
Targeted Cancer Therapy: Vital Oncogenes and a New Molecular Genetic Paradigm for Cancer Initiation Progression and Treatment


Abstract
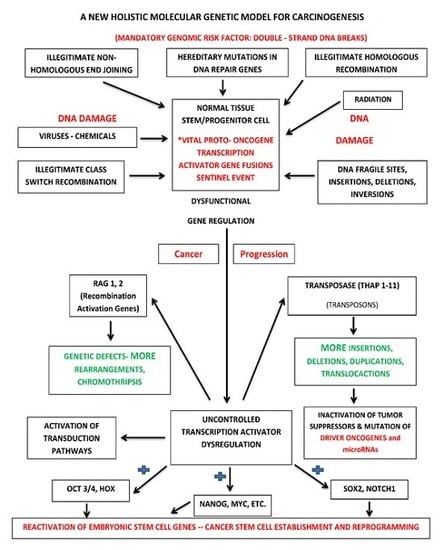
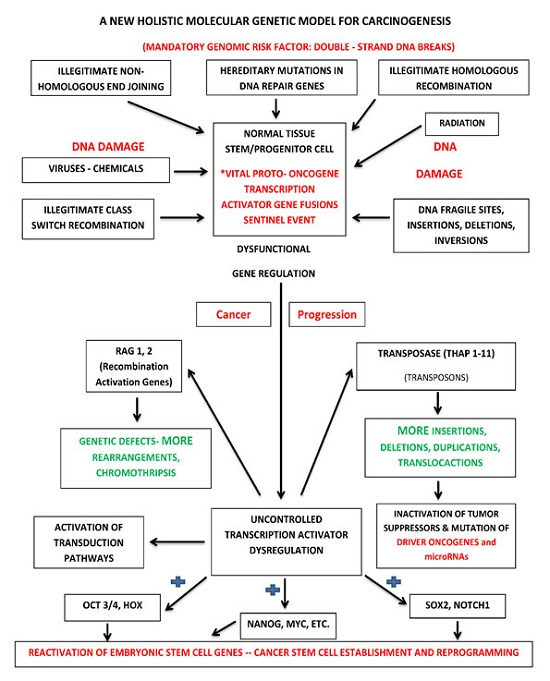
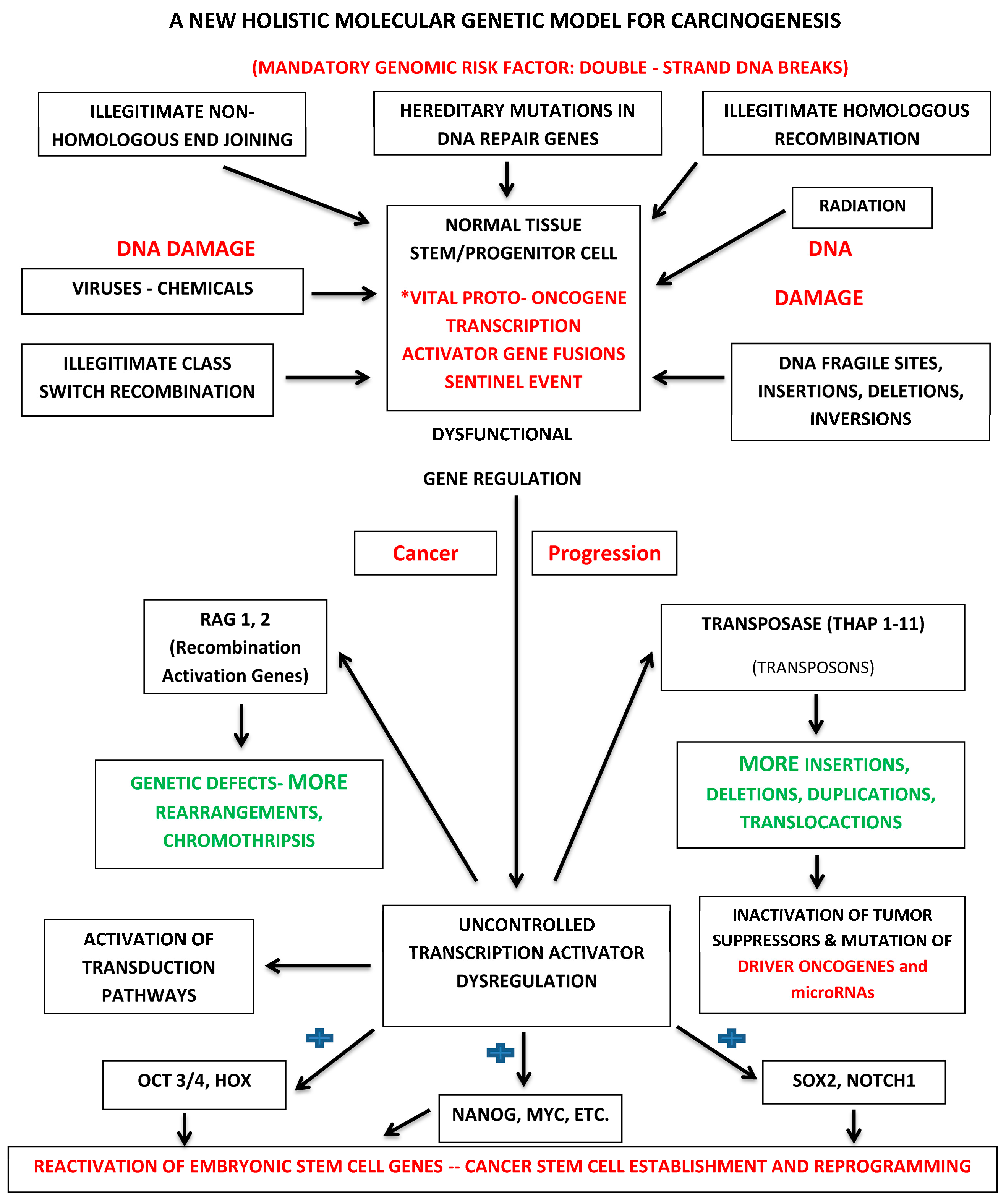
:
{kind=link}
{kind=link}
1. Introduction
2. Carcinogenesis
2.1. Facts and Theories
2.2. Cancer Stem Cells
3. The Cancer Genome
3.1. Transcription Activators
3.2. Gene Recombination
3.3. Transposons
4. Mutations in Cancer
4.1. The Genomic Landscape
4.2. Gene Fusions
5. Leukemia: A Model for Carcinogenesis
6. Carcinogenesis in Solid Cancers
6.1. Cancer Stem Cells in Solid Tumors
6.2. Fused Transcription Activators in Solid Tumors
7. Vital Oncogenes
7.1. Characteristics and Mechanisms of Action
7.2. Holistic Molecular Genetic Model for Carcinogenesis
8. Implications
8.1. Targeted Cancer Therapy
8.2. Vital Oncogenes and Cancer Molecular Diagnostics
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Croce, C.M. Oncogenes and cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Boveri, T. Concerning the origin of malignant tumours by theodor boveri. Translated and annotated by henry harris. J. Cell Sci. 2008, 121, 1–84. [Google Scholar] [CrossRef] [PubMed]
- Willis, R.E. Human gene control by vital oncogenes: Revisiting a theoretical model and its implications for targeted cancer therapy. Int. J. Mol. Sci. 2012, 13, 316–335. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Dibra, D.; Mishra, L.; Li, S. Molecular mechanisms of oncogene-induced inflammation and inflammation-sustained oncogene activation in gastrointestinal tumors: An under-appreciated symbiotic relationship. Biochim. Biophys. Acta 2014, 1846, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141. [Google Scholar] [CrossRef]
- Berenblum, I.; Shubik, P. A new, quantitative, approach to the study of the stages of chemical cartinogenesis in the mouse’s skin. Br. J. Cancer 1947, 1, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Pitot, H.C.; Dragan, Y.P. Facts and theories concerning the mechanisms of carcinogenesis. FASEB J. 1991, 5, 2280–2286. [Google Scholar] [PubMed]
- Nowell, P.C. Mechanisms of tumor progression. Cancer Res. 1986, 46, 2203–2207. [Google Scholar] [PubMed]
- Soto, A.M.; Sonnenschein, C. The somatic mutation theory of cancer: Growing problems with the paradigm? BioEssays 2004, 26, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. Warner-lambert/parke-davis award lecture. Pathological and physiological double-strand breaks: Roles in cancer, aging, and the immune system. Am. J. Pathol. 1998, 153, 1323–1332. [Google Scholar] [CrossRef]
- Mills, K.D.; Ferguson, D.O.; Alt, F.W. The role of DNA breaks in genomic instability and tumorigenesis. Immunol. Rev. 2003, 194, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Rivera, M.N.; Haber, D.A. Wilms’ tumour: Connecting tumorigenesis and organ development in the kidney. Nat. Rev. Cancer 2005, 5, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.E.; Wille, J.J., Jr.; Wier, M.L. Mechanisms for the initiation and promotion of carcinogenesis: A review and a new concept. Mayo Clin. Proc. 1984, 59, 107–117. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer 2003, 3, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tiede, B.; Massague, J.; Kang, Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res. 2007, 17, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Sprick, M.R.; Kemper, K.; Stassi, G.; Medema, J.P. Cancer stem cells—Old concepts, new insights. Cell Death Differ. 2008, 15, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Cironi, L.; Provero, P.; Suva, M.L.; Kaloulis, K.; Garcia-Echeverria, C.; Hoffmann, F.; Trumpp, A.; Stamenkovic, I. Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res. 2005, 65, 11459–11468. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Ang, B.T.; Pervaiz, S. Cancer stem cell: Target for anti-cancer therapy. FASEB J. 2007, 21, 3777–3785. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Wicha, M.S. Cancer stem cells: A step toward the cure. J. Clin. Oncol. 2008, 26, 2795–2799. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Ciurea, M.E.; Georgescu, A.M.; Purcaru, S.O.; Artene, S.A.; Emami, G.H.; Boldeanu, M.V.; Tache, D.E.; Dricu, A. Cancer stem cells: Biological functions and therapeutically targeting. Int. J. Mol. Sci. 2014, 15, 8169–8185. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulou, P.A.; Christodoulou, M.S.; Silvani, A.; Herold-Mende, C.; Passarella, D. Chemical approaches to targeting drug resistance in cancer stem cells. Drug Discov. Today 2014, 19, 1547–1562. [Google Scholar] [CrossRef] [PubMed]
