
Testosterone-Mediated Endocrine Function and TH1/TH2 Cytokine Balance after Prenatal Exposure to Perfluorooctane Sulfonate: By Sex Status

 ,
, 

Abstract
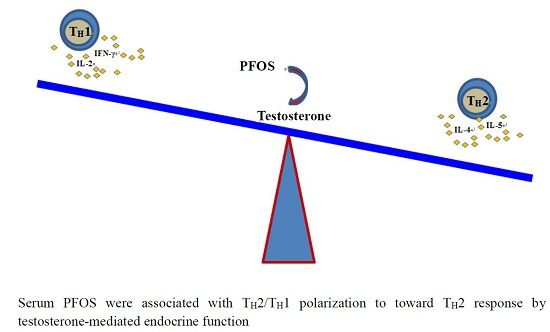
:
1. Introduction
2. Results
2.1. Body and Organ Mass/Serum PFOS Concentrations
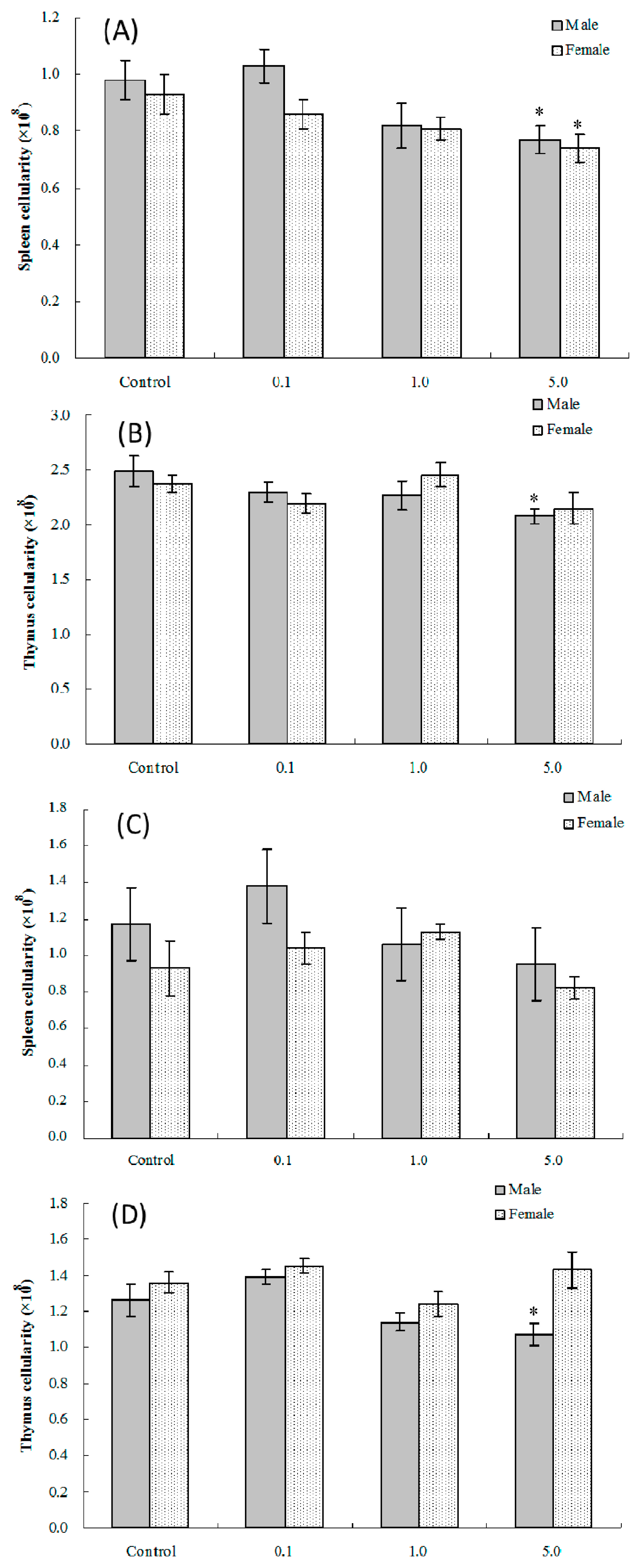
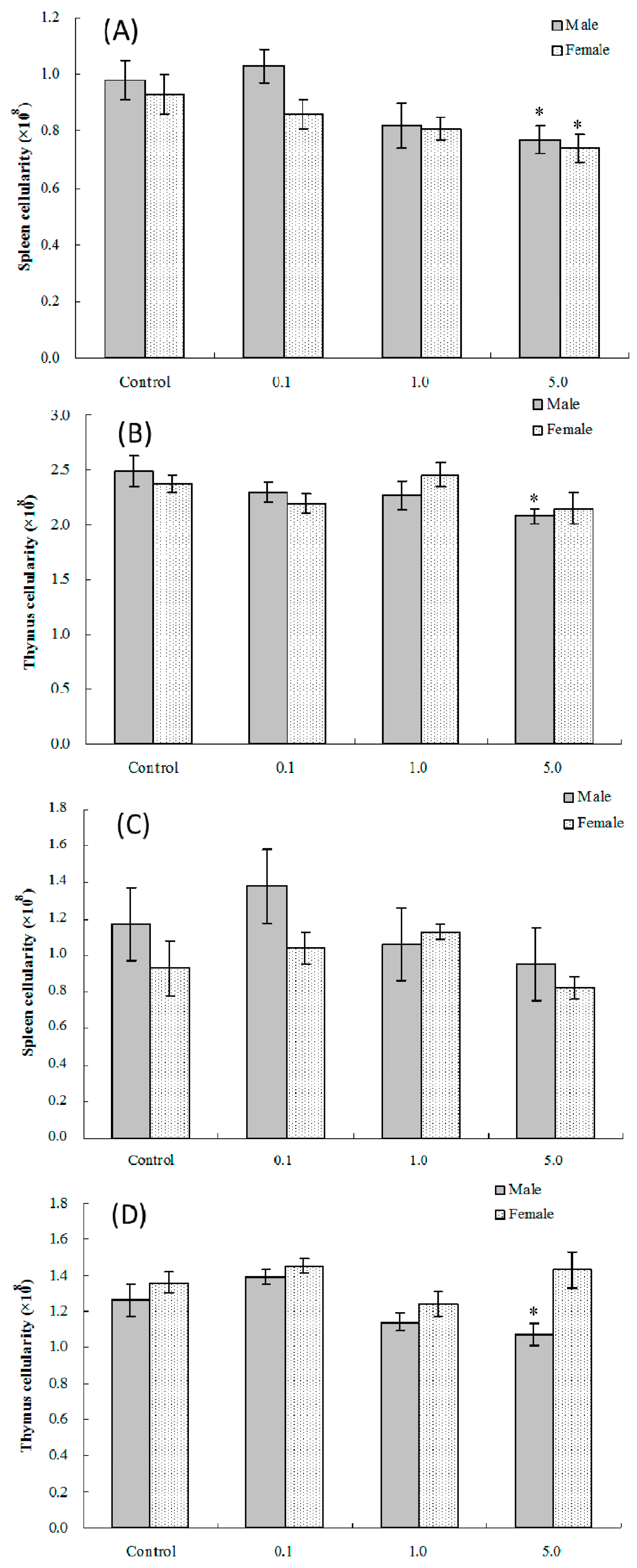
2.2. Splenic and Thymic Cellularity
2.3. Lymphocyte Immunophenotypes
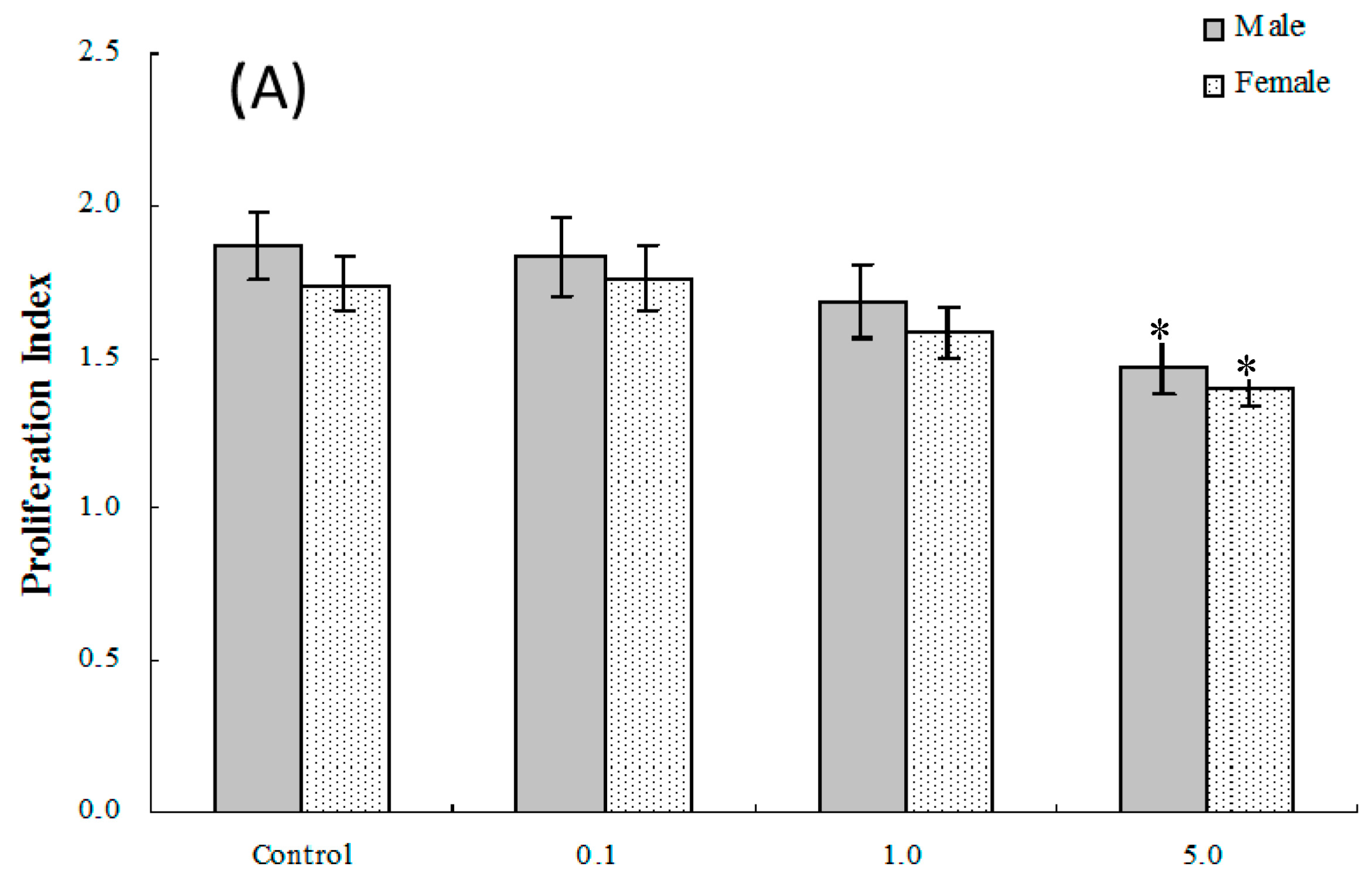
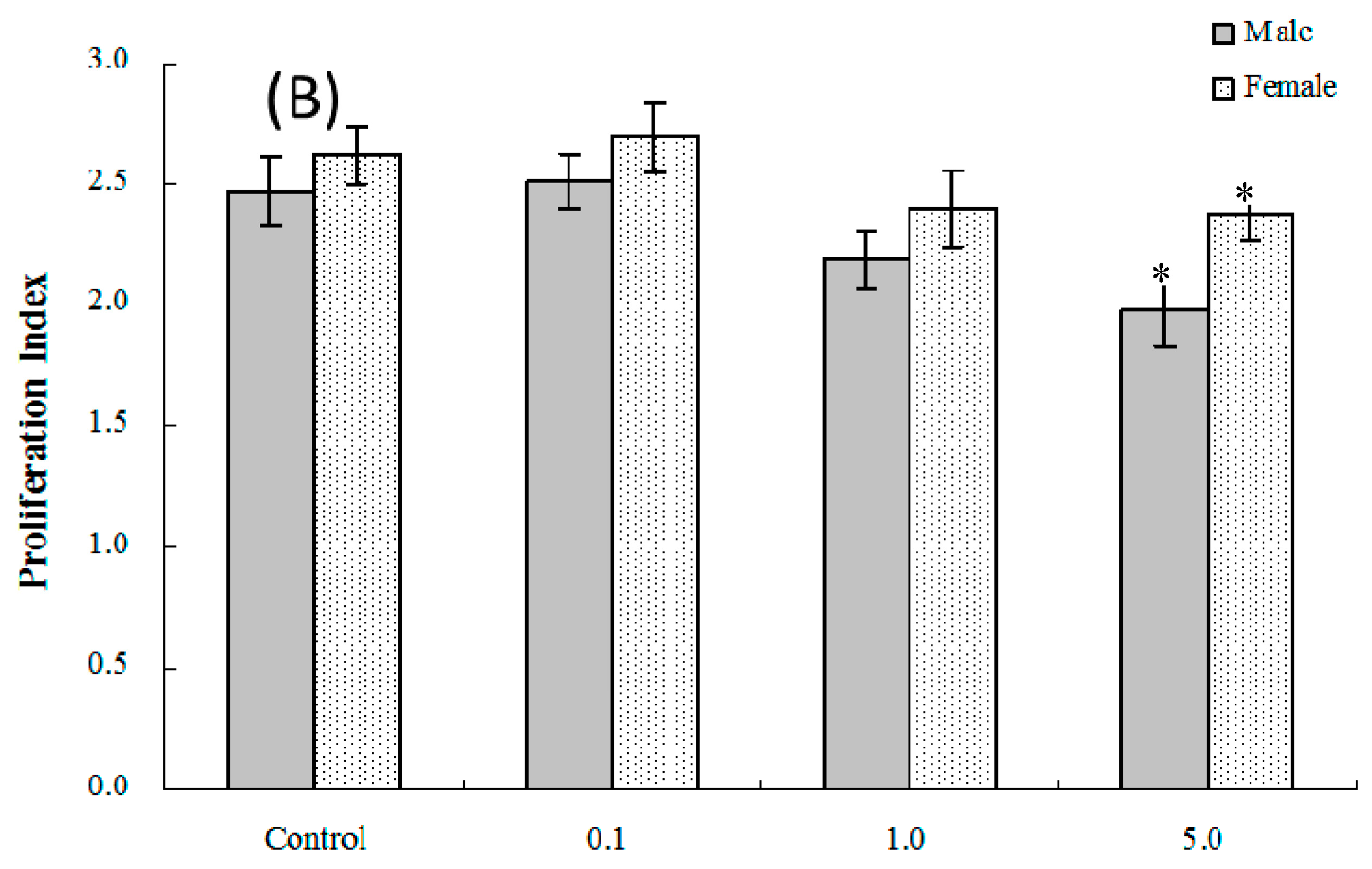
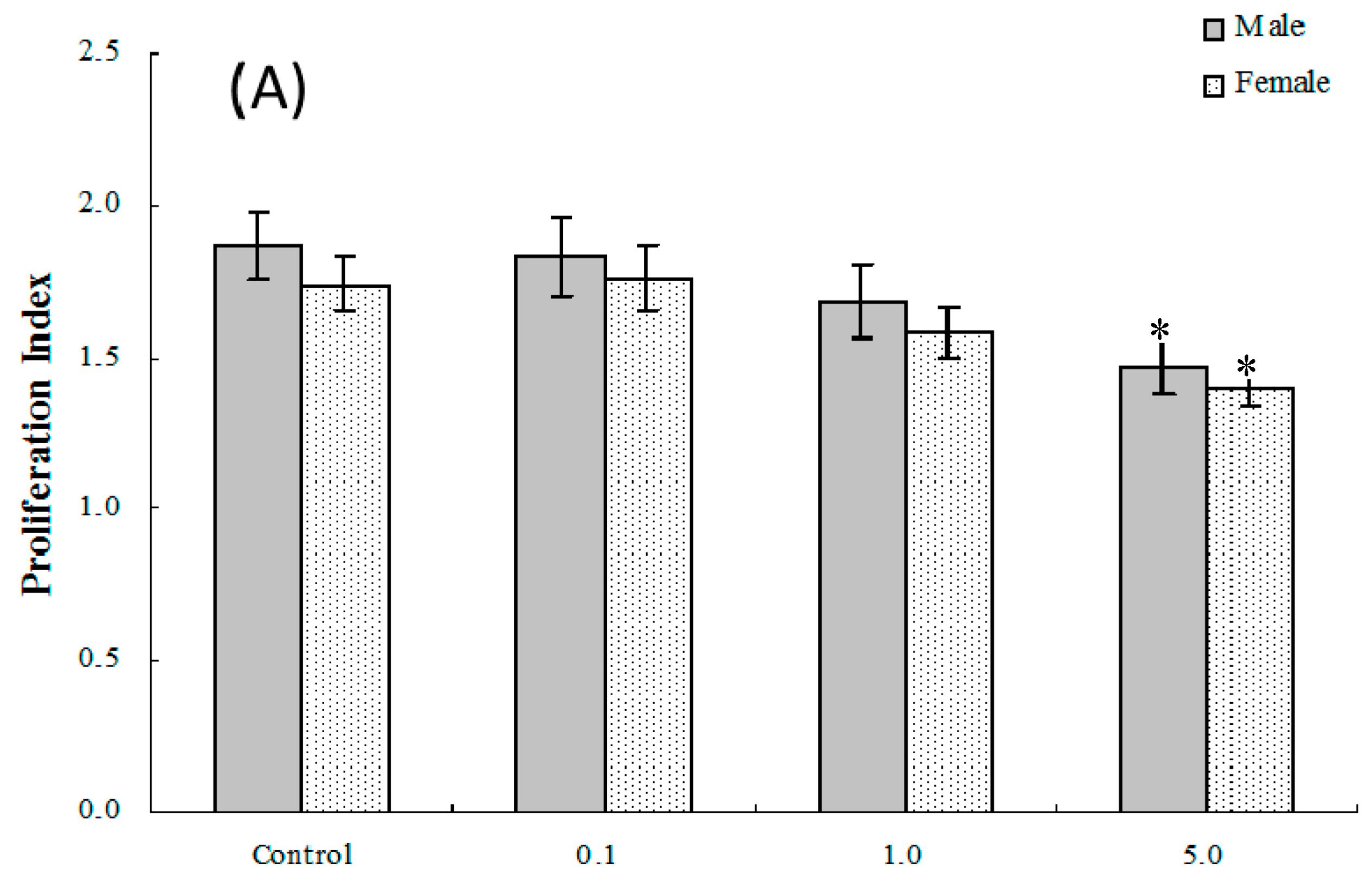
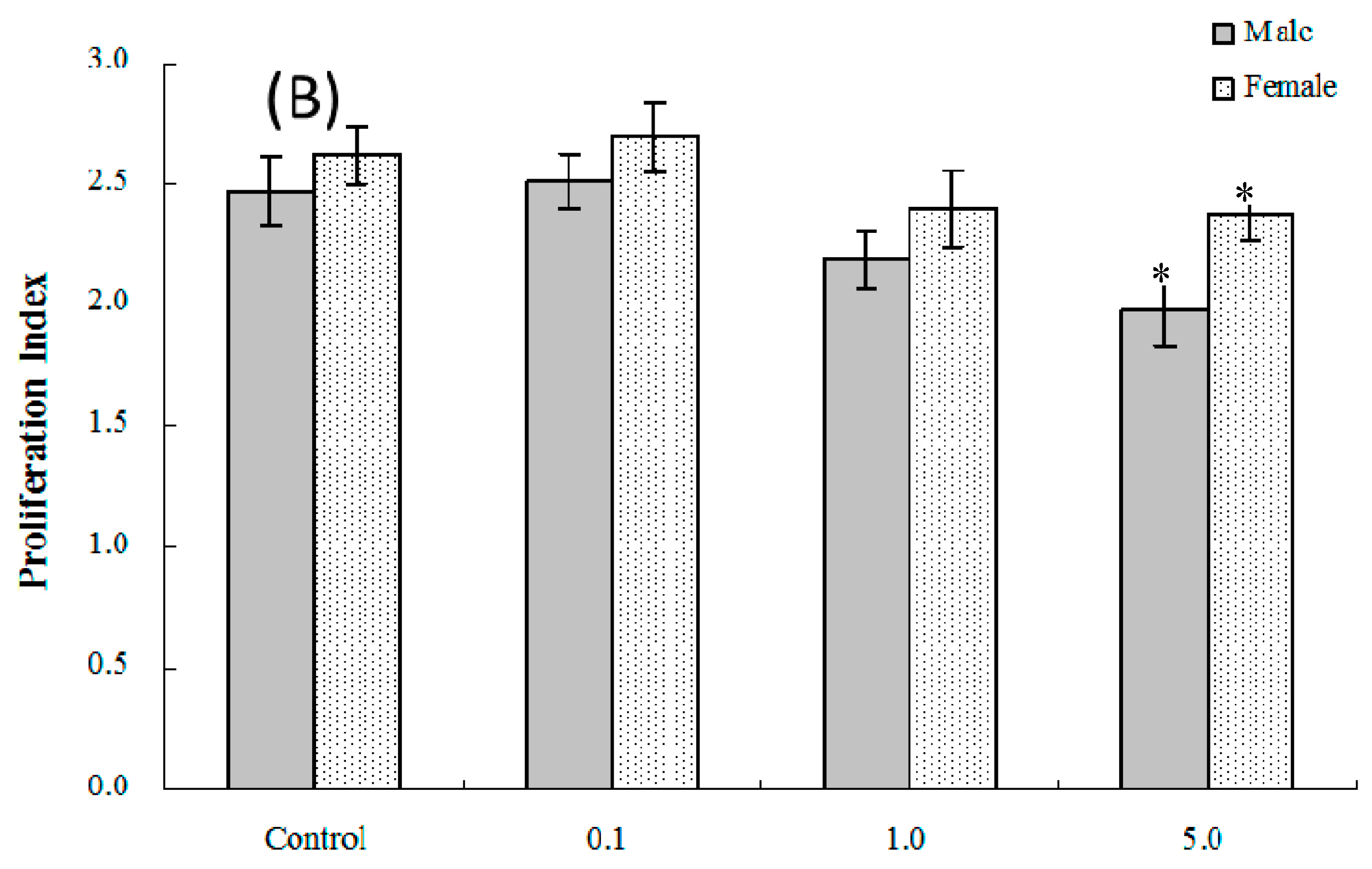
2.4. Lymphocyte Proliferation
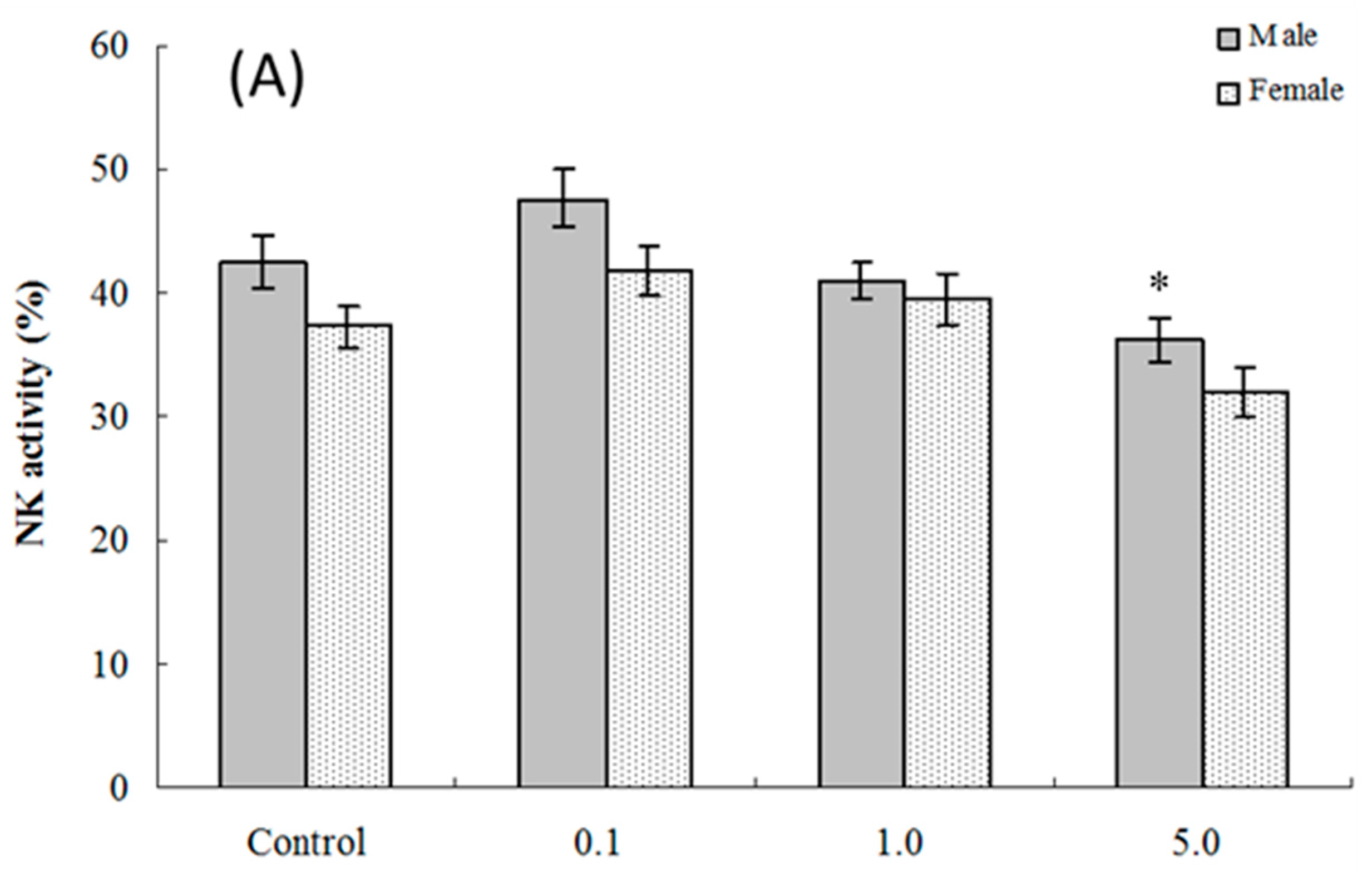
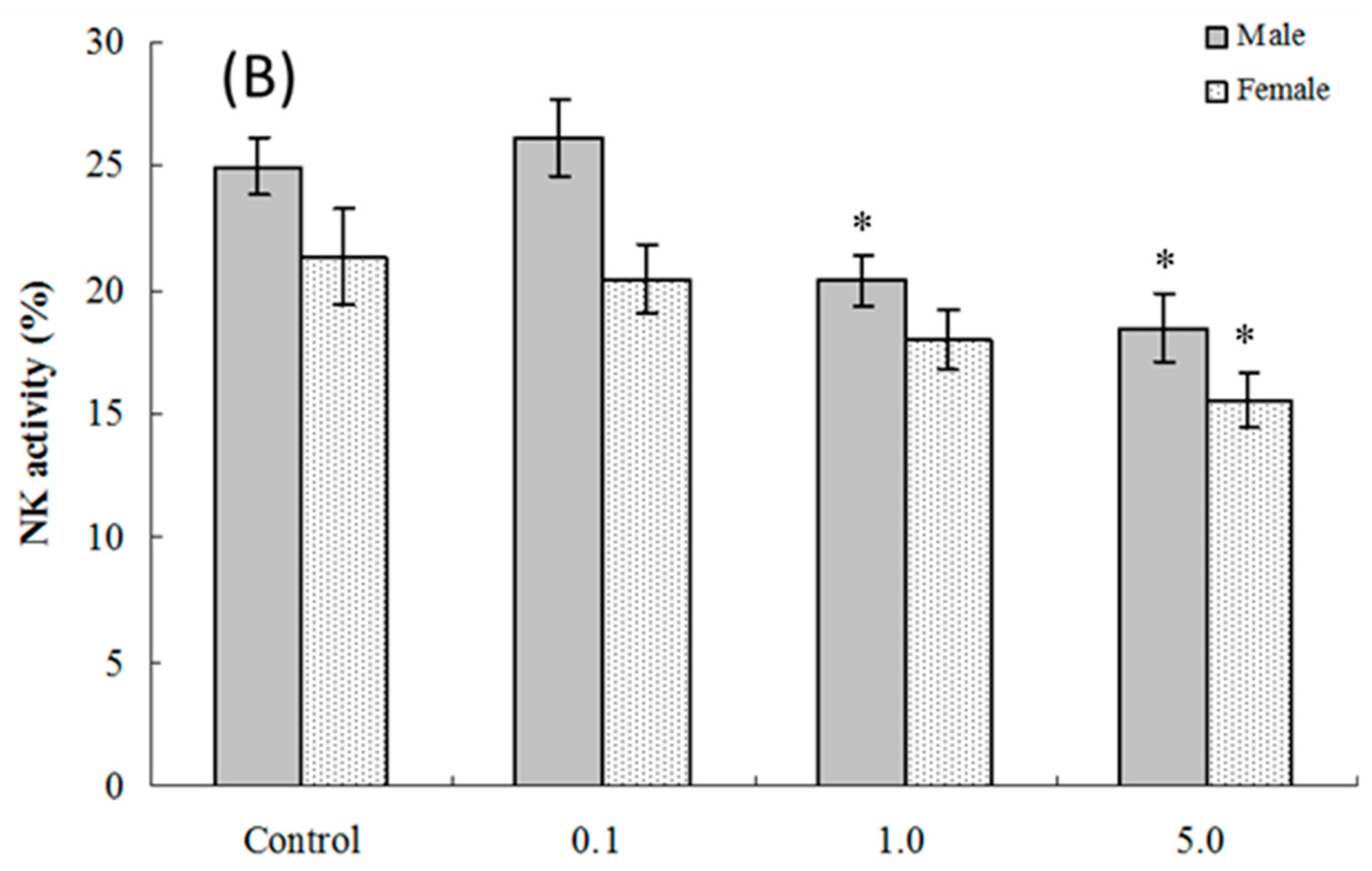
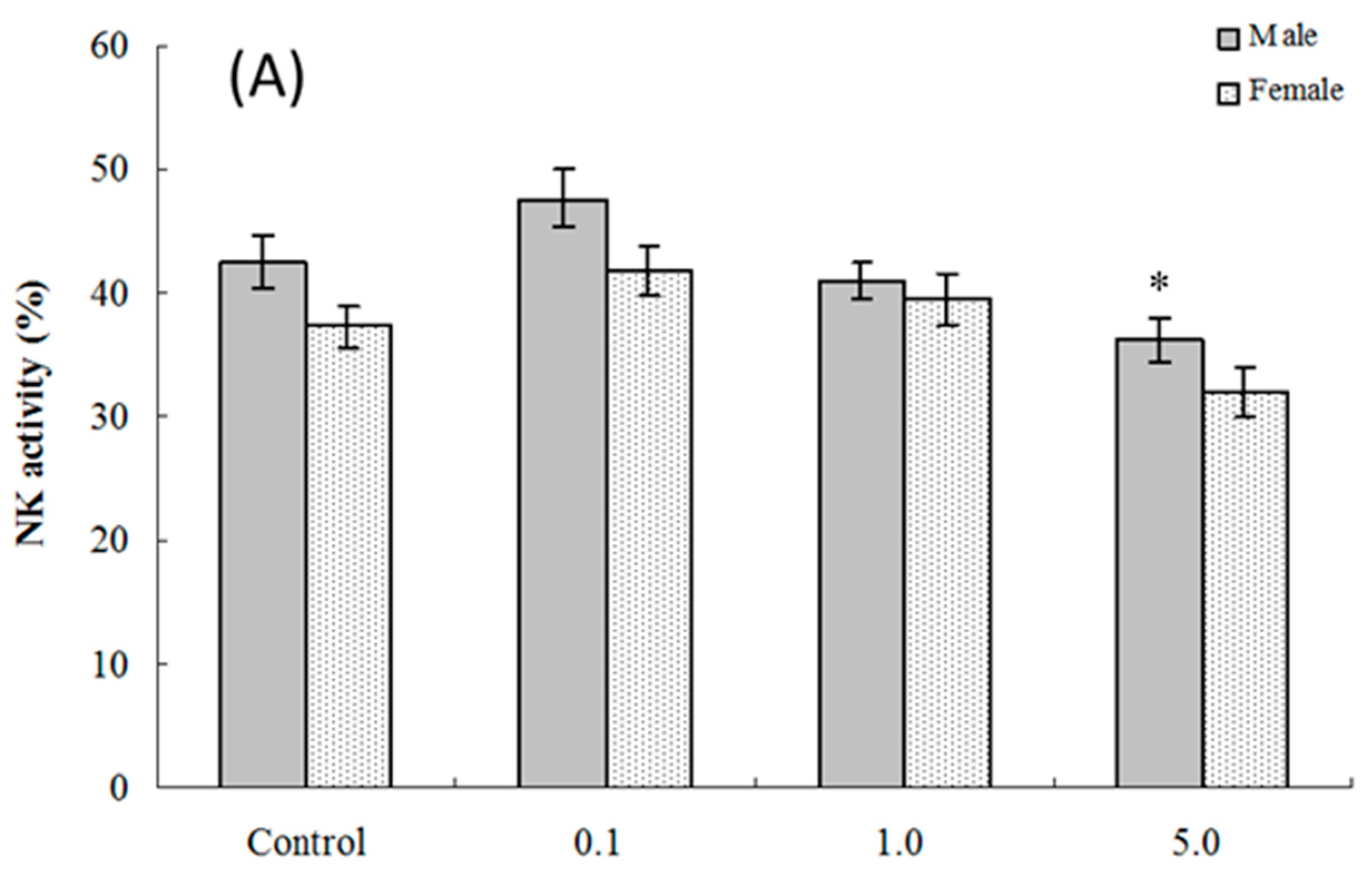
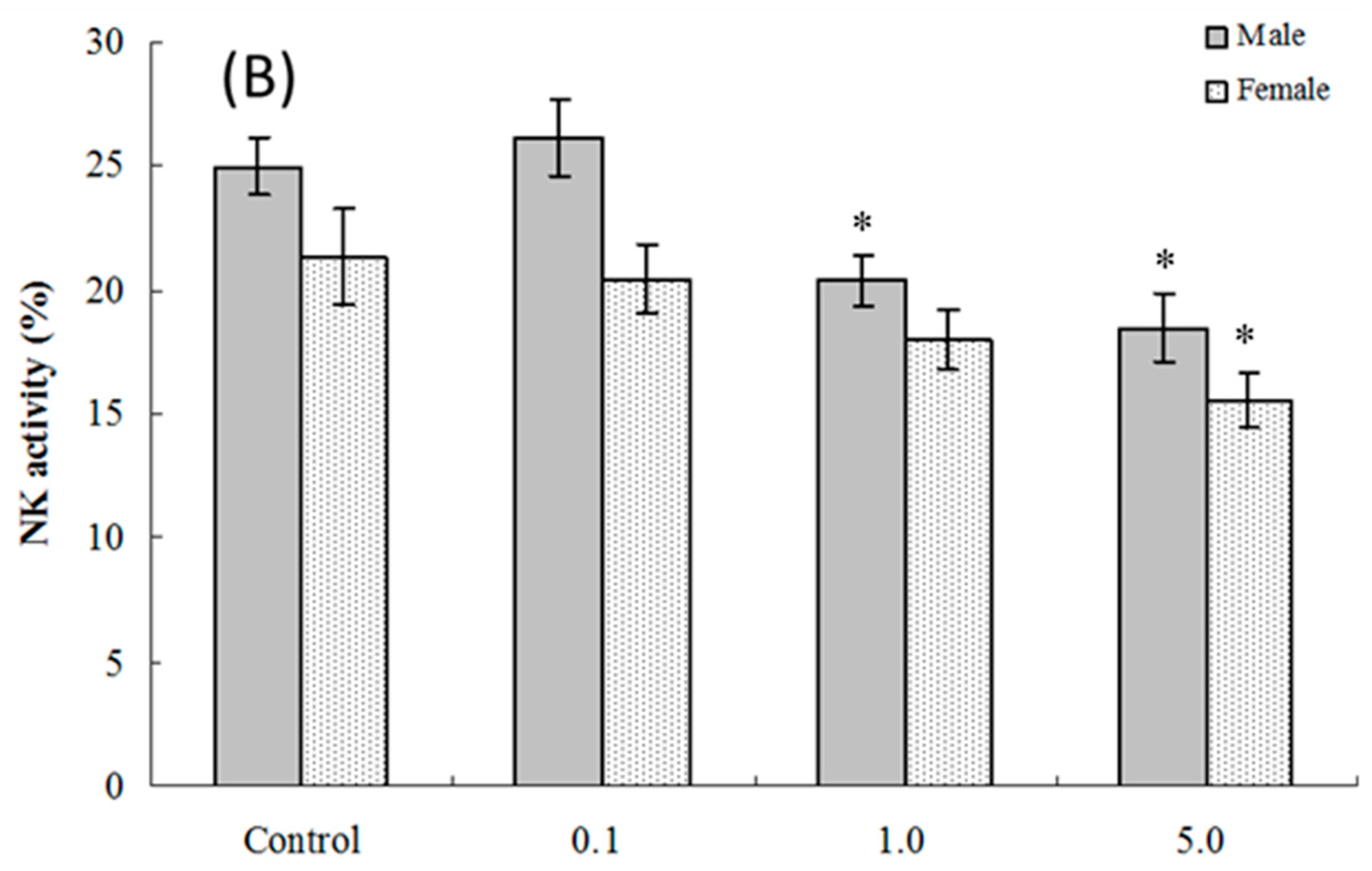
2.5. NK Cell Function
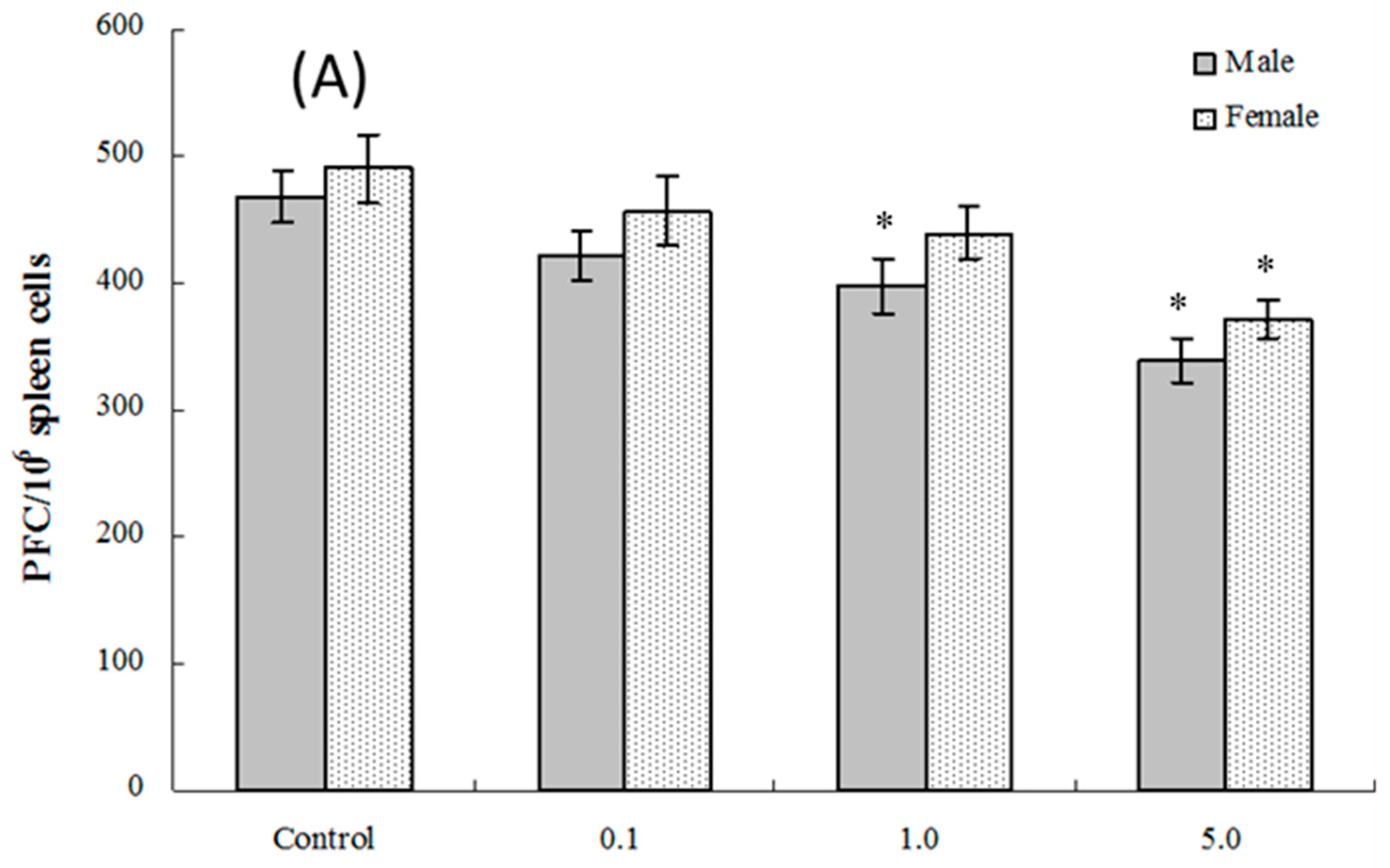
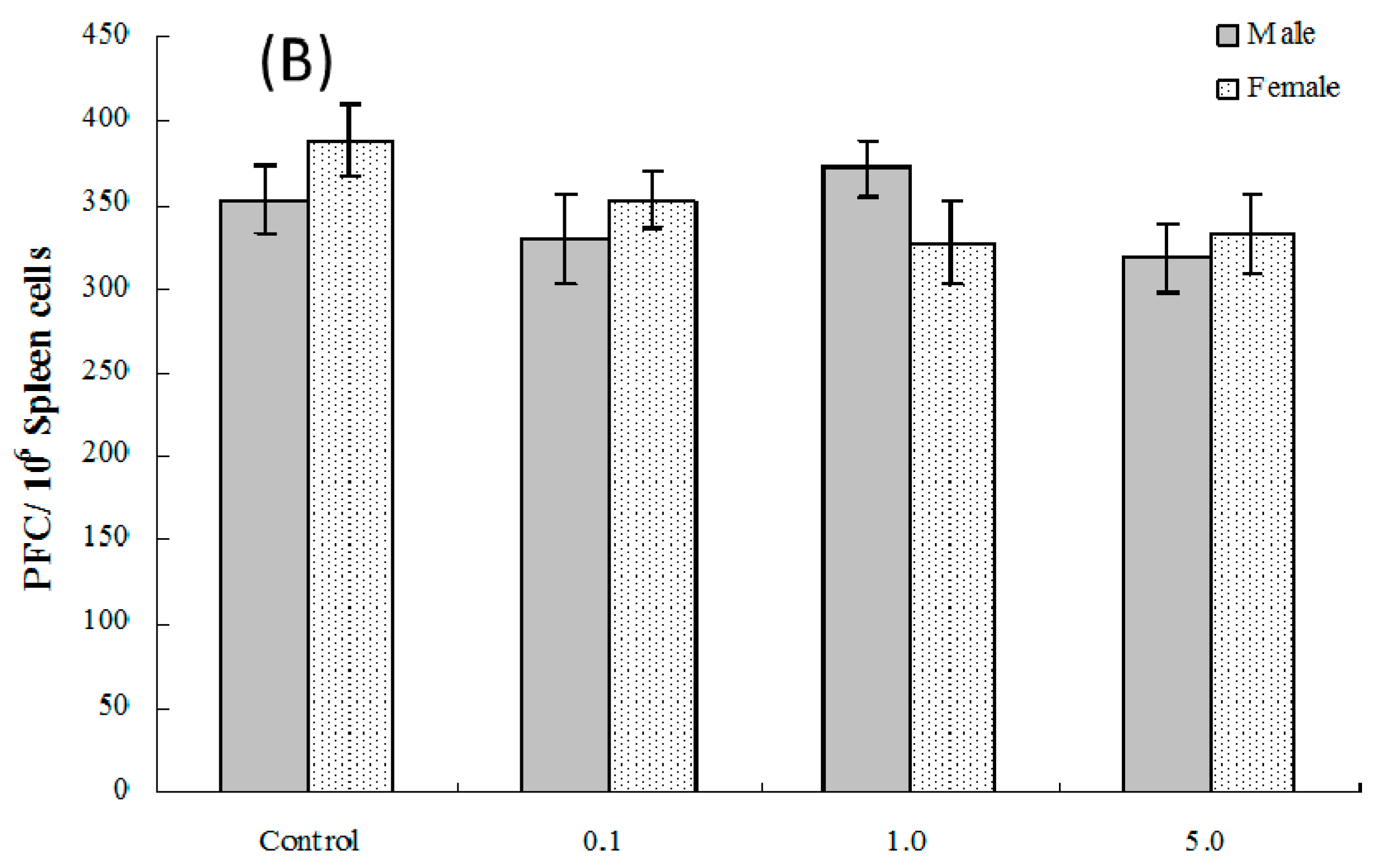
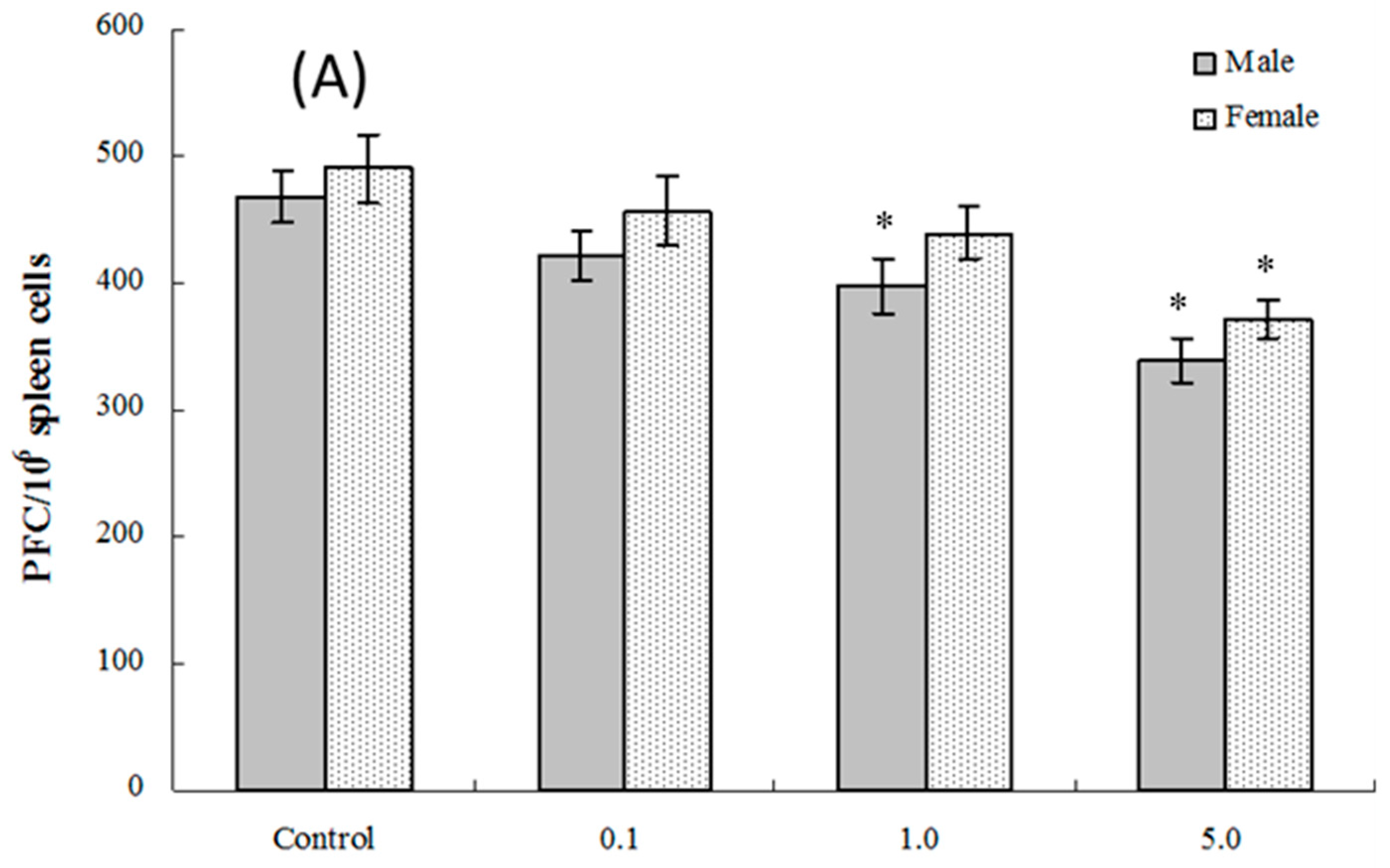
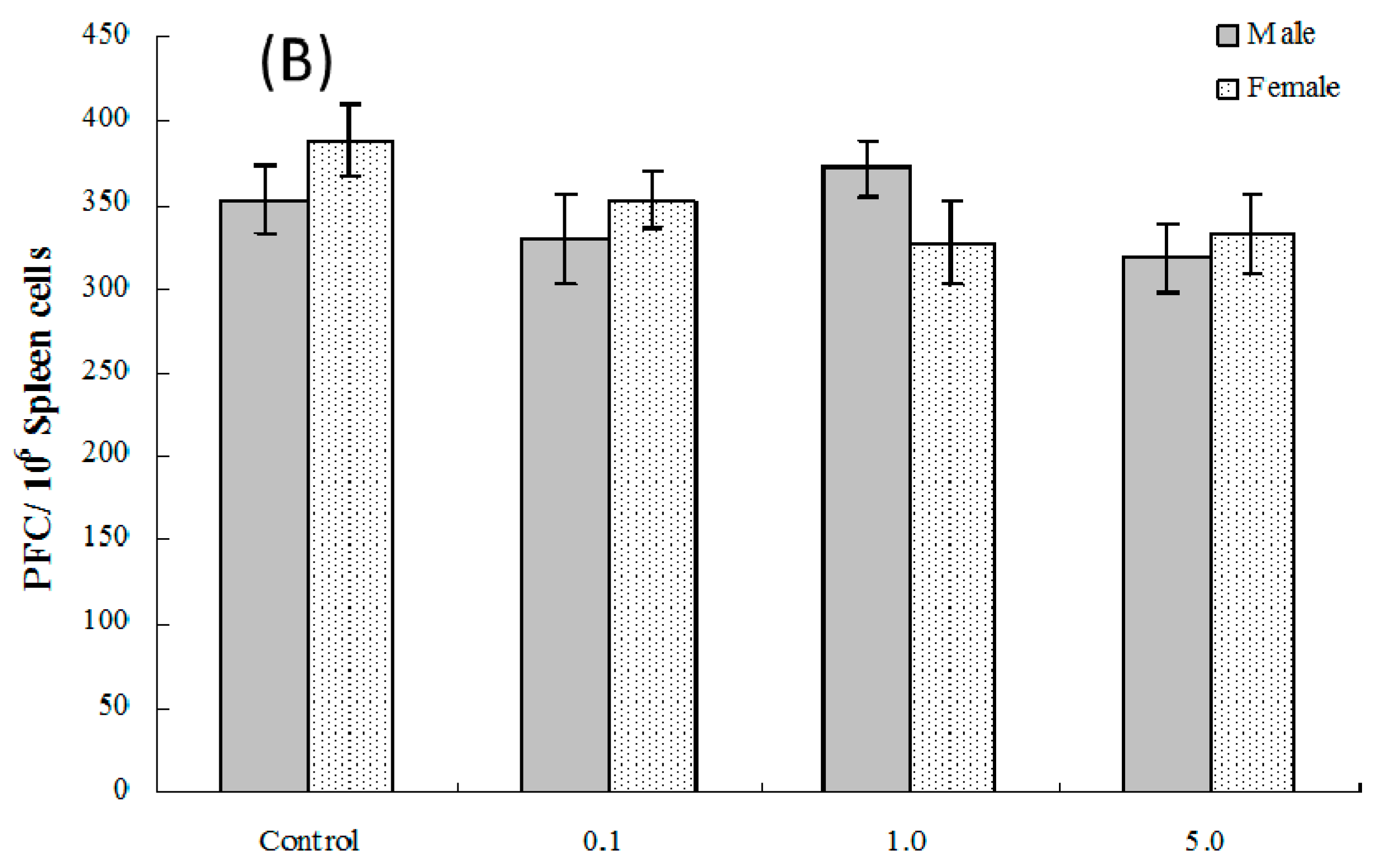
2.6. Splenic Plaque-Forming Cell Levels
2.7. Ex Vivo Splenocyte Spontaneous IL-2, IL-4, IL-10 and IFNγ Production
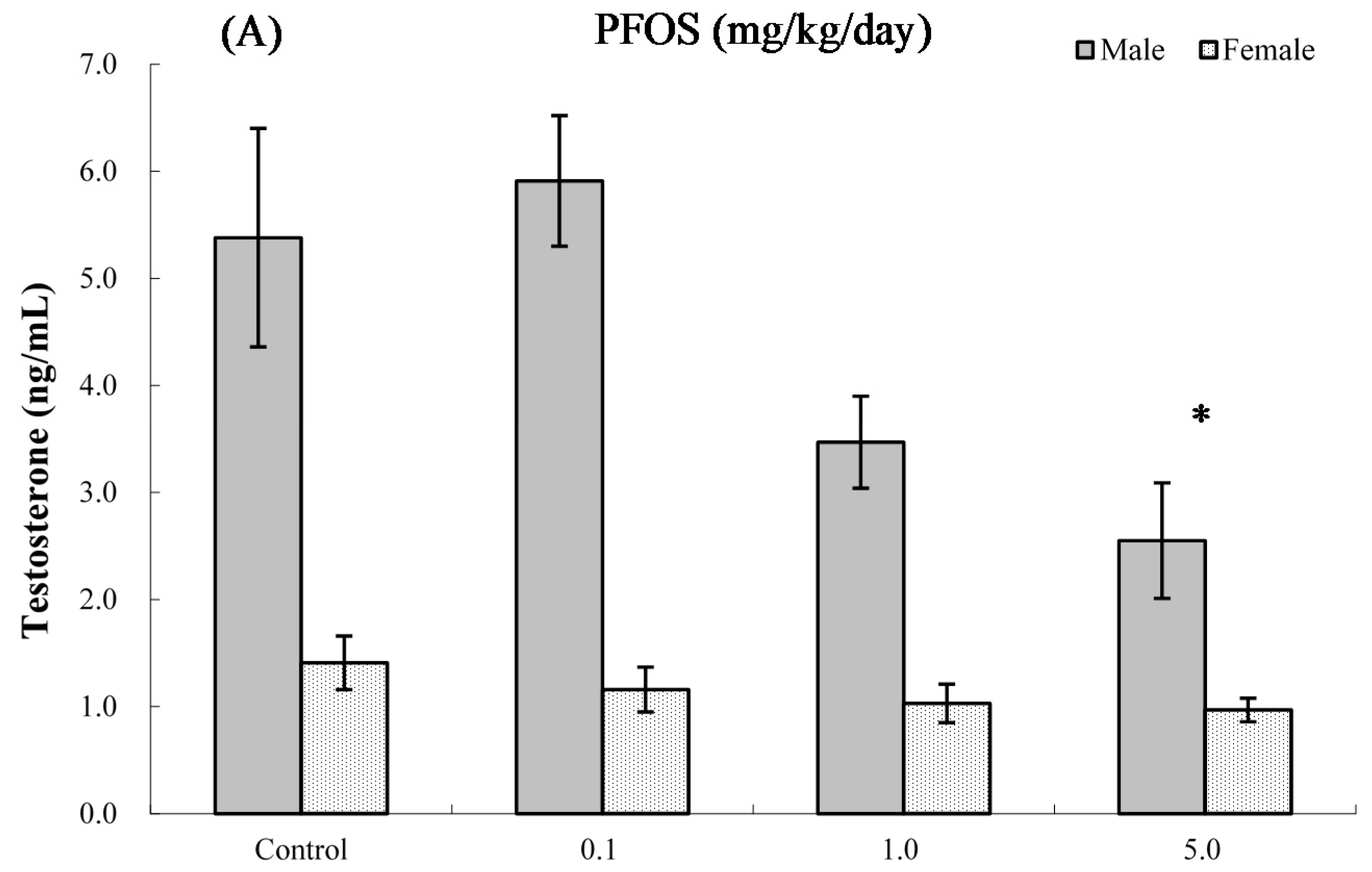
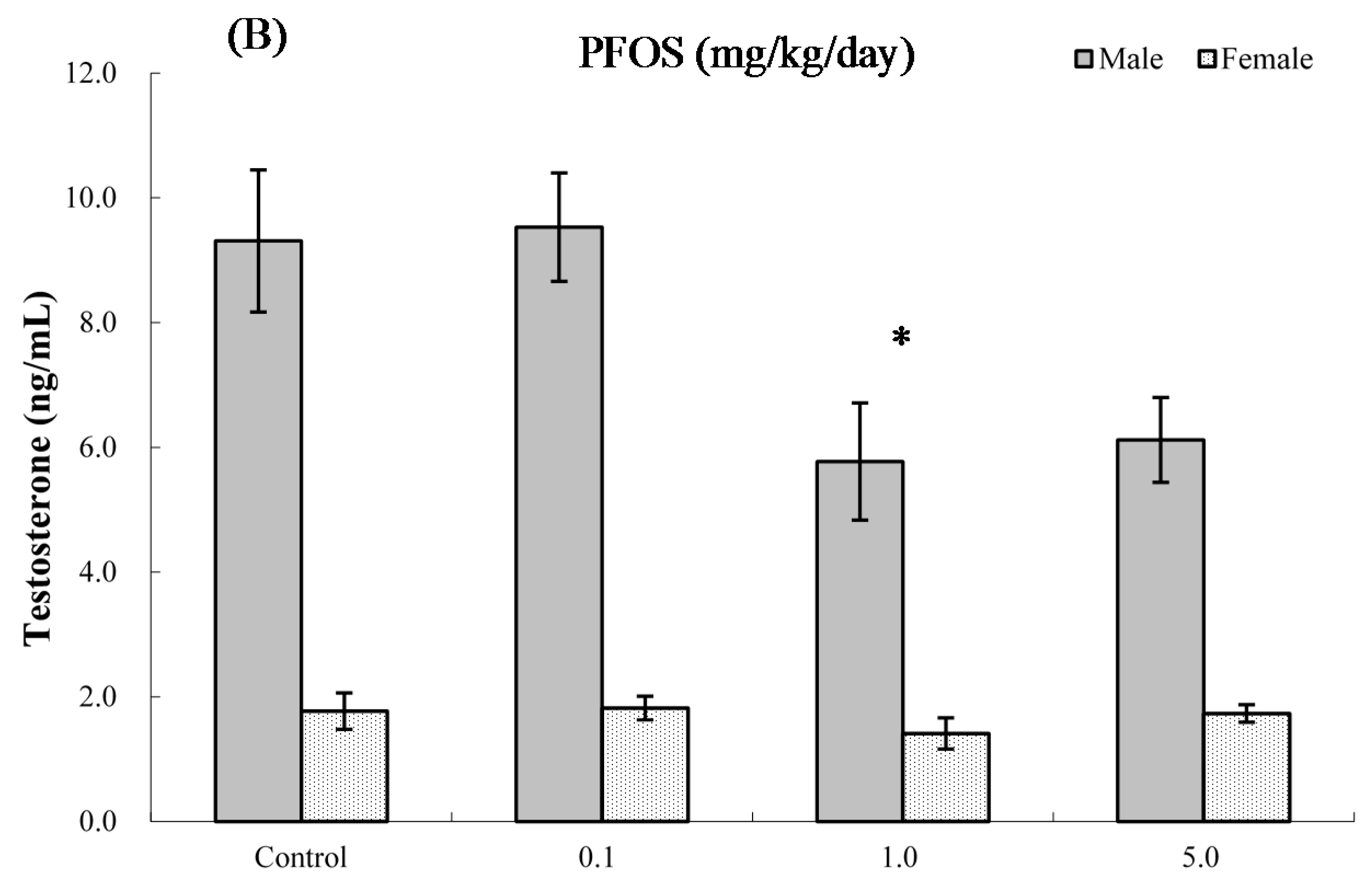
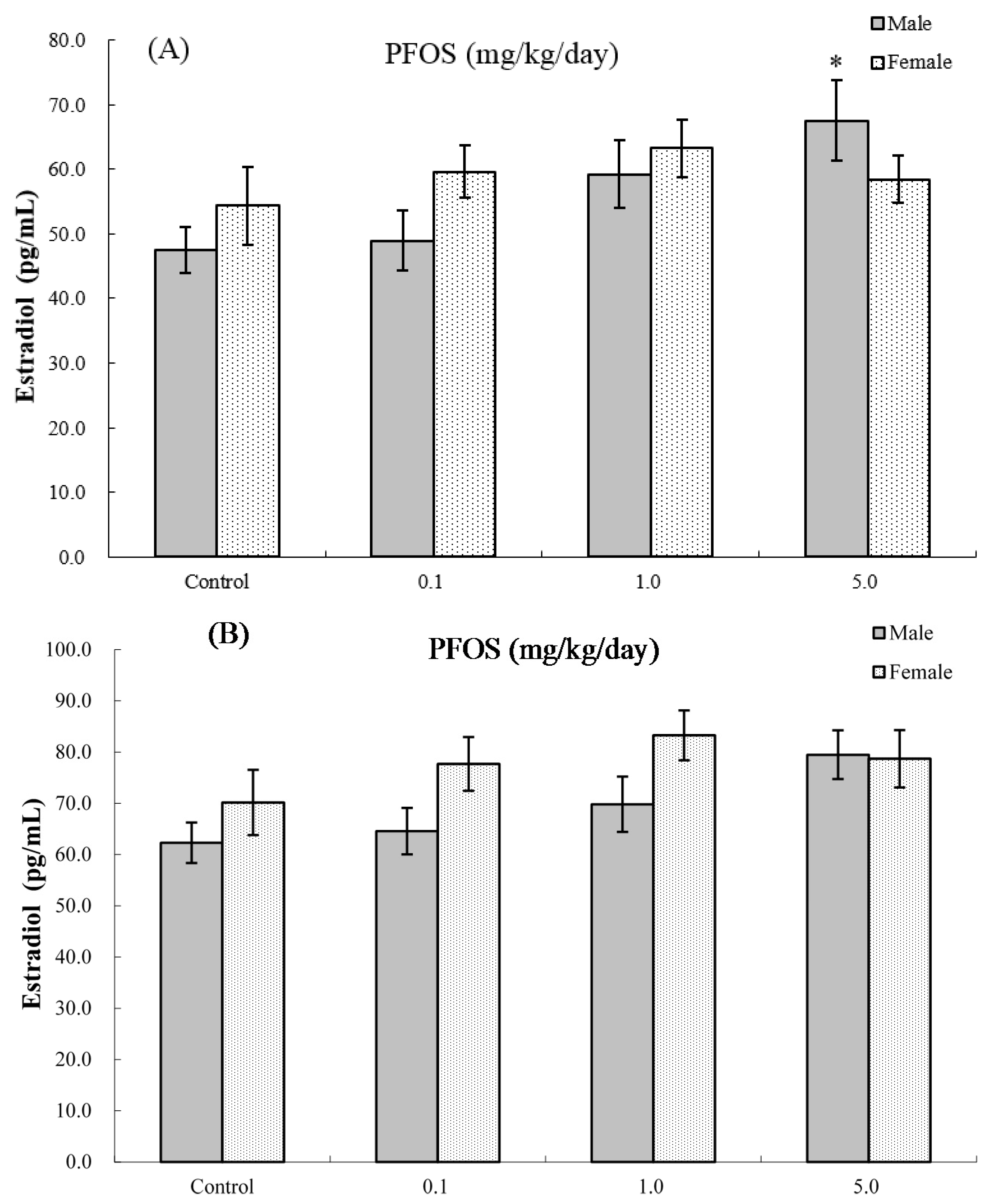
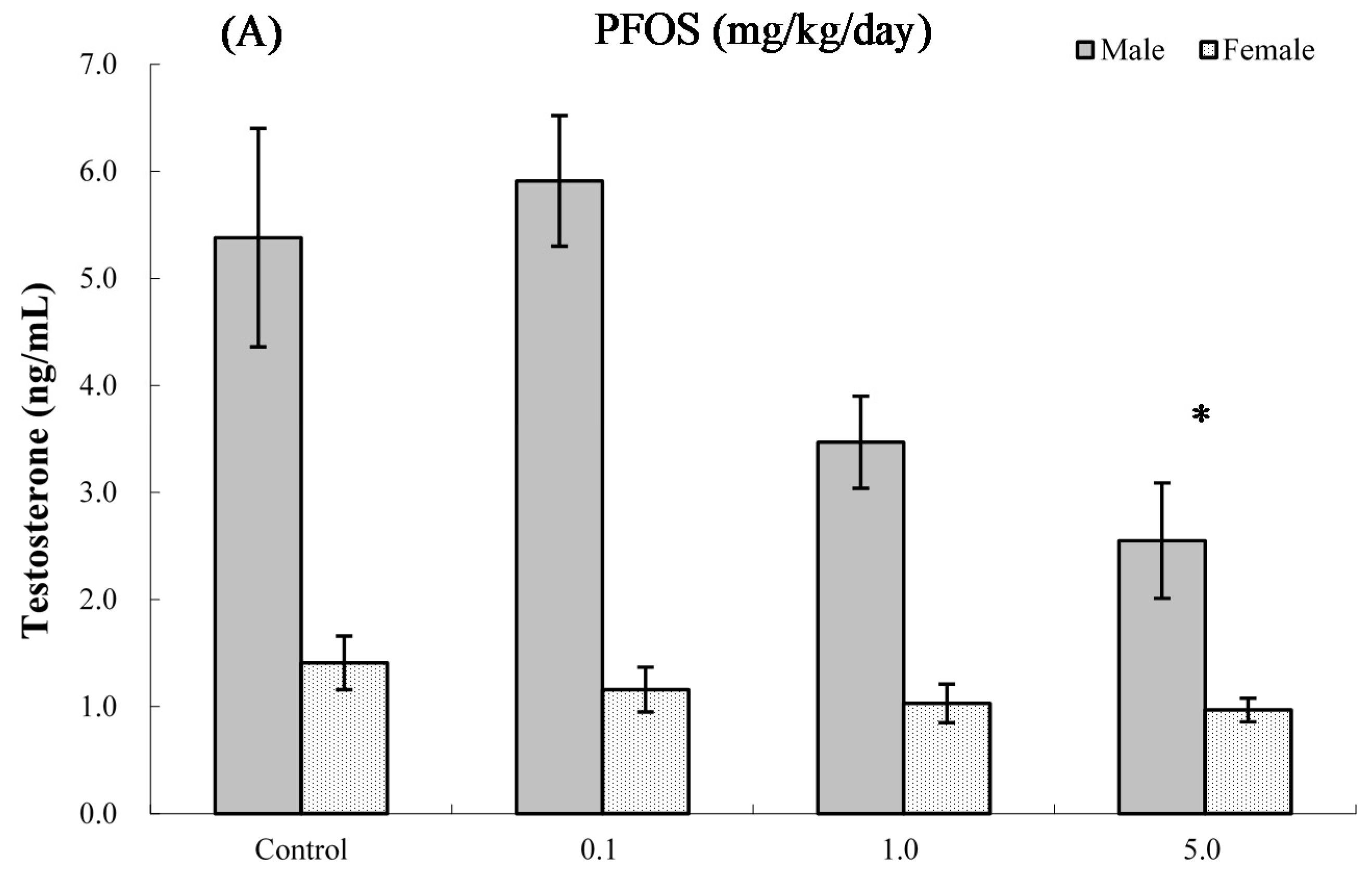
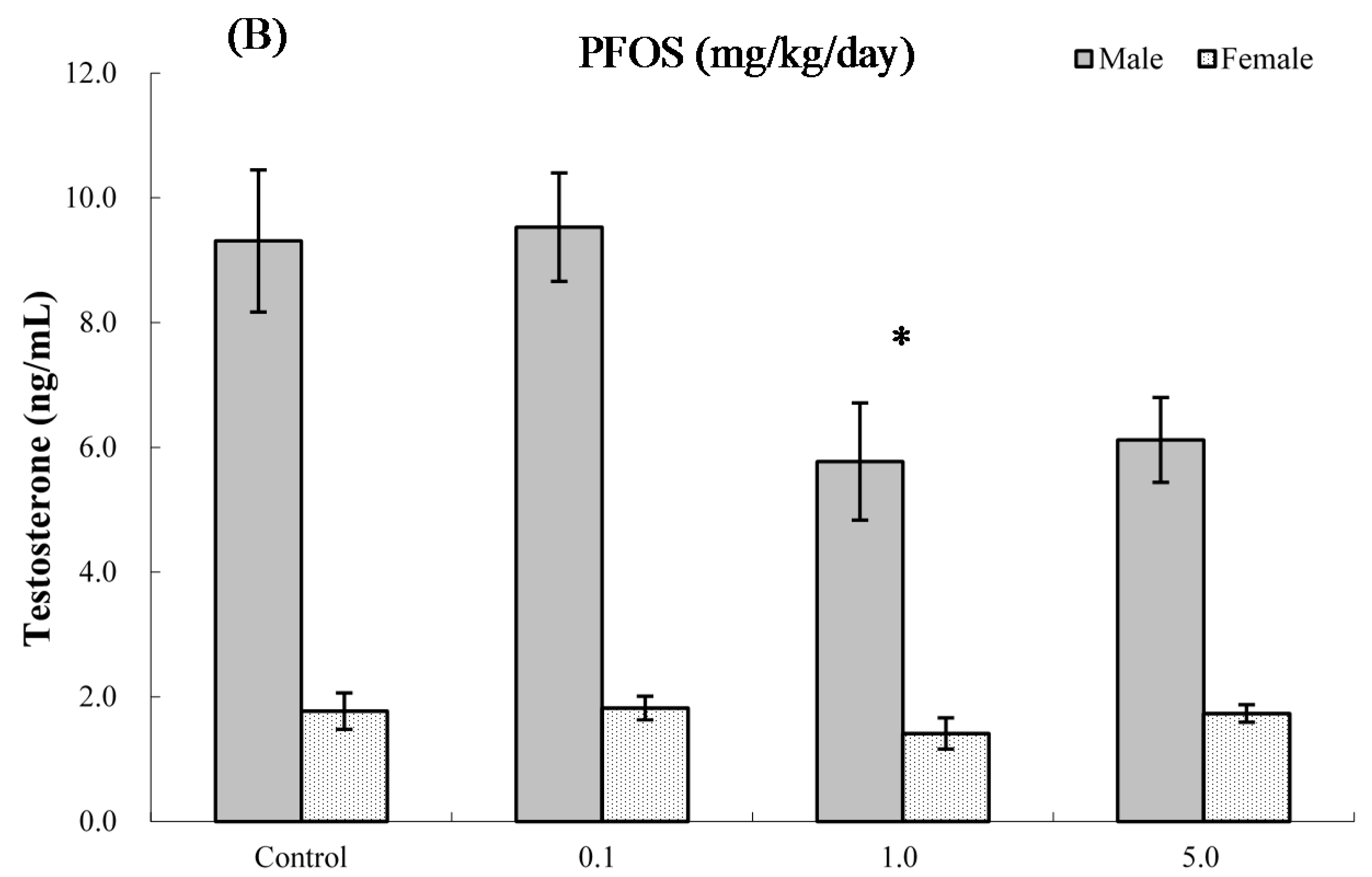
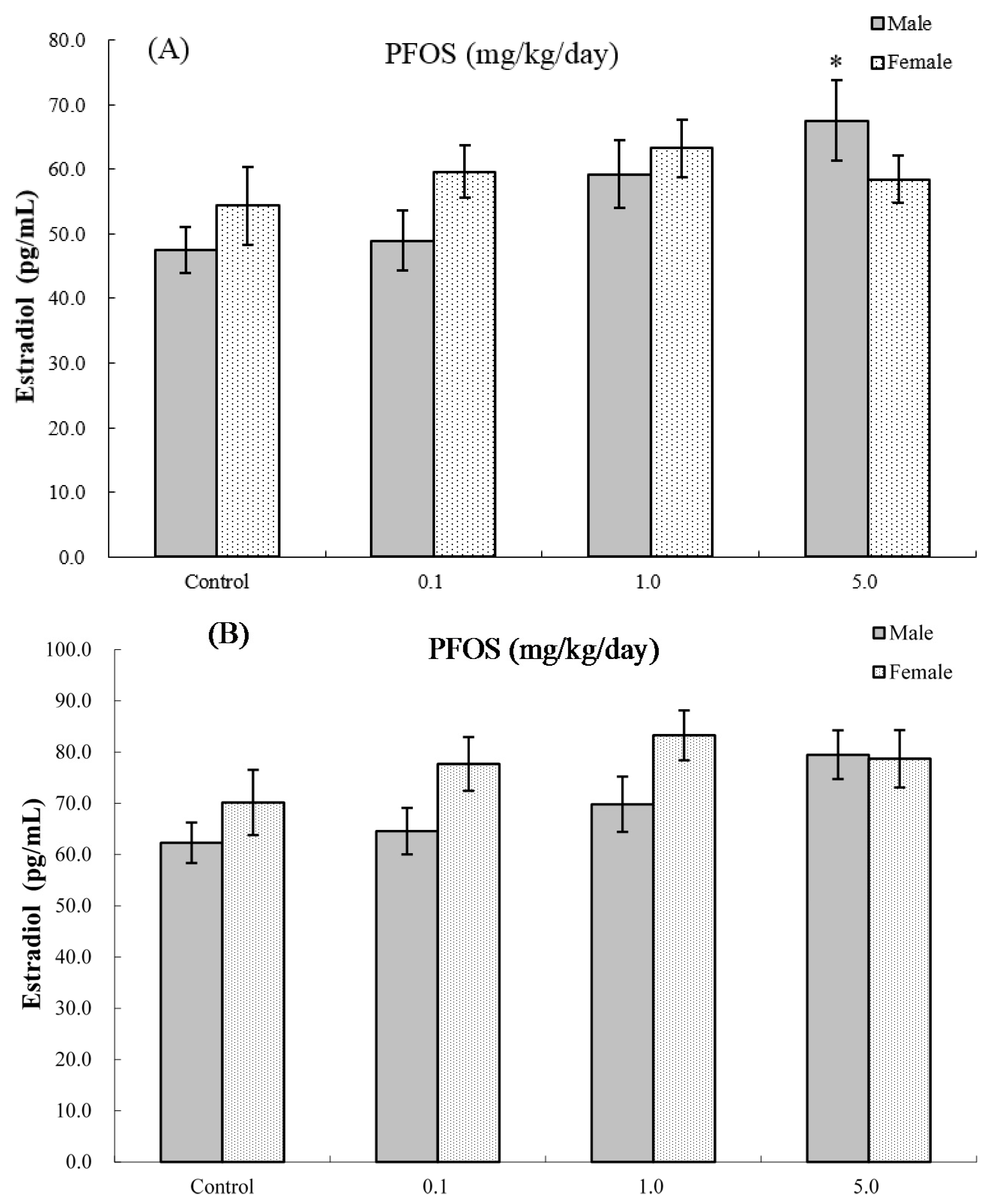
2.8. Serum Hormone Levels
2.9. Interaction between Sex and PFOS Exposure
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Chemicals, Antibodies and Supplies
4.3. Animals and Treatments
4.4. Serum Sampling, Body and Organ Mass and Organ Cellularity
4.5. Serum PFOS Analysis
4.6. Serum Hormone Levels
4.7. Lymphocyte Proliferation Assay
4.8. Splenic and Thymic CD4/CD8 Sub-Population Measurements
