol. 2012, 56, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and FAS expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Idrissova, L.; Malhi, H.; Werneburg, N.W.; LeBrasseur, N.K.; Bronk, S.F.; Fingas, C.; Tchkonia, T.; Pirtskhalava, T.; White, T.A.; Stout, M.B.; et al. Trail receptor deletion in mice suppresses the inflammation of nutrient excess. J. Hepatol. 2015, 62, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Lanthier, N. Targeting Kupffer cells in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: Why and how? World J. Hepatol. 2015, 7, 2184–2188. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.L.; Gaudin, F.; Naveau, S.; Prevot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Staels, B. Macrophage polarization in metabolic disorders: Functions and regulation. Curr. Opin. Lipidol. 2011, 22, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Huang, H.; Zhang, Z.; Wang, F.S. The role of neutrophils in the development of liver diseases. Cell. Mol. Immunol. 2014, 11, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Ibusuki, R.; Uto, H.; Arima, S.; Mawatari, S.; Setoguchi, Y.; Iwashita, Y.; Hashimoto, S.; Maeda, T.; Tanoue, S.; Kanmura, S.; et al. Transgenic expression of human neutrophil peptide-1 enhances hepatic fibrosis in mice fed a choline-deficient, l-amino acid-defined diet. Liver Int. 2013, 33, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Rensen, S.S.; Bieghs, V.; Xanthoulea, S.; Arfianti, E.; Bakker, J.A.; Shiri-Sverdlov, R.; Hofker, M.H.; Greve, J.W.; Buurman, W.A. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS ONE 2012, 7, e52411. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Oh da, Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Yoneyama, H. From NAFLD to NASH to fibrosis to HCC: Role of dendritic cell populations in the liver. Hepatology 2013, 58, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Henning, J.R.; Graffeo, C.S.; Rehman, A.; Fallon, N.C.; Zambirinis, C.P.; Ochi, A.; Barilla, R.; Jamal, M.; Deutsch, M.; Greco, S.; et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 2013, 58, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Locatelli, I.; Bruzzi, S.; Jindal, A.; Vacchiano, M.; Bozzola, C.; Albano, E. CX3CR1-expressing inflammatory dendritic cells contribute to the progression of steatohepatitis. Clin. Sci. 2015, 129, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.; Szabo, G. Immune and inflammatory pathways in NASH. Hepatol. Int. 2013, 7, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Chen, Y.; Gao, B. Natural killer cells in liver disease. Hepatology 2013, 57, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. NKT-cell subsets: Promoters and protectors in inflammatory liver disease. J. Hepatol. 2013, 59, 618–620. [Google Scholar] [CrossRef] [PubMed]
- Martin-Murphy, B.V.; You, Q.; Wang, H.; De La Houssaye, B.A.; Reilly, T.P.; Friedman, J.E.; Ju, C. Mice lacking natural killer T cell are more susceptible to metabolic alterations following high fat diet feeding. PLoS ONE 2014, 9, e80949. [Google Scholar] [CrossRef] [PubMed]