- Venters, B.J.; Pugh, B.F. How eukaryotic genes are transcribed. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 117–141. [Google Scholar] [CrossRef] [PubMed]
- Brivanlou, A.H.; Darnell, J.E., Jr. Signal transduction and the control of gene expression. Science 2002, 295, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Barberis, A.; Petrascheck, M. Transcription activation in eukaryotic cells. In eLS; John Wiley & Sons, Ltd.: New York, NY, USA, 2001. [Google Scholar]
- Ma, J. Transcriptional activators and activation mechanisms. Protein Cell 2011, 2, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Timmers, C.; Maiti, B.; Saavedra, H.I.; Sang, L.; Chong, G.T.; Nuckolls, F.; Giangrande, P.; Wright, F.A.; Field, S.J.; et al. The E2F1–3 transcription factors are essential for cellular proliferation. Nature 2001, 414, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W. Transcription factors and cancer: An overview. Toxicology 2002, 181–182, 131–141. [Google Scholar] [CrossRef]
- Lu, B.; Fang, Y.; Xu, J.; Wang, L.; Xu, F.; Xu, E.; Huang, Q.; Lai, M. Analysis of SOX9 expression in colorectal cancer. Am. J. Clin. Pathol. 2008, 130, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.S.; Fang, W.T.; Hou, Y.H.; Huang, S.F.; Yen, B.L.; Chang, J.L.; Li, S.M.; Liu, H.P.; Liu, Y.L.; Huang, C.T.; et al. Upregulation of SOX9 in lung adenocarcinoma and its involvement in the regulation of cell growth and tumorigenicity. Clin. Cancer Res. 2010, 16, 4363–4373. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Hurley, P.J.; Simons, B.W.; Marchionni, L.; Berman, D.M.; Ross, A.E.; Schaeffer, E.M. SOX9 is required for prostate development and prostate cancer initiation. Oncotarget 2012, 3, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xu, Y.; Chen, Y.; Li, X.; Mou, W.; Wang, L.; Liu, Y.; Reisfeld, R.A.; Xiang, R.; Lv, D.; et al. SOX2 gene regulates the transcriptional network of oncogenes and affects tumorigenesis of human lung cancer cells. PLoS ONE 2012, 7, e36326. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xiao, Y.; Zhang, J.; Li, J.; Liu, Y.; Zhao, Y.; Ma, C.; Luo, J.; Qiu, Y.; Huang, G.; et al. Transcription factors E2A, FOXO1 and FOXP1 regulate recombination activating gene expression in cancer cells. PLoS ONE 2011, 6, e20475. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, M.; Weaver, Z.; Barlow, C.; Coleman, A.; Pankratz, D.G.; Anderson, S.; Wynshaw-Boris, A.; Ried, T. Abnormal rearrangement within the α/δ T-cell receptor locus in lymphomas from Atm-deficient mice. Blood 2000, 96, 1940–1946. [Google Scholar] [PubMed]
- Xu, Y.; Ashley, T.; Brainerd, E.E.; Bronson, R.T.; Meyn, M.S.; Baltimore, D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996, 10, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Difilippantonio, M.J.; Zhu, J.; Chen, H.T.; Meffre, E.; Nussenzweig, M.C.; Max, E.E.; Ried, T.; Nussenzweig, A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 2000, 404, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ferguson, D.O.; Xie, W.; Manis, J.P.; Sekiguchi, J.; Frank, K.M.; Chaudhuri, J.; Horner, J.; DePinho, R.A.; Alt, F.W. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 2000, 404, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Mills, K.D.; Ferguson, D.O.; Lee, C.; Manis, J.; Fleming, J.; Gao, Y.; Morton, C.C.; Alt, F.W. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 2002, 109, 811–821. [Google Scholar] [CrossRef]