4.9. Natural Killer Cell Activity
4.10. Antibody Plaque-Forming Cell Assay
4.11. Measures of Spontaneous ex Vivo IL-2, IL-4, IL-10 and IFNγ
4.12. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| POP | Persistent organic pollutant |
| PFOS | Perfluorooctane sulfonate |
| PFASs | polyfluoroalkyl substances |
| SRBC | Sheep red blood cells |
| GPC | Guinea pig complement |
| LDH | Lactate dehydrogenase |
| PFC | Plaque-forming cells |
| NK | Natural killer |
| TH | T-helper |
| IFNγ | Interferon-γ |
| IL-2 | Interleukin-2 |
| IL-4 | Interleukin-4 |
| IL-10 | Interleukin-10 |
References
- Stockholm Convention on Persistent Organic Pollutants. Governments Unite to Step-Up Reduction on Global DDT Reliance and Add Nine New Chemicals under International Treaty; Convention Secretariat: Geneva, Switzerland; Stockholm, Sweden, 2009. [Google Scholar]
- Löfstedt Gilljam, J.; Leonel, J.; Cousins, I.T.; Benskinm, J.P. Is ongoing sulfluramid use in South America a significant source of perfluorooctanesulfonate (PFOS)? Production inventories, environmental fate, and local occurrence. Environ. Sci. Technol. 2016, 50, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, A.B.; Strynar, M.J.; Libelo, E.L. Polyfluorinated compounds: Past, present, and future. Environ. Sci. Technol. 2011, 45, 7954–7961. [Google Scholar] [CrossRef] [PubMed]
- Taxvig, C.; Rosenmai, A.K.; Vinggaard, A.M. Polyfluorinated alkyl phosphate ester surfactants-current knowledge and knowledge gaps. Basic Clin. Pharmacol. Toxicol. 