- Kremer, M.; Thomas, E.; Milton, R.J.; Perry, A.W.; van Rooijen, N.; Wheeler, M.D.; Zacks, S.; Fried, M.; Rippe, R.A.; Hines, I.N. Kupffer cell and interleukin-12-dependent loss of natural killer T cell in hepatosteatosis. Hepatology 2010, 51, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Oo, Y.H.; Pereira, T.A.; Karaca, G.F.; Jung, Y.; Omenetti, A.; Witek, R.P.; Choi, S.S.; Guy, C.D.; Fearing, C.M.; et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010, 51, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, K.; Shimizu, Y. Role of NKT cells in the pathogenesis of NAFLD. Int. J. Hepatol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M.; Nash, C.R.N. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Verdelho Machado, M.; Diehl, A.M. Role of Hedgehog Signaling Pathway in NASH. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Song, K.; Han, C.; Chen, W.; Wang, Y.; Dash, S.; Lim, K.; Wu, T. Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease. Hepatology 2015, 63, 1155–1169. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P.; et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Pereira, T.; Boursier, J.; Swiderska-Syn, M.; Karaca, G.; Xie, G.; Guy, C.D.; Bohinc, B.; Lindblom, K.R.; et al. Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase-2 deficient mice with non-alcoholic steatohepatitis. Gut 2015, 64, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Pereira, T.A.; Xie, G.; Premont, R.; Cortez-Pinto, H.; Diehl, A.M. Accumulation of duct cell with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J. Hepatol. 2015, 63, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.D.; Suzuki, A.; Abdelmalek, M.F.; Burchette, J.L.; Diehl, A.M. Treatment response in the PIVENS trial is associated with decreased hedgehog pathway activity. Hepatology 2015, 61, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors, RXR, and the big bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.D.; Traussnigg, S.A.; Trauner, M. Nuclear receptor modulation for the treatment of nonalcoholic fatty liver disease. Semin. Liver Dis. 2016, 36, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Tailleux, A.; Wouters, K.; Staels, B. Roles of PPARs in NAFLD: Potential therapeutic targets. Biochim. Biophys. Acta 2012, 1821, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Rao, M.S. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G852–G858. [Google Scholar] [CrossRef] [PubMed]
- Ip, E.; Farrell, G.; Hall, P.; Robertson, G.; Leclercq, I. Administration of the potent PPARα agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 2004, 39, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Nicol, C.J.; Ito, S.; Bility, M.T.; Kennett, M.J.; Ward, J.M.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-β/δ protects against chemically induced liver toxicity in mice. Hepatology 2008, 47, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Sakaida, I.; Tsuchiya, M.; Omori, K.; Takami, T.; Okita, K. Pioglitazone prevents hepatic steatosis, fibrosis, and enzyme-altered lesions in rat liver cirrhosis induced by a choline-deficient l-amino acid-defined diet. Biochem. Biophys. Res. Commun. 2004, 315, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Duval, C.; Muller, M.; Kersten, S. PPARs, obesity, and inflammation. PPAR Res. 2007, 2007. [Google Scholar] [CrossRef] [PubMed]
- Lutchman, G.; Modi, A.; Kleiner, D.E.; Promrat, K.; Heller, T.; Ghany, M.; Borg, B.; Loomba, R.; Liang, T.J.; Premkumar, A.; et al. The effects of discontinuing pioglitazone in patients with nonalcoholic steatohepatitis. Hepatology 2007, 46, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Eto, M.; Ito, Y.; Mochizuki, S.; Son, B.K.; Ogawa, S.; Iijima, K.; Kaneki, M.; Kozaki, K.; Toba, K.; et al. Suppressive role of PPARγ-regulated endothelial nitric oxide synthase in adipocyte lipolysis. PLoS ONE 2015, 10, e0136597. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -β, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Yasuda, K.; Lamba, J.K.; Assem, M.; Davila, J.; Strom, S.; Schuetz, E.G. PXR (NR1I2): Splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators. Toxicol. Appl. Pharmacol. 2004, 199, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Haughton, E.L.; Tucker, S.J.; Marek, C.J.; Durward, E.; Leel, V.; Bascal, Z.; Monaghan, T.; Koruth, M.; Collie-Duguid, E.; Mann, D.A.; et al. Pregnane X receptor activators inhibit human hepatic stellate cell transdifferentiation in vitro. Gastroenterology 2006, 131, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Castano, G.O.; Burgueno, A.L.; Gianotti, T.F.; Rosselli, M.S.; Pirola, C.J. The nuclear receptor PXR gene variants are associated with liver injury in nonalcoholic fatty liver disease. Pharmacogenet. Genom. 2010, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Breuker, C.; Planque, C.; Rajabi, F.; Nault, J.C.; Couchy, G.; Zucman-Rossi, J.; Evrard, A.; Kantar, J.; Chevet, E.; Bioulac-Sage, P.; et al. Characterization of a novel PXR isoform with potential dominant-negative properties. J. Hepatol. 2014, 61, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Monostory, K.; Dvorak, Z. Steroid regulation of drug-metabolizing cytochromes P450. Curr. Drug Metab. 2011, 12, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Spruiell, K.; Richardson, R.M.; Cullen, J.M.; Awumey, E.M.; Gonzalez, F.J.; Gyamfi, M.A. Role of pregnane X receptor in obesity and glucose homeostasis in male mice. J. Biol. Chem. 2014, 289, 3244–3261. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, H.; Garzel, B.; Yang, H.; Sueyoshi, T.; Li, Q.; Shu, Y.; Zhang, J.; Hu, B.; Heyward, S.; et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol. Pharmacol. 2015, 87, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Cui, W.; Woody, S.K.; Staudinger, J.L. Pregnane X receptor modulates the inflammatory response in primary cultures of hepatocytes. Drug Metab. Dispos. 2015, 43, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Naito, S.; Yokoi, T. Tissue-specific MRNA expression profiles of human nuclear receptor subfamilies. Drug Metab. Pharmacokinet. 2004, 19, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Beilke, L.D.; Aleksunes, L.M.; Holland, R.D.; Besselsen, D.G.; Beger, R.D.; Klaassen, C.D.; Cherrington, N.J. Constitutive androstane receptor-mediated changes in bile acid composition contributes to hepatoprotection from lithocholic acid-induced liver injury in mice. Drug Metab. Dispos. 2009, 37, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.D.; Lickteig, A.J.; Augustine, L.M.; Ranger-Moore, J.; Jackson, J.P.; Ferguson, S.S.; Cherrington, N.J. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab. Dispos. 2009, 37, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; He, J.; Zhai, Y.; Wada, T.; Xie, W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J. Biol. Chem. 2009, 284, 25984–25992. [Google Scholar] [CrossRef] [PubMed]