- Sadofsky, M.J. The RAG proteins in V(D)J recombination: More than just a nuclease. Nucl. Acids Res. 2001, 29, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Hiom, K.; Melek, M.; Gellert, M. DNA transposition by the RAG1 and RAG2 proteins: A possible source of oncogenic translocations. Cell 1998, 94, 463–470. [Google Scholar] [CrossRef]
- Rothkamm, K.; Kuhne, M.; Jeggo, P.A.; Lobrich, M. Radiation-induced genomic rearrangements formed by nonhomologous end-joining of DNA double-strand breaks. Cancer Res. 2001, 61, 3886–3893. [Google Scholar] [PubMed]
- Zucman-Rossi, J.; Legoix, P.; Victor, J.M.; Lopez, B.; Thomas, G. Chromosome translocation based on illegitimate recombination in human tumors. Proc. Natl. Acad. Sci. USA 1998, 95, 11786–11791. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Mizukami, T.; Kohno, T. Molecular process producing oncogene fusion in lung cancer cells by illegitimate repair of DNA double-strand breaks. Biomolecules 2015, 5, 2464–2476. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, W.P.; Hoogstraat, M.; Paling, O.; Tavakoli-Yaraki, M.; Renkens, I.; Vermaat, J.S.; van Roosmalen, M.J.; van Lieshout, S.; Nijman, I.J.; Roessingh, W.; et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol. 2011, 12, R103. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.A.; Wilson, R.K. Chromothripsis and human disease: Piecing together the shattering process. Cell 2012, 148, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.; Jallepalli, P.V. Chromothripsis: Chromosomes in crisis. Dev. Cell 2012, 23, 908–917. [Google Scholar] [CrossRef]
- Sorzano, C.O.; Pascual-Montano, A.; Sanchez de Diego, A.; Martinez, A.C.; van Wely, K.H. Chromothripsis: Breakage-fusion-bridge over and over again. Cell Cycle 2013, 12, 2016–2023. [Google Scholar]
- Biémont, C. A brief history of the status of transposable elements: From junk DNA to major players in evolution. Genetics 2010, 186, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Kidwell, M.G.; Lisch, D.R. Perspective: Transposable elements, parasitic DNA, and genome evolution. Evolution 2001, 55, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Nekrutenko, A.; Li, W.H. Transposable elements are found in a large number of human protein-coding genes. Trends Genet. 2001, 17, 619–621. [Google Scholar] [CrossRef]
- Jordan, I.K.; Rogozin, I.B.; Glazko, G.V.; Koonin, E.V. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 2003, 19, 68–72. [Google Scholar] [CrossRef]
- Fodde, R.; Smits, R.; Clevers, H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Nishisho, I.; Horii, A.; Miyoshi, Y.; Utsunomiya, J.; Kinzler, K.W.; Vogelstein, B.; Nakamura, Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992, 52, 643–645. [Google Scholar] [PubMed]
- Iskow, R.C.; McCabe, M.T.; Mills, R.E.; Torene, S.; Pittard, W.S.; Neuwald, A.F.; van Meir, E.G.; Vertino, P.M.; Devine, S.E. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell 2010, 141, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J., 3rd; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Bielas, J.H.; Beckman, R.A. Cancers exhibit a mutator phenotype: Clinical implications. Cancer Res. 2008, 68, 3551–3557. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Human cancers express mutator phenotypes: Origin, consequences and targeting. Nat. Rev. Cancer 2011, 11, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Watson, I.R.; Takahashi, K.; Futreal, P.A.; Chin, L. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013, 14, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. Chromosome translocations: Dangerous liaisons revisited. Nat. Rev. Cancer 2001, 1, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Aman, P. Fusion oncogenes in tumor development. Semin. Cancer Biol. 2005, 15, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Hotfilder, M.; Röttgers, S.; Rosemann, A.; Schrauder, A.; Schrappe, M.; Pieters, R.; Jürgens, H.; Harbott, J.; Vormoor, J. Leukemic stem cells in childhood high-risk ALL/t(9;22) and t(4;11) are present in primitive lymphoid-restricted CD34+CD19− cells. Cancer Res. 2005, 65, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Cironi, L.; Provero, P.; Suva, M.L.; Stehle, J.C.; Baumer, K.; Guillou, L.; Stamenkovic, I. Expression of the fus-chop fusion protein in primary mesenchymal progenitor cells gives rise to a model of myxoid liposarcoma. Cancer Res. 2006, 66, 7016–7023. [Google Scholar] [CrossRef] [PubMed]