2014, 115, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.J.; Webster, T.F.; Watkins, D.J.; Strynar, M.J.; Kato, K.; Calafat, A.M.; Vieira, V.M.; McClean, M.D. Polyfluorinated compounds in dust from homes, offices, and vehicles as predictors of concentrations in office workers’ serum. Environ. Int. 2013, 60C, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; Anitole, K.; Hodes, C.; Lai, D.; Pfahles-Hutchens, A.; Seed, J. Perfluoroalkyl acids: A review of monitoring and toxicological findings. Toxicol. Sci. 2007, 99, 366–394. [Google Scholar] [CrossRef] [PubMed]
- Prosser, R.S.; Mahon, K.; Sibley, P.K.; Poirier, D.; Watson-Leung, T. Bioaccumulation of perfluorinated carboxylates and sulfonates and polychlorinated biphenyls in laboratory-cultured Hexagenia spp., Lumbriculus variegatus and Pimephales promelas from field-collected sediments. Sci. Total Environ. 2016, 543, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Su, T.C.; Kuo, C.C.; Hwang, J.J.; Lien, G.W.; Chen, M.F.; Chen, P.C. Serum perfluorinated chemicals, glucose homeostasis and the risk of diabetes in working-aged Taiwanese adults. Environ. Int. 2015, 88, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Dietz, R.; Gustavson, K.; Sonne, C.; Desforges, J.P.; Rigét, F.F.; Pavlova, V.; McKinney, M.A.; Letcher, R.J. Physiologically-based pharmacokinetic modelling of immune, reproductive and carcinogenic effects from contaminant exposure in polar bears (Ursus maritimus) across the Arctic. Environ. Res. 2015, 140, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Barber, J.L.; Berger, U.; Chaemfa, C.; Huber, S.; Jahnke, A.; Temme, C.; Jones, K.C. Analysis of per- and polyfluorinated alkyl substances in air samples from Northwest Europe. J. Environ. Monit. 2007, 9, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Fromme, H.; Tittlemier, S.A.; Völkel, W.; Wilhelm, M.; Twardella, D. Perfluorinated compounds—Exposure assessment for general population in Western countries. Int. J. Hyg. Environ. Health 2009, 212, 239–270. [Google Scholar] [CrossRef] [PubMed]