- Sberna, A.L.; Assem, M.; Xiao, R.; Ayers, S.; Gautier, T.; Guiu, B.; Deckert, V.; Chevriaux, A.; Grober, J.; Le Guern, N.; et al. Constitutive androstane receptor activation decreases plasma apolipoprotein B-containing lipoproteins and atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yan, J.; Xu, M.; Ren, S.; Xie, W. CAR suppresses hepatic gluconeogenesis by facilitating the ubiquitination and degradation of PGC1α. Mol. Endocrinol. 2015, 29, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Lee, J.S.; Park, Y.Y.; Yang, F.; Xu, G.; Huang, W.; Finegold, M.J.; Moore, D.D. Activating CAR and β-catenin induces uncontrolled liver growth and tumorigenesis. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mackowiak, B.; Brayman, T.G.; Mitchell, M.; Zhang, L.; Huang, S.M.; Wang, H. Genome-wide analysis of human constitutive androstane receptor (CAR) transcriptome in wild-type and CAR-knockout HepaRG cells. Biochem. Pharmacol. 2015, 98, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Kunne, C.; Acco, A.; Duijst, S.; de Waart, D.R.; Paulusma, C.C.; Gaemers, I.; Oude Elferink, R.P. FXR-dependent reduction of hepatic steatosis in a bile salt deficient mouse model. Biochim. Biophys. Acta 2014, 1842, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Pineda Torra, I.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.C.; Staels, B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor α gene via activation of the farnesoid X receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Palladino, G.; Fiorucci, S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in zucker (FA/FA) obese rats. J. Lipid Res. 2010, 51, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Huang, Y.; Yan, L.; Gao, M.; Liu, D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm. Res. 2013, 30, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid x receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Jahn, D.; Rau, M.; Wohlfahrt, J.; Hermanns, H.M.; Geier, A. Non-alcoholic steatohepatitis: From pathophysiology to novel therapies. Dig. Dis. 2016, 34, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Suh, J.M.; Reilly, S.M.; Yu, E.; Osborn, O.; Lackey, D.; Yoshihara, E.; Perino, A.; Jacinto, S.; Lukasheva, Y.; et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 2015, 21, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Degirolamo, C.; Modica, S.; Vacca, M.; Di Tullio, G.; Morgano, A.; D’Orazio, A.; Kannisto, K.; Parini, P.; Moschetta, A. Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid x receptor reactivation. Hepatology 2015, 61, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A. Toll-like receptors and adaptor molecules in liver disease: Update. Hepatology 2008, 48, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Vanni, E.; Bugianesi, E. The gut-liver axis in nonalcoholic fatty liver disease: Another pathway to insulin resistance? Hepatology 2009, 49, 1790–1792. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Douhara, A.; Moriya, K.; Yoshiji, H.; Noguchi, R.; Namisaki, T.; Kitade, M.; Kaji, K.; Aihara, Y.; Nishimura, N.; Takeda, K.; et al. Reduction of endotoxin attenuates liver fibrosis through suppression of hepatic stellate cell activation and remission of intestinal permeability in a rat non-alcoholic steatohepatitis model. Mol. Med. Rep. 2015, 11, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Meier, D.T.; Wueest, S.; Rytka, J.; Boller, S.; Wielinga, P.Y.; Schraenen, A.; Lemaire, K.; Debray, S.; van Lommel, L.; et al. Toll-like receptor 2-deficient mice are protected from insulin resistance and β cell dysfunction induced by a high-fat diet. Diabetologia 2010, 53, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Al-Daghri, N.M.; Clerici, M.; Al-Attas, O.; Forni, D.; Alokail, M.S.; Alkharfy, K.M.; Sabico, S.; Mohammed, A.K.; Cagliani, R.; Sironi, M. A nonsense polymorphism (R392X) in TLR5 protects from obesity but predisposes to diabetes. J. Immunol. 2013, 190, 3716–3720. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Gabele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Scholmerich, J.; Hellerbrand, C.; Obermeier, F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Velayudham, A.; Hritz, I.; Petrasek, J.; Levin, I.; Lippai, D.; Catalano, D.; Mandrekar, P.; Dolganiuc, A.; Kurt-Jones, E.; et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G433–G441. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Norkina, O.; Kodys, K.; Catalano, D.; Bakis, G.; Marshall, C.; Mandrekar, P.; Szabo, G. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology 2007, 133, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Inferences, questions and possibilities in toll-like receptor signalling. Nature 2004, 430, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenes. Tissue Repair 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Li, F.Y.; Lam, K.S.; Li, H.; Jia, W.; Wang, Y.; Man, K.; Lo, C.M.; Li, X.; Xu, A. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut 2012, 61, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Bala, S. Alcoholic liver disease and the gut-liver axis. World J. Gastroenterol. 2010, 16, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; de Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Loke, J.; Zheng, F.; Hong, F.; Yea, S.; Fukata, M.; Tarocchi, M.; Abar, O.T.; Huang, H.; Sninsky, J.J.; et al. Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of toll-like receptor 4 to hepatic stellate cell responses. Hepatology 2009, 49, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Dasu, M.R.; Jialal, I. Free fatty acids in the presence of high glucose amplify monocyte inflammation via toll-like receptors. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E145–E154. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Petrilli, V.; van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through NALP3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Szabo, G.; Csak, T. Inflammasomes in liver diseases. J. Hepatol. 2012, 57, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Flask, C.A.; Papouchado, B.G.; Feldstein, A.E.; Nagy, L.E. Caspase-1 as a central regulator of high fat diet-induced non-alcoholic steatohepatitis. PLoS ONE 2013, 8, e56100. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Joosten, L.A.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van den Berg, S.A.; Rensen, P.C.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Membrez, M.; Ammon-Zufferey, C.; Philippe, D.; Aprikian, O.; Monnard, I.; Mace, K.; Darimont, C. Interleukin-18 protein level is upregulated in adipose tissue of obese mice. Obesity 2009, 17, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Pillai, A.; Ganz, M.; Lippai, D.; Petrasek, J.; Park, J.K.; Kodys, K.; Dolganiuc, A.; Kurt-Jones, E.A.; Szabo, G. Both bone marrow-derived and non-bone marrow-derived cells contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis. Liver Int. 2014, 34, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.; Bukong, T.N.; Csak, T.; Saha, B.; Park, J.K.; Ambade, A.; Kodys, K.; Szabo, G. Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice. J. Transl. Med. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; McGeough, M.D.; Pena, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Huebener, P.; Pradere, J.P.; Hernandez, C.; Gwak, G.Y.; Caviglia, J.M.; Mu, X.; Loike, J.D.; Jenkins, R.E.; Antoine, D.J.; Schwabe, R.F. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Investig. 