- Castro-Giner, F.; Ratcliffe, P.; Tomlinson, I. The mini-driver model of polygenic cancer evolution. Nat. Rev. Cancer 2015, 15, 680–685. [Google Scholar] [CrossRef] [PubMed]
- McFarland, C.D.; Mirny, L.A.; Korolev, K.S. Tug-of-war between driver and passenger mutations in cancer and other adaptive processes. Proc. Natl. Acad. Sci. USA 2014, 111, 15138–15143. [Google Scholar] [CrossRef] [PubMed]
- McFarland, C.D.; Korolev, K.S.; Kryukov, G.V.; Sunyaev, S.R.; Mirny, L.A. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. USA 2013, 110, 2910–2915. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, F.H.; Bernards, R. Drug resistance to targeted therapies: Deja vu all over again. Mol. Oncol. 2014, 8, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.W.; Vogelstein, B.; Nowak, M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Merid, S.K.; Goranskaya, D.; Alexeyenko, A. Distinguishing between driver and passenger mutations in individual cancer genomes by network enrichment analysis. BMC Bioinform. 2014, 15, 308. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Dobson, J.R.; Oesper, L.; Vandin, F. Identifying driver mutations in sequenced cancer genomes: Computational approaches to enable precision medicine. Genome Med. 2014, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.; Liu, L.L.; Leder, K.; Riester, M.; Iwasa, Y.; Lengauer, C.; Michor, F. An evolutionary approach for identifying driver mutations in colorectal cancer. PLoS Comput. Biol. 2015, 11, e1004350. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Kandoth, C.; Reimand, J.; Lawrence, M.S.; Getz, G.; Bader, G.D.; Ding, L.; et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013, 3, 2650. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Acute lymphoblastic leukemia in children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Mrózek, K.; Dodge, R.K.; Carroll, A.J.; Edwards, C.G.; Arthur, D.C.; Pettenati, M.J.; Patil, S.R.; Rao, K.W.; Watson, M.S.; et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: Results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002, 100, 4325–4336. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Corral, J.; Lavenir, I.; Impey, H.; Warren, A.J.; Forster, A.; Larson, T.A.; Bell, S.; McKenzie, A.N.; King, G.; Rabbitts, T.H. An MLL-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: A method to create fusion oncogenes. Cell 1996, 85, 853–861. [Google Scholar] [CrossRef]
- Kuo, Y.-H.; Landrette, S.F.; Heilman, S.A.; Perrat, P.N.; Garrett, L.; Liu, P.P.; Le Beau, M.M.; Kogan, S.C.; Castilla, L.H. Cbfβ-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell 2006, 9, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Mrózek, K.; Marcucci, G.; Paschka, P.; Whitman, S.P.; Bloomfield, C.D. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: Are we ready for a prognostically prioritized molecular classification? Blood 2007, 109, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Haferlach, T.; Döhner, H. Molecular genetics of adult acute myeloid leukemia: Prognostic and therapeutic implications. J. Clin. Oncol. 2011, 29, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Mendler, J.H.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Becker, H.; Metzeler, K.H.; Schwind, S.; Whitman, S.P.; Khalife, J.; Kohlschmidt, J.; et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J. Clin. Oncol. 2012, 30, 3109–3118. [Google Scholar] [CrossRef]
- Tang, J.-L.; Hou, H.-A.; Chen, C.-Y.; Liu, C.-Y.; Chou, W.-C.; Tseng, M.-H.; Huang, C.-F.; Lee, F.-Y.; Liu, M.-C.; Yao, M.; et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: Prognostic implication and interaction with other gene alterations. Blood 2009, 114, 5352–5361. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Kowarz, E.; Hofmann, J.; Renneville, A.; Zuna, J.; Trka, J.; Ben Abdelali, R.; Macintyre, E.; de Braekeleer, E.; de Braekeleer, M.; et al. New insights to the MLL recombinome of acute leukemias. Leukemia 2009, 23, 1490–1499. [Google Scholar] [CrossRef]