- Burris, J.M.; Lundberg, J.K.; Olsen, G.W.; Simpson, C.; Mandel, J.H. Determination of Serum Half-Lives of Several Fluorochemicals. Interim Report No. 2. St. Paul, MN: 3M Company. U.S. EPA Docket AR-226-1086; US Environmental Protection Agency: Washington, DC, USA, 2002. [Google Scholar]
- Apelberg, B.J.; Witter, F.R.; Herbstman, J.B.; Calafat, A.M.; Halden, R.U.; Needham, L.L.; Goldman, L.R. Cord serum concentrations of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in relation to weight and size at birth. Environ. Health Perspect. 2007, 115, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.H.; Tung, K.Y.; Tsai, C.H.; Liu, M.M.; Wang, D.; Liu, W.; Jin, Y.H.; Hsieh, W.S.; Lee, Y.L.; Chen, P.C. Serum polyfluoroalkyl concentrations, asthma outcomes, and immunological markers in a case-control study of Taiwanese children. Environ. Health Perspect. 2013, 121, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Wong, L.Y.; Jia, L.T.; Kuklenyik, Z.; Calafat, A.M. Trends in exposure to polyfuoroalkyl chemicals in US population: 1999–2008. Environ. Sci. Technol. 2011, 45, 8037–8045. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.M.; Bennett, D.H.; Calafat, A.M.; Kato, K.; Strynar, M.; Andersen, E.; Moran, R.E.; Tancredi, D.J.; Tulve, N.S.; Hertz-Picciotto, I. Serum concentrations of perfluorinated compounds (PFC) among selected populations of children and adults in California. Environ. Res. 2015, 136, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.W.; Qian, Z.; Emo, B.; Vaughn, M.; Bao, J.; Qin, X.D.; Zhu, Y.; Li, J.; Lee, Y.L.; Dong, G.H. Association of polyfluoroalkyl chemical exposure with serum lipids in children. Sci. Total Environ. 