2015, 125, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial damps cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J. Mitochondria: Sovereign of inflammation? Eur. J. Immunol. 2011, 41, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Carp, H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J. Exp. Med. 1982, 155, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C.; et al. Necrotic cells trigger a sterile inflammatory response through the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 20388–20393. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Coddou, C.; Yan, Z.; Obsil, T.; Huidobro-Toro, J.P.; Stojilkovic, S.S. Activation and regulation of purinergic P2X receptor channels. Pharmacol. Rev. 2011, 63, 641–683. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F. Liaisons dangereuses: P2X7 and the inflammasome. Trends Pharmacol. Sci. 2007, 28, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Stros, M. HMGB proteins: Interactions with DNA and chromatin. Biochim. Biophys. Acta 2010, 1799, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E. HMGB1 loves company. J. Leukoc. Biol. 2009, 86, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M. Clinical relevance of adipokines. Diabetes Metab. J. 2012, 36, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Bertolani, C. Adipokines in liver diseases. Hepatology 2009, 50, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Nonalcoholic fatty liver disease: The pathogenetic roles of insulin resistance and adipocytokines. Curr. Mol. Med. 2009, 9, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Tsiaousi, E. The role of adiponectin in the pathogenesis and treatment of non-alcoholic fatty liver disease. Diabetes Obes. Metab. 2010, 12, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. The multi-hit process and the antagonistic roles of tumor necrosis factor-α and adiponectin in non alcoholic fatty liver disease. Hippokratia 2009, 13, 127. [Google Scholar] [PubMed]
- Tilg, H.; Hotamisligil, G.S. Nonalcoholic fatty liver disease: Cytokine-adipokine interplay and regulation of insulin resistance. Gastroenterology 2006, 131, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.S.; Dalamaga, M.; Kim, S.Y.; Polyzos, S.A.; Hamnvik, O.P.; Magkos, F.; Paruthi, J.; Mantzoros, C.S. Leptin’s role in lipodystrophic and nonlipodystrophic insulin-resistant and diabetic individuals. Endocr. Rev. 2013, 34, 377–412. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Dattaroy, D.; Pourhoseini, S.; Das, S.; Alhasson, F.; Seth, R.K.; Nagarkatti, M.; Michelotti, G.A.; Diehl, A.M.; Chatterjee, S. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G298–G312. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Guan, W.; Qiao, H.; Cheng, Y.; Li, Z.; Zhai, X.; Zhou, Y. Gata binding protein 2 mediates leptin inhibition of PPARγ1 expression in hepatic stellate cells and contributes to hepatic stellate cell activation. Biochim. Biophys. Acta 2014, 1842, 2367–2377. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, G.; Russo, L.; Castaneda, T.R.; Pfeiffer, V.; Ghadieh, H.E.; Ghanem, S.S.; Wu, J.; Faulkner, L.D.; Ergun, S.; McInerney, M.F.; et al. Leptin resistance contributes to obesity in mice with null mutation of carcinoembryonic antigen-related cell adhesion molecule 1. J. Biol. Chem. 2016, 291, 11124–11132. [Google Scholar] [CrossRef] [PubMed]