- Look, A.T. Oncogenic transcription factors in the human acute leukemias. Science 1997, 278, 1059–1064. [Google Scholar] [CrossRef]
- Scandura, J.M.; Boccuni, P.; Cammenga, J.; Nimer, S.D. Transcription factor fusions in acute leukemia: Variations on a theme. Oncogene 2002, 21, 3422–3444. [Google Scholar] [CrossRef]
- McNeil, S.; Zeng, C.; Harrington, K.S.; Hiebert, S.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. The t(8;21) chromosomal translocation in acute myelogenous leukemia modifies intranuclear targeting of the AML1/CBFA2 transcription factor. Proc. Natl. Acad. Sci. USA 1999, 96, 14882–14887. [Google Scholar] [CrossRef] [PubMed]
- Licht, J.D. AML1 and the AML1-ETO fusion protein in the pathogenesis of t(8;21) AML. Oncogene 2001, 20, 5660–5679. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.C.; Sun, X.; Berguido, F.J.; Jakubowiak, A.; Nimer, S.D. The t(8;21) fusion protein, AML1/ETO, transforms NIH3T3 cells and activates AP-1. Oncogene 1999, 18, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Riggi, N.; Stehle, J.C.; Baumer, K.; Tercier, S.; Joseph, J.M.; Suva, D.; Clement, V.; Provero, P.; Cironi, L.; et al. Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res. 2009, 69, 1776–1781. [Google Scholar] [CrossRef] [PubMed]
- Naka, N.; Takenaka, S.; Araki, N.; Miwa, T.; Hashimoto, N.; Yoshioka, K.; Joyama, S.; Hamada, K.; Tsukamoto, Y.; Tomita, Y.; et al. Synovial sarcoma is a stem cell malignancy. Stem Cells 2010, 28, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Stratford, E.W.; Castro, R.; Wennerstrom, A.; Holm, R.; Munthe, E.; Lauvrak, S.; Bjerkehagen, B.; Myklebost, O. Liposarcoma cells with aldefluor and CD133 activity have a cancer stem cell potential. Clin. Sarcoma Res. 2011, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.-H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G.; Patrawala, L.; Calhoun, T.; Bhatia, B.; Choy, G.; Schneider-Broussard, R.; Jeter, C. Prostate cancer stem/progenitor cells: Identification, characterization, and implications. Mol. Carcinog. 2007, 46, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; de Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer stem cell hierarchy in glioblastoma multiforme. Front. Surg. 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Kaye, F.J. Mutation-associated fusion cancer genes in solid tumors. Mol. Cancer Ther. 2009, 8, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, C.P.; Sun, S.; Varma, S.; Shain, A.H.; Giacomini, M.M.; Balagtas, J.; Sweeney, R.T.; Lai, E.; del Vecchio, C.A.; Forster, A.D.; et al. Breakpoint analysis of transcriptional and genomic profiles uncovers novel gene fusions spanning multiple human cancer types. PLoS Genet. 2013, 9, e1003464. [Google Scholar] [CrossRef] [PubMed]
- Stenman, G.; Andersson, M.K.; Andren, Y. New tricks from an old oncogene: Gene fusion and copy number alterations of MYB in human cancer. Cell Cycle 2010, 9, 2986–2995. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G. The role of chimeric paired box transcription factors in the pathogenesis of pediatric rhabdomysarcoma. Cancer Res. 1999, 59, S1711–S1715. [Google Scholar]
- Davicioni, E.; Finckenstein, F.G.; Shahbazian, V.; Buckley, J.D.; Triche, T.J.; Anderson, M.J. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006, 66, 6936–6946. [Google Scholar] [CrossRef] [PubMed]
- Perez-Losada, J.; Pintado, B.; Gutierrez-Adan, A.; Flores, T.; Banares-Gonzalez, B.; del Campo, J.C.; Martin-Martin, J.F.; Battaner, E.; Sanchez-Garcia, I. The chimeric FUS/TLS-CHOP fusion protein specifically induces liposarcomas in transgenic mice. Oncogene 2000, 19, 2413–2422. [Google Scholar] [CrossRef] [PubMed]