2015, 512, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Lopez-Espinosa, M.J.; Armstong, B.; Stein, C.R.; Fletcher, T. Relationships of perfluorooctanoate and perfluorooctane sulfonate serum concentrations between mother-child pairs in a population with perfluorooctanoate exposure from drinking water. Environ. Health Perspect. 2012, 120, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Weldon, R.H.; Armstrong, B.G.; Gibson, L.J.; Lopez-Espinosa, M.J.; Shin, H.M.; Fletcher, T. Breastfeeding: A potential excretion route for mothers and implications for infant exposure to perfluoroalkyl acids. Environ. Health Perspect. 2014, 122, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Okada, F.; Ito, R.; Kato, S.; Sasaki, S.; Nakajima, S.; Uno, A.; Saijo, Y.; Sata, F.; Yoshimura, Y.; Kishi, R.; Nakazawa, H. Perfluorooctane sulfonate (PFOS) and related perfluorinated compounds in human maternal and cord blood samples: Assessment of PFOS exposure in a susceptible population during pregnancy. Environ. Health Perspect. 2004, 112, 1204–1207. [Google Scholar] [CrossRef] [PubMed]
- Beesoon, S.; Webster, G.M.; Shoeib, M.; Harner, T.; Benskin, J.P.; Martin, J.W. Isomer profiles of perfluorochemicals in matched maternal, cord, and house dust samples: Manufacturing sources and transplacental transfer. Environ. Health Perspect. 2011, 119, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Ode, A.; Rylander, L.; Lindh, C.H.; Kallen, K.; Jonsson, B.A.; Gustafsson, P.; Olofsson, P.; Ivarsson, S.A.; Righell-Hydbom, A. Determinants of maternal and fetal exposure and temporal trends of perfluorinated compounds. Environ. Sci. Pollut. Res. Int. 2013, 20, 7970–7978. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Qin, X. Assessment of fetal exposure and maternal elimination of perfluoroalkyl substances. Environ. Sci. Process. Impacts 2014, 16, 1878–1881. [Google Scholar] [CrossRef] [PubMed]