- Poy, M.N.; Yang, Y.; Rezaei, K.; Fernstrom, M.A.; Lee, A.D.; Kido, Y.; Erickson, S.K.; Najjar, S.M. CEACAM1 regulates insulin clearance in liver. Nat. Genet. 2002, 30, 270–276. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, A.M.; Heinrich, G.; Dai, T.; Bowman, T.A.; Patel, P.R.; Lee, S.J.; Hong, E.G.; Jung, D.Y.; Assmann, A.; Kulkarni, R.N.; et al. Carcinoembryonic antigen-related cell adhesion molecule 1: A link between insulin and lipid metabolism. Diabetes 2008, 57, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Yoda-Murakami, M.; Taniguchi, M.; Takahashi, K.; Kawamata, S.; Saito, K.; Choi-Miura, N.H.; Tomita, M. Change in expression of GBP28/adiponectin in carbon tetrachloride-administrated mouse liver. Biochem. Biophys. Res. Commun. 2001, 285, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Heiker, J.T.; Kosel, D.; Beck-Sickinger, A.G. Molecular mechanisms of signal transduction via adiponectin and adiponectin receptors. Biol. Chem. 2010, 391, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Qu, A.; Matsubara, T.; Chanturiya, T.; Jou, W.; Gavrilova, O.; Shah, Y.M.; Gonzalez, F.J. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 2011, 60, 2484–2495. [Google Scholar] [CrossRef] [PubMed]
- Otani, H. Oxidative stress as pathogenesis of cardiovascular risk associated with metabolic syndrome. Antioxid. Redox Signal. 2011, 15, 1911–1926. [Google Scholar] [CrossRef] [PubMed]
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 2012, 23, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Nonlinear distribution of adiponectin in patients with nonalcoholic fatty liver disease limits its use in linear regression analysis. J. Clin. Gastroenterol. 2010, 44, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Van der Poorten, D.; Samer, C.F.; Ramezani-Moghadam, M.; Coulter, S.; Kacevska, M.; Schrijnders, D.; Wu, L.E.; McLeod, D.; Bugianesi, E.; Komuta, M.; et al. Hepatic fat loss in advanced nonalcoholic steatohepatitis: Are alterations in serum adiponectin the cause? Hepatology 2013, 57, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Bozaoglu, K.; Bolton, K.; McMillan, J.; Zimmet, P.; Jowett, J.; Collier, G.; Walder, K.; Segal, D. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 2007, 148, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Divoux, A.; Poitou, C.; Basdevant, A.; Bouillot, J.L.; Bedossa, P.; Tordjman, J.; Eckel, J.; Clement, K. Chemerin correlates with markers for fatty liver in morbidly obese patients and strongly decreases after weight loss induced by bariatric surgery. J. Clin. Endocrinol. Metab. 2010, 95, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.C.; Issa, M.; Goralski, K.B.; Sinal, C.J. Chemerin exacerbates glucose intolerance in mouse models of obesity and diabetes. Endocrinology 2010, 151, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Oppenheim, J.J. Chemerin reveals its chimeric nature. J. Exp. Med. 2008, 205, 2187–2190. [Google Scholar] [CrossRef] [PubMed]
- Krautbauer, S.; Wanninger, J.; Eisinger, K.; Hader, Y.; Beck, M.; Kopp, A.; Schmid, A.; Weiss, T.S.; Dorn, C.; Buechler, C. Chemerin is highly expressed in hepatocytes and is induced in non-alcoholic steatohepatitis liver. Exp. Mol. Pathol. 2013, 95, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.L.; Onnerfalt, J.; Xu, J.; Molin, G.; Ahrne, S.; Thorngren-Jerneck, K. The microbiota of the gut in preschool children with normal and excessive body weight. Obesity 2012, 20, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Wishnok, J.S.; Blusztajn, J.K. Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 1983, 225, 320–324. [Google Scholar] [PubMed]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Prawitt, J.; Abdelkarim, M.; Stroeve, J.H.; Popescu, I.; Duez, H.; Velagapudi, V.R.; Dumont, J.; Bouchaert, E.; van Dijk, T.H.; Lucas, A.; et al. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes 2011, 60, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Noverr, M.C.; Huffnagle, G.B. Does the microbiota regulate immune responses outside the gut? Trends Microbiol. 2004, 12, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Sabate, J.M.; Jouet, P.; Harnois, F.; Mechler, C.; Msika, S.; Grossin, M.; Coffin, B. High prevalence of small intestinal bacterial overgrowth in patients with morbid obesity: A contributor to severe hepatic steatosis. Obes. Surg. 2008, 18, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor α in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Minemura, M.; Shimizu, Y. Gut microbiota and liver diseases. World J. Gastroenterol. 2015, 21, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H. Gut-liver axis in liver cirrhosis: How to manage leaky gut and endotoxemia. World J. Hepatol. 2015, 7, 425–442. [Google Scholar] [CrossRef] [PubMed]
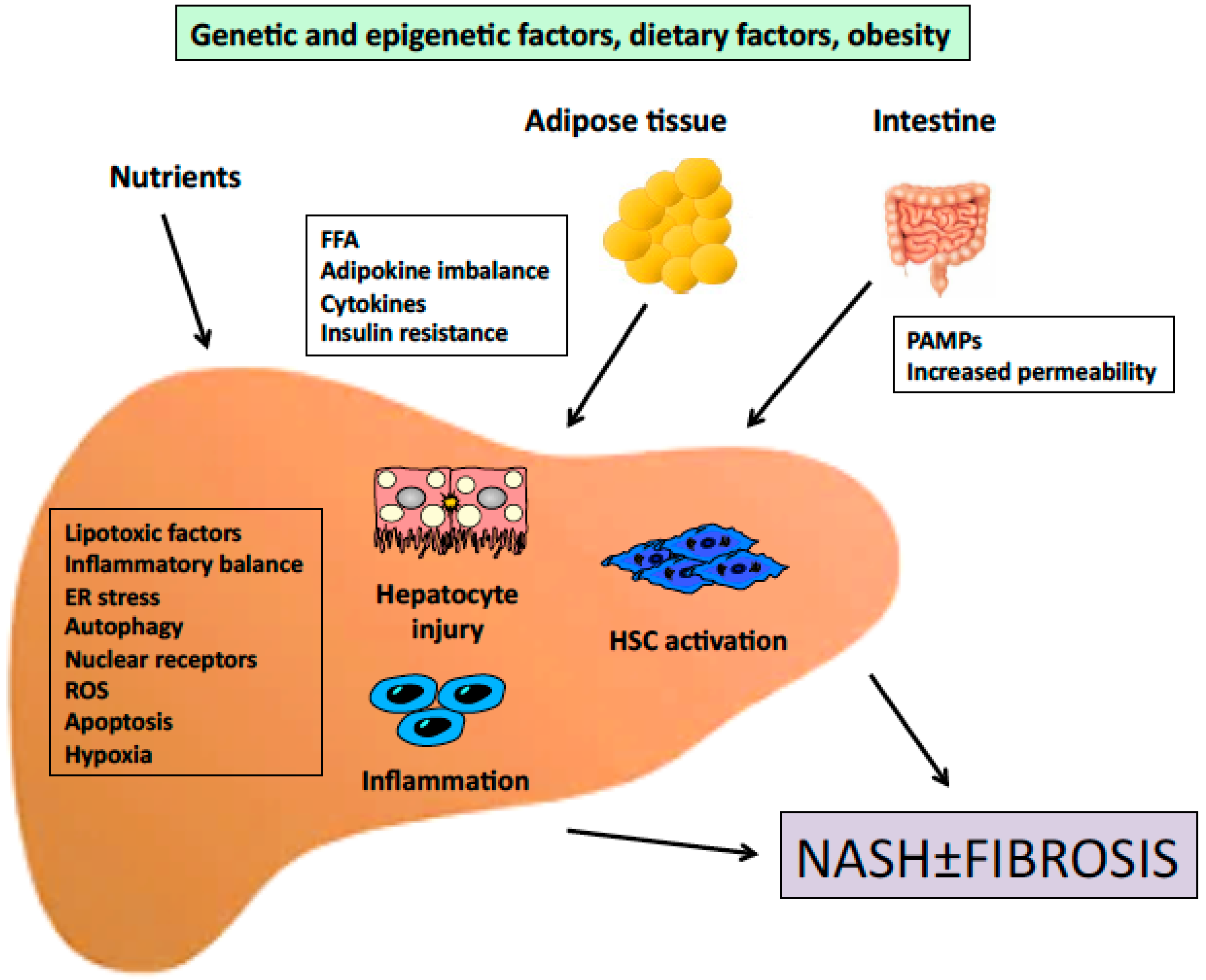
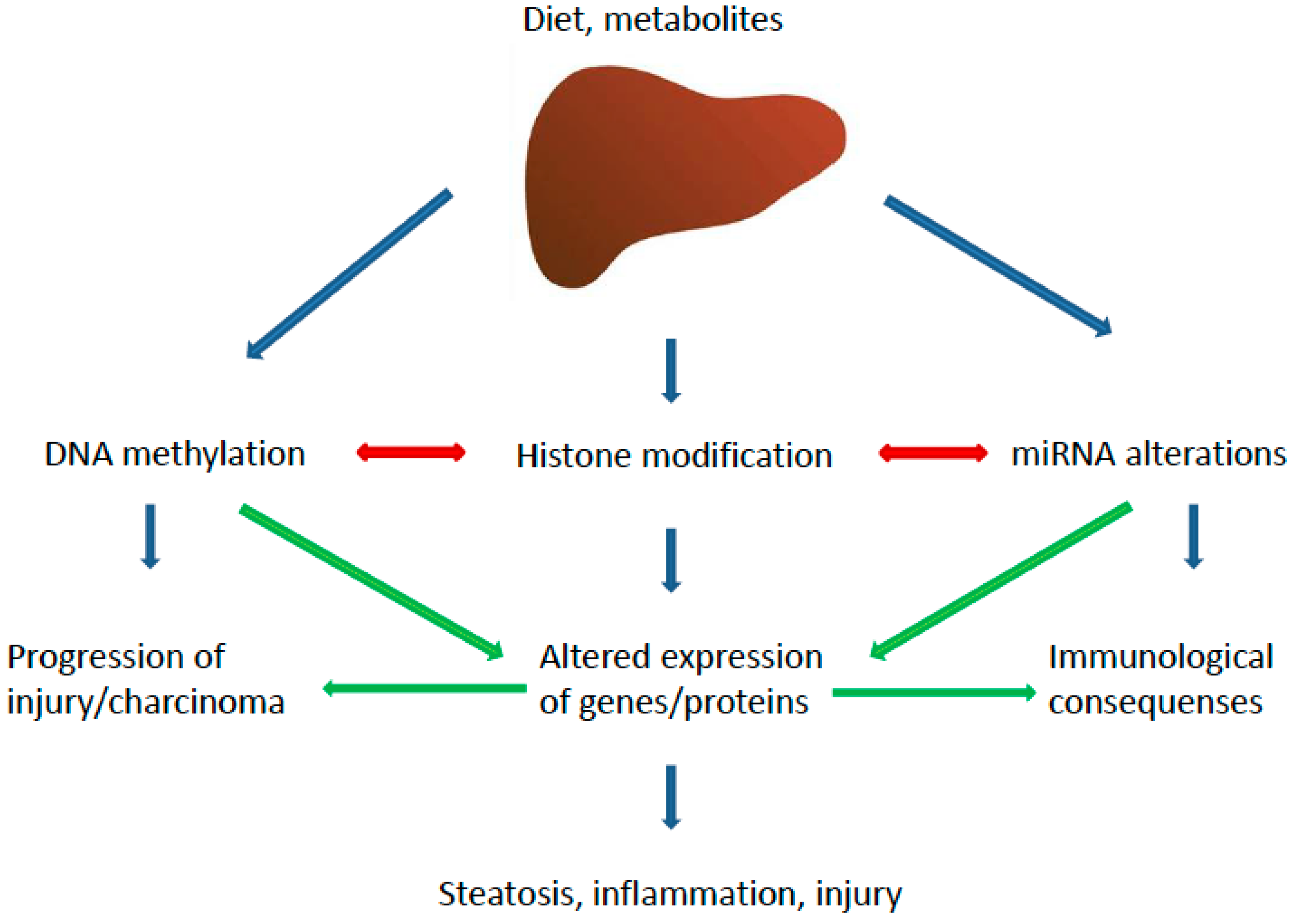
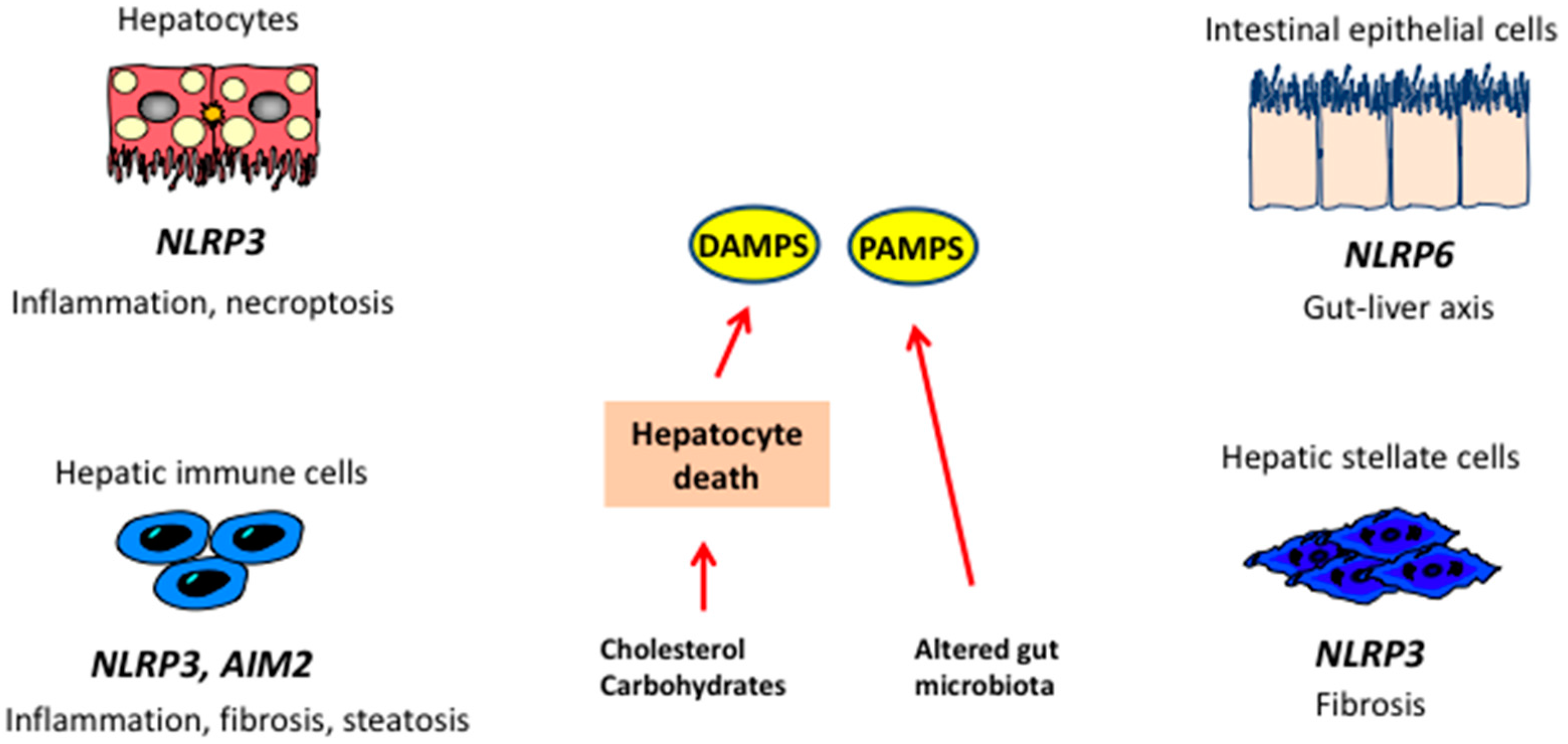

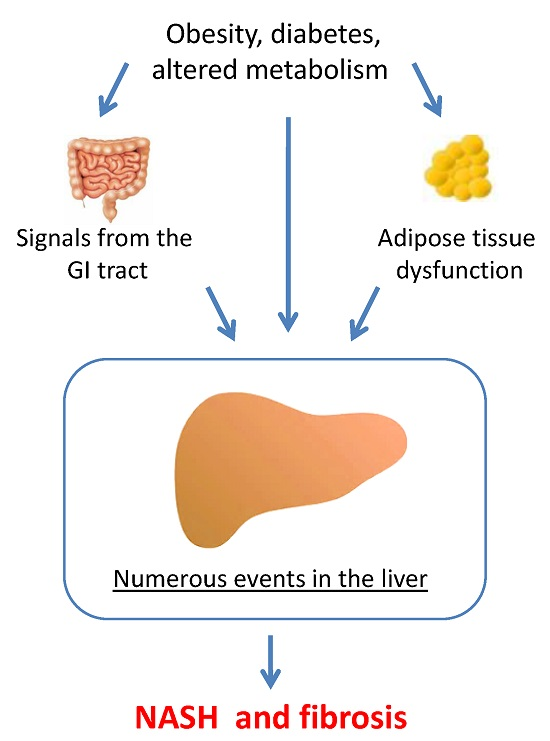{kind=link}
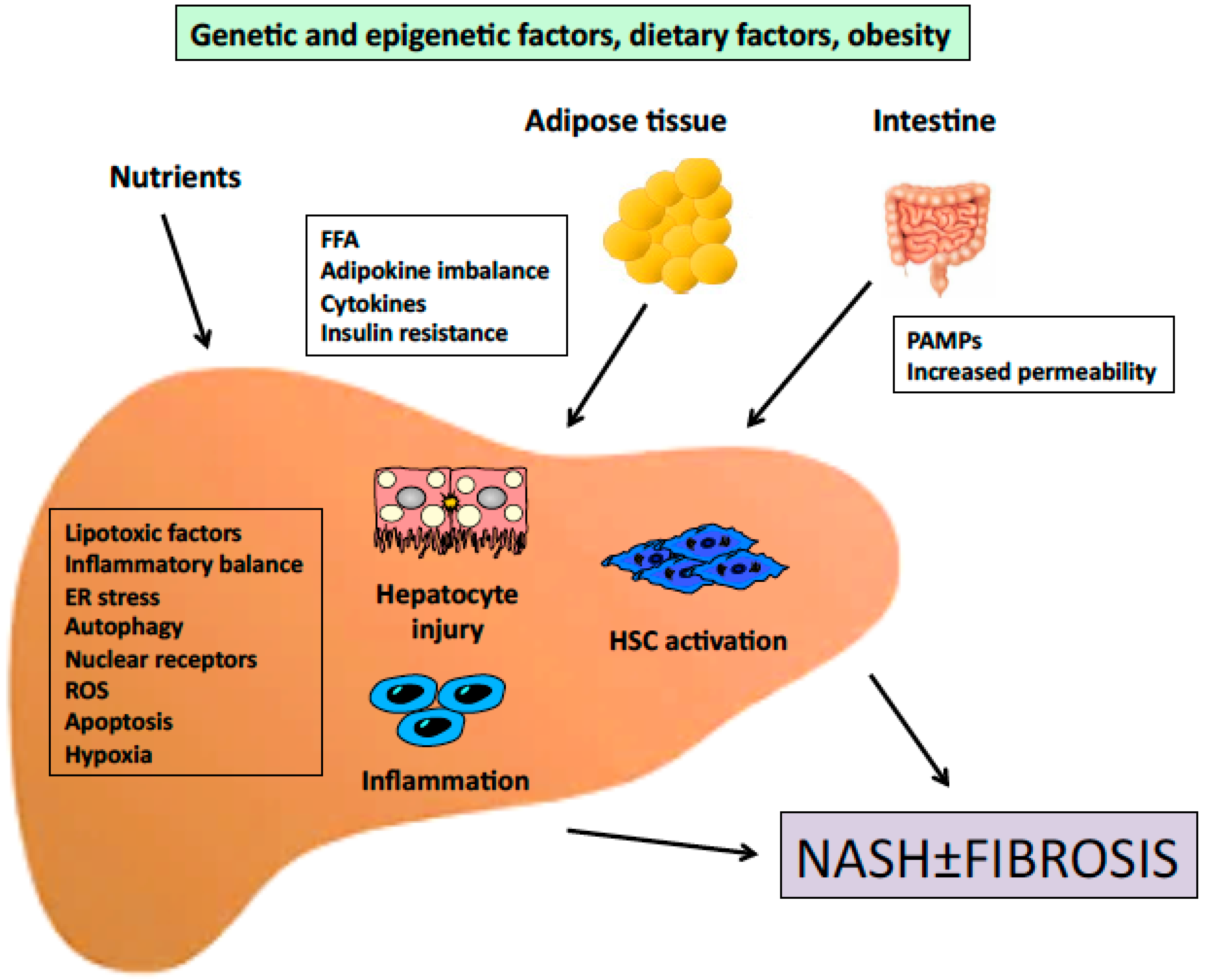{kind=link}
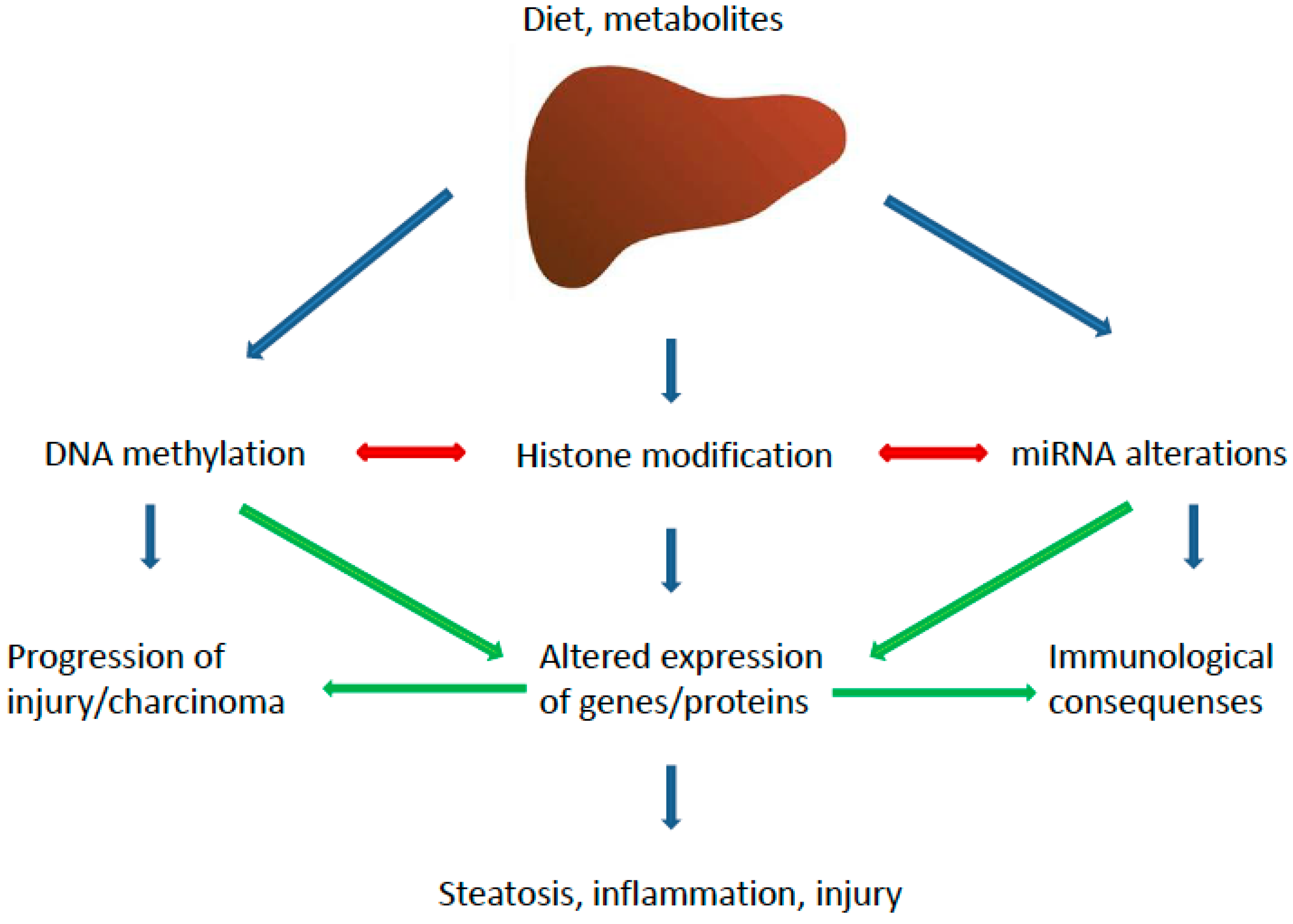{kind=link}
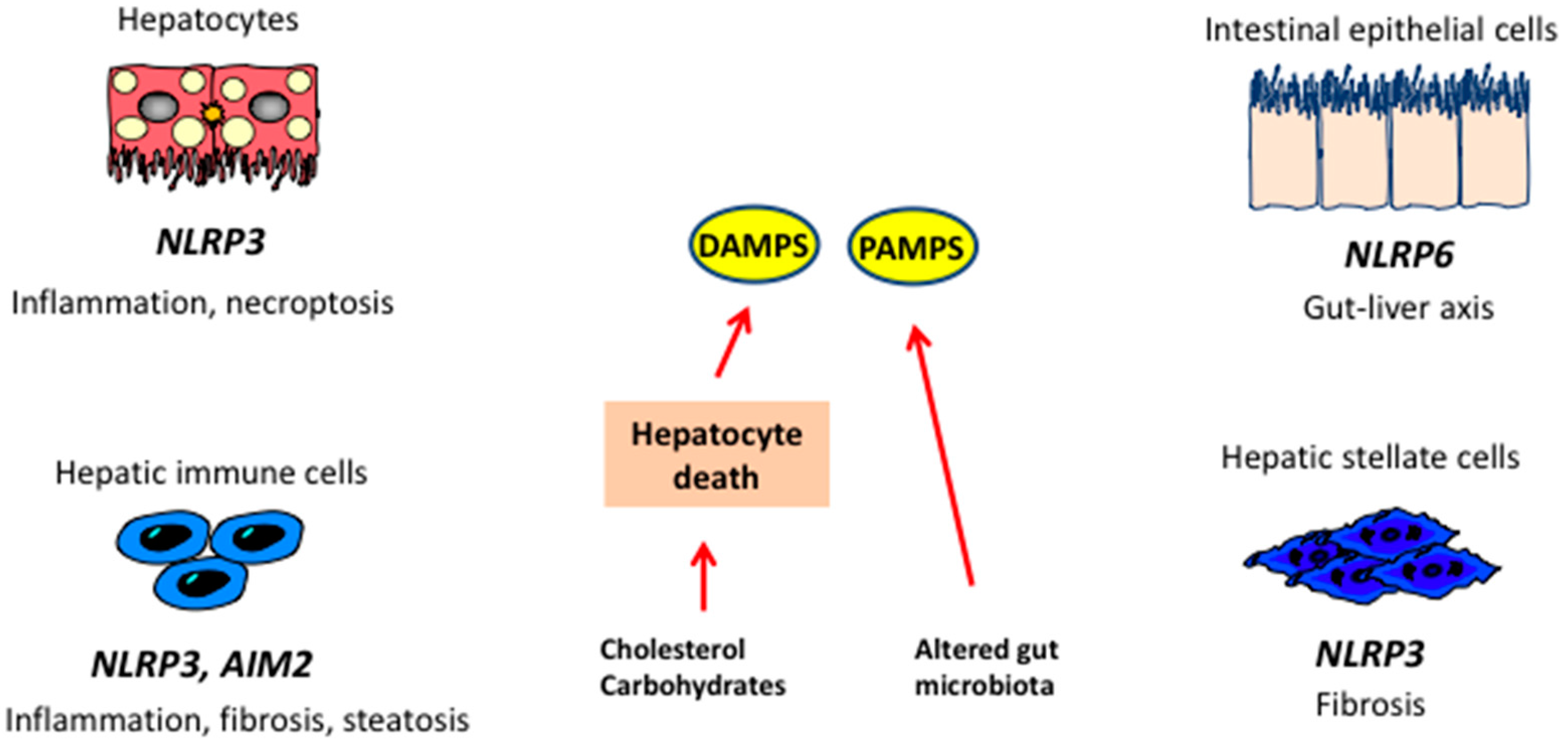{kind=link}
| miR | Δ | Disease | Model | Role | Validated/Predicted Target | Reference |
|---|---|---|---|---|---|---|
| 33a | ↑ | NASH | Mouse Liver | Cholesterol and bile acid homeostasis | CYP7A1, SREPB2 | [68] |
| 34a | ↑ | NAFLD/NASH | Human Biopsies | Lipid homeostasis | SIRT1-AMPK-HMGCR | [67,69] |
| 103a2 | ↑ | NAFLD | Human Biopsies | Insulin signaling, metabolism, inflammation | ? | [30,70] |
| 160b | ↑ | NAFLD | Human Biopsies | Insulin signaling | ? | [30] |
| 122 | ↓ | NASH | HFD mice/Human Biopsies | Lipid and cholesterol metabolism | HMGCR, FAS, SREBP1/2, ACC | [66,71] |
| 301a-3p | ↑ | Steatosis/NAFLD/NASH | Human Biopsies | ? | ? | [69] |
| 375 | ↓ | NAFLD/NASH/Cirrhosis | Human Biopsies | ? | ? | [69] |
| 576-5p | ↑ | NAFLD | Human Biopsies | Insulin signaling, metabolic homeostasis, inflammation | mTOR signaling, ephrin B signaling, ROS production, RAC1 | [70] |
| 892a | ↑ | NAFLD | Human Biopsies | Kupffer cell activation ? | ? | [70] |
| I137 | ↑ | NAFLD | Human Biopsies | ? | ? | [70] |
| 1282 | ↓ | NAFLD | Human Biopsies | Insulin signaling, metabolism, inflammation | ? | [70] |
| 3663-5p | ↑ | NAFLD | Human Biopsies | Insulin signaling, metabolism, inflammation | ? | [70] |
| 3924 | ↑ | NAFLD | Human Biopsies | Insulin signaling, metabolism, inflammation | ? | [70] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. Int. J. Mol. Sci. 2016, 17, 1575. https://doi.org/10.3390/ijms17091575
Caligiuri A, Gentilini A, Marra F. Molecular Pathogenesis of NASH. International Journal of Molecular Sciences. 2016; 17(9):1575. https://doi.org/10.3390/ijms17091575
Chicago/Turabian StyleCaligiuri, Alessandra, Alessandra Gentilini, and Fabio Marra. 2016. "Molecular Pathogenesis of NASH" International Journal of Molecular Sciences 17, no. 9: 1575. https://doi.org/10.3390/ijms17091575
APA StyleCaligiuri, A., Gentilini, A., & Marra, F. (2016). Molecular Pathogenesis of NASH. International Journal of Molecular Sciences, 17(9), 1575. https://doi.org/10.3390/ijms17091575