- Edwards, P.A.; Howarth, K.D. Are breast cancers driven by fusion genes? Breast Cancer Res. 2012, 14, 303. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; McBride, D.J.; Lin, M.L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Asmann, Y.W.; Necela, B.M.; Kalari, K.R.; Hossain, A.; Baker, T.R.; Carr, J.M.; Davis, C.; Getz, J.E.; Hostetter, G.; Li, X.; et al. Detection of redundant fusion transcripts as biomarkers or disease-specific therapeutic targets in breast cancer. Cancer Res. 2012, 72, 1921–1928. [Google Scholar] [CrossRef] [PubMed]
- Rubin, M.A.; Maher, C.A.; Chinnaiyan, A.M. Common gene rearrangements in prostate cancer. J. Clin. Oncol. 2011, 29, 3659–3668. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.D. The myb and myc nuclear oncogenes as transcriptional activators. Curr. Opin. Cell Biol. 1990, 2, 502–508. [Google Scholar] [CrossRef]
- Boxer, L.M.; Dang, C.V. Translocations involving c-myc and c-myc function. Oncogene 2001, 20, 5595–5610. [Google Scholar] [CrossRef] [PubMed]
- Fredericks, W.J.; Galili, N.; Mukhopadhyay, S.; Rovera, G.; Bennicelli, J.; Barr, F.G.; Rauscher, F.J., III. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol. Cell. Biol. 1995, 15, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Bennicelli, J.L.; Edwards, R.H.; Barr, F.G. Mechanism for transcriptional gain of function resulting from chromosomal translocation in alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 1996, 93, 5455–5459. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Simon, J.M.; Iglesia, M.D.; Wu, S.B.; McFadden, A.W.; Lieb, J.D.; Davis, I.J. Tumor-specific retargeting of an oncogenic transcription factor chimera results in dysregulation of chromatin and transcription. Genome Res. 2012, 22, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Zwerner, J.P.; Joo, J.; Warner, K.L.; Christensen, L.; Hu-Lieskovan, S.; Triche, T.J.; May, W.A. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2007, 27, 3282–3291. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. Gli1 is a direct transcriptional target of ews-fli1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Muller-Tidow, C.; Steffen, B.; Cauvet, T.; Tickenbrock, L.; Ji, P.; Diederichs, S.; Sargin, B.; Kohler, G.; Stelljes, M.; Puccetti, E.; et al. Translocation products in acute myeloid leukemia activate the wnt signaling pathway in hematopoietic cells. Mol. Cell. Biol. 2004, 24, 2890–2904. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Skytting, B.; Nilsson, G.; Grimer, R.J.; Mangham, C.D.; Fisher, C.; Shipley, J.; Bjerkehagen, B.; Myklebost, O.; Larsson, O. The SYT-SSX1 fusion type of synovial sarcoma is associated with increased expression of cyclin A and D1. A link between t(X;18)(p11.2; q11.2) and the cell cycle machinery. Oncogene 2002, 21, 5791–5796. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.W.; Bose, S.; Gora-Tybor, J.; Yan, X.H.; Goldman, J.M.; Melo, J.V. Selective induction of leukemia-associated fusion genes by high-dose ionizing radiation. Cancer Res. 1998, 58, 421–425. [Google Scholar] [PubMed]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction—A rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Settleman, J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007, 21, 3214–3231. [Google Scholar] [CrossRef] [PubMed]
- Torti, D.; Trusolino, L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: Promises and perils. EMBO Mol. Med. 2011, 3, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, R.; Shao, W.; Sellers, W.R. Oncogene addiction: Pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015, 16, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Buchdunger, E.; Druker, B.J. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005, 105, 2640–2653. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Shen, A.; Ding, J.; Geng, M. Molecularly targeted cancer therapy: Some lessons from the past decade. Trends Pharmacol. Sci. 2014, 35, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Hare, T.; Eide, C.A.; Deininger, M.W.N. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Apperley, J.F. Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007, 8, 1018–1029. [Google Scholar] [CrossRef]