- Peden-Adams, M.M.; Keller, J.M.; EuDaly, J.G.; Berger, J.; Gilkeson, G.S.; Keil, D.E. Suppression of humoral immunity in mice following exposure to perfluorooctane sulfonate. Toxicol. Sci. 2008, 104, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.H.; Zhang, Y.H.; Zheng, L.; Liu, W.; Jin, Y.H.; He, Q.C. Chronic effects of perfluorooctanesulfonate exposure on immunotoxicity in adult male C57BL/6 mice. Arch. Toxicol. 2009, 83, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.H.; Wang, J.; Zhang, Y.H.; Liu, M.M.; Wang, D.; Zheng, L.; Jin, Y.H. Induction of p53-mediated apoptosis in splenocytes and thymocytes of C57BL/6 mice exposed to perfluorooctane sulfonate (PFOS). Toxicol. Appl. Pharmacol. 2012, 264, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.R.; Abedi, M.R.; Nelson, B.D.; DePierre, J.W.; Abedi-Valugerdi, M. Dietary exposure to perfluorooctanoate or perfluorooctane sulfonate induces hypertrophy in centrilobular hepatocytes and alters the hepatic immune status in mice. Int. Immunopharmacol. 2010, 10, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.T.; Adami, H.O.; Boffetta, P.; Wedner, H.J.; Mandel, J.S. A critical review of perfluorooctanoate and perfluorooctanesulfonate exposure and immunological health conditions in humans. Crit. Rev. Toxicol. 2016, 46, 279–331. [Google Scholar] [CrossRef] [PubMed]
- Peden-Adams, M.M.; Stuckey, J.; EuDaly, J.; Keil, D.E. Oral exposure to perfluorooctane sulfonate (PFOS) for 28 days suppresses immunological function in B6C3F1 mice. Toxicol. Sci. 2006, 90, A256. [Google Scholar]