- Lackner, M.R.; Wilson, T.R.; Settleman, J. Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol. 2012, 8, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Jänne, P.A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Karamouzis, M.V.; Gorgoulis, V.G.; Papavassiliou, A.G. Transcription factors and neoplasia: Vistas in novel drug design. Clin. Cancer Res. 2002, 8, 949–961. [Google Scholar] [PubMed]
- Derheimer, F.A.; Chang, C.W.; Ljungman, M. Transcription inhibition: A potential strategy for cancer therapeutics. Eur. J. Cancer 2005, 41, 2569–2576. [Google Scholar] [CrossRef] [PubMed]
- Mees, C.; Nemunaitis, J.; Senzer, N. Transcription factors: Their potential as targets for an individualized therapeutic approach to cancer. Cancer Gene Ther. 2009, 16, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Stellrecht, C.M.; Chen, L.S. Transcription inhibition as a therapeutic target for cancer. Cancers 2011, 3, 4170–4190. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.E.; Toniolo, P.A.; Frank, D.A. Targeting transcription factors: Promising new strategies for cancer therapy. Curr. Opin. Oncol. 2013, 25, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Gonda, T.J.; Ramsay, R.G. Directly targeting transcriptional dysregulation in cancer. Nat. Rev. Cancer 2015, 15, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut Espinosa, C.E.; Slack, F.J. The role of micrornas in cancer. Yale J. Biol. Med. 2006, 79, 131–140. [Google Scholar] [PubMed]
- Jansson, M.D.; Lund, A.H. Microrna and cancer. Mol. Oncol. 2012, 6, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.U.; Miyazaki, H.; Ochiya, T. The role of micrornas in the regulation of cancer stem cells. Front. Genet. 2014, 4, 295. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. Somamir: A database for somatic mutations impacting microrna function in cancer. Nucl. Acids Res. 2013, 41, D977–D982. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Cui, Y. Somamir 2.0: A database of cancer somatic mutations altering microrna–cerna interactions. Nucl. Acids Res. 2016, 44, D1005–D1010. [Google Scholar] [CrossRef] [PubMed]
- Ziebarth, J.D.; Bhattacharya, A.; Cui, Y. Integrative analysis of somatic mutations altering microrna targeting in cancer genomes. PLoS ONE 2012, 7, e47137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, K.; Liao, Z.; Li, Y.; Yang, H.; Chen, C.; Zhou, Y.A.; Tao, Y.; Guo, M.; Ren, T.; et al. Promoter mutation of tumor suppressor MicroRNA-7 is associated with poor prognosis of lung cancer. Mol. Clin. Oncol. 2015, 3, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Greil, J.; Heidenreich, O. Targeting leukemic fusion proteins with small interfering RNAs: Recent advances and therapeutic potentials. Acta Pharmacol. Sin. 2006, 27, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Koehler, A.N. A complex task? Direct modulation of transcription factors with small molecules. Curr. Opin. Chem. Biol. 2010, 14, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Mapp, A.K.; Pricer, R.; Sturlis, S. Targeting transcription is no longer a quixotic quest. Nat. Chem. Biol. 2015, 11, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein ews-fli1 interaction with RNA helicase a inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Grohar, P.J.; Woldemichael, G.M.; Griffin, L.B.; Mendoza, A.; Chen, Q.R.; Yeung, C.; Currier, D.G.; Davis, S.; Khanna, C.; Khan, J.; et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J. Natl. Cancer Inst. 2011, 103, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.; Wolf, J.; Thomas, R.K. Lessons learned from lung cancer genomics: The emerging concept of individualized diagnostics and treatment. J. Clin. Oncol. 2013, 31, 1858–1865. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.C.; Zhang, W. Fusion genes in solid tumors: An emerging target for cancer diagnosis and treatment. Chin. J. Cancer 2013, 32, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.A.; Kumar-Sinha, C.; Cao, X.; Kalyana-Sundaram, S.; Han, B.; Jing, X.; Sam, L.; Barrette, T.; Palanisamy, N.; Chinnaiyan, A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009, 458, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Howarth, K.D.; Blood, K.A.; Ng, B.L.; Beavis, J.C.; Chua, Y.; Cooke, S.L.; Raby, S.; Ichimura, K.; Collins, V.P.; Carter, N.P.; et al. Array painting reveals a high frequency of balanced translocations in breast cancer cell lines that break in cancer-relevant genes. Oncogene 2008, 27, 3345–3359. [Google Scholar] [CrossRef] [PubMed]