- Wang, I.J.; Hsieh, W.S.; Chen, C.Y.; Fletcher, T.; Lien, G.W.; Chiang, H.L.; Wu, T.N.; Chen, P.C. The effect of prenatal perfluorinated chemicals exposures on pediatric atopy. Environ. Res. 2011, 111, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Keil, D.E.; Mehlmann, T.; Butterworth, L.; Peden-Adams, M.M. Gestational exposure to perfluorooctane sulfonate suppresses immune function in B6C3F1 mice. Toxicol. Sci. 2008, 103, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Katakura, M.; Sato, Y.; Kawashima, Y. Sex hormone-regulated renal transport of perfluorooctanoic acid. Chem. Biol. Interact 2002, 139, 301–316. [Google Scholar] [CrossRef]
- Bao, J.; Lee, Y.L.; Chen, P.C.; Jin, Y.H.; Dong, G.H. Perfluoroalkyl acids in blood serum samples from children in Taiwan. Environ. Sci. Pollut. Res. Int. 2014, 21, 7650–7655. [Google Scholar] [CrossRef] [PubMed]
- Maras, M.; Vanparys, C.; Muylle, F.; Robbens, J.; Berger, U.; Barber, J.L.; Blus, R.; de Coen, W. Estrogen-like properties of fluoro-telomer alcohols as revealed by MCF-7 breast cancer cell proliferation. Environ. Health Perspect. 2006, 114, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, H.; Ishida, H.; Matsuoka, M.; Tominaga, N.; Arizono, K. Estrogenic effects of fluorotelomer alcohols for human estrogen receptor isoforms and in vitro. Environ. Toxicol. Chem. 2013, 32, 353–360. [Google Scholar] [CrossRef]
- Fort, D.; Rodgers, R.L.; Guiney, P.D.; Weeks, J.A. Effects of perfluorooctanesulfonate (PFOS) exposure on steroidogenesis in juvenile Xenopus (Silurana) tropicalis. In Proceedings of the 28th Annual Meeting on Society of Environmental Toxicology and Chemistry, Milwaukee, WI, USA, 11–15 November 2007; p. 231.