- MacConaill, L.E. Existing and emerging technologies for tumor genomic profiling. J. Clin. Oncol. 2013, 31, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Stephens, P.J.; Pleasance, E.D.; O’Meara, S.; Li, H.; Santarius, T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Hardy, C.; et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008, 40, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Edgren, H.; Murumagi, A.; Kangaspeska, S.; Nicorici, D.; Hongisto, V.; Kleivi, K.; Rye, I.H.; Nyberg, S.; Wolf, M.; Borresen-Dale, A.L.; et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011, 12, R6. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Van Der Vaart, M.; Pretorius, P.J. Circulating DNA. Ann. N. Y. Acad. Sci. 2008, 1137, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H. Circulating nucleic acids as biomarkers in breast cancer. Breast Cancer Res. 2013, 15, 211. [Google Scholar] [CrossRef] [PubMed]
- Tatischeff, I. Cell-derived microvesicles and antitumoral multidrug resistance. C. R. Biol. 2012, 335, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, C.; Cavallini, L.; Spinelli, C.; Yang, J.; Reis-Sobreiro, M.; de Candia, P.; Minciacchi, V.R.; di Vizio, D. Focus on extracellular vesicles: New frontiers of cell-to-cell communication in cancer. Int. J. Mol. Sci. 2016, 17, 175. [Google Scholar] [CrossRef] [PubMed]
- Merino, C.; Zuñiga, F.A.; Escudero, C.A.; Ormazabanl, V.; Reyes, C.; Salomon, C.; Aguayo, C. Mesenchymal stem cell-derived extracellular vesicles promote angiogenesis: New alternatives for application to human health. Front. Physiol. 2016, 7, 24. [Google Scholar]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.-J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Chennakrishnaiah, S.; Audemard, E.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys. Res. Commun. 2014, 451, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; Horswell, S.; Mitter, R.; Sakarya, O.; Constantin, T.; Salari, R.; Kirkizlar, E.; Sigurjonsson, S.; Pelham, R.; et al. Detection of ubiquitous and heterogeneous mutations in cell-free DNA from patients with early-stage non-small-cell lung cancer. Ann. Oncol. 2016, 27, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Frenel, J.S.; Carreira, S.; Goodall, J.; Roda, D.; Perez-Lopez, R.; Tunariu, N.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; NavaRodrigues, D.; et al. Serial next generation sequencing of circulating cell free DNA evaluating tumour clone response to molecularly targeted drug administration. Clin. Cancer Res. 2015, 21, 4586–4596. [Google Scholar] [CrossRef] [PubMed]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.S.B.; Kopetz, E.S.; et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willis, R.E. Targeted Cancer Therapy: Vital Oncogenes and a New Molecular Genetic Paradigm for Cancer Initiation Progression and Treatment. Int. J. Mol. Sci. 2016, 17, 1552. https://doi.org/10.3390/ijms17091552
Willis RE. Targeted Cancer Therapy: Vital Oncogenes and a New Molecular Genetic Paradigm for Cancer Initiation Progression and Treatment. International Journal of Molecular Sciences. 2016; 17(9):1552. https://doi.org/10.3390/ijms17091552
Chicago/Turabian StyleWillis, Rudolph E. 2016. "Targeted Cancer Therapy: Vital Oncogenes and a New Molecular Genetic Paradigm for Cancer Initiation Progression and Treatment" International Journal of Molecular Sciences 17, no. 9: 1552. https://doi.org/10.3390/ijms17091552
APA StyleWillis, R. E. (2016). Targeted Cancer Therapy: Vital Oncogenes and a New Molecular Genetic Paradigm for Cancer Initiation Progression and Treatment. International Journal of Molecular Sciences, 17(9), 1552. https://doi.org/10.3390/ijms17091552