- Seacat, A.M.; Thomford, P.J.; Hansen, K.J.; Olsen, G.W.; Case, M.T.; Butenhoff, J.L. Subchronic toxicity studies on perfluorooctanesulfonate potassium salt in cynomolgus monkeys. Toxicol. Sci. 2002, 68, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Ankley, G.T.; Kuehl, D.W.; Kahl, M.D.; Jensen, K.M.; Linnum, A.; Leino, R.L.; Villeneuvet, D.A. Reproductive and developmental toxicity and bioconcentration of perfluorooctanesulfonate in a partial life-cycle test with the fathead minnow (Pimephales promelas). Environ. Toxicol. Chem. 2005, 24, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Hu, J.; Huang, H.; Qin, Y.; Han, X.; Wu, D.; Song, L.; Xia, Y.; Wang, X. Perfluorooctane sulfonate (PFOS) affects hormone receptor activity, steroidogenesis, and expression of endocrine-related genes in vitro and in vivo. Environ. Toxicol. Chem. 2013, 32, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Henry, N.D.; Fair, P.A. Comparison of in vitro cytotoxicity, estrogenicity and anti-estrogenicity of triclosan, perfluorooctane sulfonate and perfluorooctanoic acid. J. Appl. Toxicol. 2013, 33, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Biegel, L.B.; Liu, R.C.; Hurtt, M.E.; Cook, J.C. Effects of ammonium perfluorooctanoate on Leydig cell function: in vitro, in vivo, and ex vivo studies. Toxicol. Appl. Pharmacol. 1995, 134, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, L.S.; Bonefeld-Jørgensen, E.C. Perfluorinated compounds affect the function of sex hormone receptors. Environ. Sci. Pollut. Res. Int. 2013, 20, 8031–8044. [Google Scholar] [CrossRef] [PubMed]
- López-Doval, S.; Salgado, R.; Fernández-Pérez, B.; Lafuente, A. Possible role of serotonin and neuropeptide Y on the disruption of the reproductive axis activity by perfluorooctane sulfonate. Toxicol. Lett. 2015, 233, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Rosenmai, A.K.; Nielsen, F.K.; Pedersen, M.; Hadrup, N.; Trier, X.; Christensen, J.H.; Vinggaard, A.M. Fluorochemicals used in food packaging inhibit male sex hormone synthesis. Toxicol. Appl. Pharmacol. 2013, 266, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhang, H.; Liu, Y.; Xu, M.; Dai, J. Alterations in gene expression and testosterone synthesis in the tests of male rats exposed to perfluorododecanoic acid. Toxicol. Sci. 2007, 98, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tan, Y.S.; Haslam, S.Z.; Yang, C. Perfluorooctanoic acid effects on steroid hormone and growth factor levels mediate stimulation of peripubertalmammary gland development in C57BL/6 mice. Toxicol. Sci. 2010, 115, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Liu, J.; Li, H.; Zhang, C.; Han, P.; Zhang, Y.; Yuan, X.; Ge, R.S.; Chu, Y. Exposure to perfluorooctane sulfonate in utero reduces testosterone production in rat fetal Leydig cells. PLoS ONE 2014, 9, e78888. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.C.; Murray, S.M.; Frame, S.R.; Hurtt, M.E. Induction of Leydig cell adenomas by ammonium perfluorooctanoate: A possible endocrine-related mechanism. Toxicol. Appl. Pharmacol. 1992, 113, 209–217. [Google Scholar] [CrossRef]
- Haghmorad, D.; Amini, A.A.; Mahmoudi, M.B.; Rastin, M.; Hosseini, M.; Mahmoudi, M. Pregnancy level of estrogen attenuates experimental autoimmune encephalomyelitis in both ovariectomized and pregnant C57BL/6 mice through expansion of Treg and Th2 cells. J. Neuroimmunol. 2014, 277, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C. Synergistic effect of various regulatory factors in TH1/TH2 balance: Immuno–therapeutic approaches in asthma. Int. J. Biomed. Sci. 2008, 4, 8–13. [Google Scholar] [PubMed]
- Cohen, J.H.; Danel, L.; Cordier, G.; Saez, S.; Revillard, J.P. Sex steroid receptors in peripheral T-cells: Absence of androgen receptors and restriction of estrogen receptors to OKT8+ cells. J. Immunol. 1983, 131, 2767–2771. [Google Scholar] [PubMed]
- Kovacs, W.J.; Olsen, N.J. Androgen receptors in human thymocytes. J. Immunol. 1987, 139, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Shackleton, C.H.; Pahwa, S.; White, P.C.; Speiser, P.W. Prominent sex steroid metabolism in human lymphocytes. Mol. Cell. Endocrinol. 1998, 138, 61–69. [Google Scholar] [CrossRef]
- Huber, S.A.; Kupperman, J.; Newell, M.K. Hormonal regulation of CD4 T-cell responses in coxsackievirus B3-induced myocarditis in mice. J. Virol. 1999, 73, 4689–4695. [Google Scholar] [PubMed]
- Knox, S.S.; Jackson, T.; Javins, B.; Frisbee, S.J.; Shankar, A.; Ducatman, A.M. Implications of early menopause in women exposed to perfluorocarbons. J. Clin. Endocrinol. Metab. 2011, 96, 1747–1753. [Google Scholar] [CrossRef] [PubMed]
- Joensen, U.N.; Veyrand, B.; Antignac, J.P.; Blomberg Jensen, M.; Petersen, J.H.; Marchand, P.; Skakkebaek, N.E.; Andersson, A.M.; le Bizec, B.; Jørgensen, N. PFOS (perfluorooctanesulfonate) in serum is negatively associated with testosterone levels, but not with semen quality, in healthy men. Hum. Reprod. 2013, 28, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Espinosa, M.J.; Fletcher, T.; Armstrong, B.; Genser, B.; Dhatariya, K.; Mondal, D.; Ducatman, A.; Leonardi, G. Association of Perfluorooctanoic Acid (PFOA) and Perfluorooctane Sulfonate (PFOS) with age of puberty among children living near a chemical plant. Environ. Sci. Technol. 2011, 45, 8160–8166. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Dong, G.H.; Jin, Y.H.; He, Q.C. Immunotoxic changes associated with a 7-day oral exposure to perfluorooctanesulfonate (PFOS) in adult male C57BL/6 mice. Arch. Toxicol. 2009, 83, 679–689. [Google Scholar] [CrossRef] [PubMed]
- OECD (Organization for Economic Co-Operation and Development). Hazard Assessment of Perfluorooctane Sulfonate (PFOS) and Its Salts. 2002. Available online: http://www.oecd.org/dataoecd/23/18/2382880.pdf (accessed on 27 July 2011).
- Zhou, Z.; Shi, Y.; Vestergren, R.; Wang, T.; Liang, Y.; Cai, Y. Highly elevated serum concentrations of perfluoroalkyl substances in fishery employees from Tangxun Lake, China. Environ. Sci. Technol. 2014, 48, 3864–3874. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; Thibodeaux, J.R.; Hanson, R.G.; Rogers, J.M.; Grey, B.E.; Stanton, M.E.; Butenhoff, J.L.; Stevenson, L.A. Exposure to perfluorooctane sulfonate during pregnancy in rat and mouse II: Post-natal evaluation. Toxicol. Sci. 2003, 74, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.J.; Clemen, L.A.; Ellefson, M.E.; Johnson, H.O. Compound-specific, quantitative characterization of organic fluorochemicals in biological matrices. Environ. Sci. Technol. 2001, 35, 766–770. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | PFOS Gestational Exposure Level (mg/kg/day) | ||||
|---|---|---|---|---|---|
| 0.0 (Control) | 0.1 | 1.0 | 5.0 | ||
| 4 weeks of age | |||||
| Body weight (g) | Male | 16.13 ± 0.63 | 16.59 ± 0.75 | 14.70 ± 0.58 | 14.37 ± 0.62 |
| Female | 14.33 ± 0.37 | 14.16 ± 0.49 | 13.52 ± 0.31 | 13.28 ± 0.40 | |
| Spleen b | Male | 0.53 ± 0.04 | 0.49 ± 0.03 | 0.45 ± 0.05 | 0.42 ± 0.02 a |
| Female | 0.51 ± 0.03 | 0.48 ± 0.02 | 0.47 ± 0.04 | 0.45 ± 0.02 | |
| Thymus b | Male | 0.61 ± 0.05 | 0.54 ± 0.04 | 0.58 ± 0.03 | 0.49 ± 0.02 a |
| Female | 0.57 ± 0.03 | 0.59 ± 0.02 | 0.54 ± 0.02 | 0.51 ± 0.01 | |
| Kidney b | Male | 1.70 ± 0.05 | 1.74 ± 0.03 | 1.82 ± 0.04 | 1.76 ± 0.05 |
| Female | 1.67 ± 0.04 | 1.61 ± 0.05 | 1.74 ± 0.03 | 1.65 ± 0.04 | |
| Liver b | Male | 7.21 ± 0.14 | 6.97 ± 0.21 | 7.75 ± 0.17 | 8.16 ± 0.24 a |
| Female | 6.88 ± 0.15 | 6.65 ± 0.25 | 7.04 ± 0.22 | 7.59 ± 0.30 a | |
| Serum PFOS concentration (mg/L) | |||||
| Male | 0.05 ± 0.01 | 6.38 ± 0.35 | 47.03 ± 3.23 a | 118.40 ± 6.27 a | |
| Female | 0.04 ± 0.01 | 5.16 ± 0.27 | 41.81 ± 3.62 a | 107.53 ± 4.51 a | |
| 8 weeks of age | |||||
| Body weight (g) | Male | 23.04 ± 0.75 | 24.96 ± 0.64 | 25.12 ± 0.96 | 23.48 ± 0.43 |
| Female | 21.51 ± 0.91 | 22.84 ± 0.67 | 20.65 ± 0.59 | 19.37 ± 0.73 | |
| Spleen b | Male | 0.42 ± 0.03 | 0.37 ± 0.05 | 0.43 ± 0.02 | 0.32 ± 0.03 |
| Female | 0.45 ± 0.02 | 0.46 ± 0.03 | 0.41 ± 0.03 | 0.43 ± 0.04 | |
| Thymus b | Male | 0.30 ± 0.01 | 0.27 ± 0.02 | 0.29 ± 0.01 | 0.24 ± 0.03 a |
| Female | 0.33 ± 0.02 | 0.31 ± 0.03 | 0.36 ± 0.02 | 0.32 ± 0.01 | |
| Kidney b | Male | 1.59 ± 0.04 | 1.61 ± 0.03 | 1.68 ± 0.03 | 1.54 ± 0.06 |
| Female | 1.43 ± 0.11 | 1.57 ± 0.05 | 1.38 ± 0.04 | 1.46 ± 0.03 | |
| Liver b | Male | 6.60 ± 0.18 | 6.41 ± 0.14 | 7.05 ± 0.21 | 6.39 ± 0.17 |
| Female | 5.77 ± 0.21 | 5.94 ± 0.13 | 6.03 ± 0.07 | 5.82 ± 0.09 | |
| Serum PFOS concentration (mg/L) | |||||
| Male | 0.04 ± 0.01 | 3.79 ± 0.26 | 37.53 ± 3.96 a | 82.66 ± 4.18 a | |
| Female | 0.04 ± 0.01 | 3.04 ± 0.17 | 31.17 ± 2.59 a | 71.68 ± 4.49 a | |
| Sex | PFOS (mg/kg/day) | CD8+ (Cells × 106) | DP (Cells × 105) | CD4+ (Cells × 107) | B220 (Cells × 107) |
|---|---|---|---|---|---|
| 4 weeks of age | |||||
| Male | 0.0 (Control) | 10.62 ± 0.61 | 5.24 ± 0.94 | 2.21 ± 0.18 | 5.98 ± 0.41 |
| 0.1 | 10.86 ± 0.55 | 5.63 ± 0.37 | 2.06 ± 0.13 | 5.65 ± 0.35 | |
| 1.0 | 9.98 ± 0.43 | 6.76 ± 0.68 | 1.85 ± 0.25 | 5.14 ± 0.26 | |
| 5.0 | 8.49 ± 0.36 a | 6.48 ± 0.42 | 1.64 ± 0.09 a | 4.73 ± 0.21 a | |
| Female | 0.0 (Control) | 9.18 ± 0.67 | 4.51 ± 0.53 | 1.94 ± 0.33 | 5.44 ± 0.31 |
| 0.1 | 9.44 ± 0.58 | 5.76 ± 0.46 | 1.67 ± 0.32 | 5.62 ± 0.25 | |
| 1.0 | 8.59 ± 0.34 | 5.14 ± 0.70 | 1.74 ± 0.46 | 5.09 ± 0.19 | |
| 5.0 | 8.80 ± 0.75 | 4.28 ± 0.64 | 1.43 ± 0.27 | 4.47 ± 0.27 a | |
| 8 weeks of age | |||||
| Male | 0.0 | 5.69 ± 0.36 | 1.81 ± 0.60 | 1.77 ± 0.09 | 6.43 ± 0.25 |
| 0.1 | 6.07 ± 0.25 | 1.77 ± 0.28 | 1.54 ± 0.11 | 6.17 ± 0.28 | |
| 1.0 | 6.31 ± 0.30 | 2.14 ± 0.26 | 1.60 ± 0.08 | 5.90 ± 0.24 | |
| 5.0 | 5.78 ± 0.42 | 1.62 ± 0.19 | 1.37 ± 0.10 a | 5.74 ± 0.30 | |
| Female | 0.0 | 5.40 ± 0.34 | 1.69 ± 0.24 | 1.63 ± 0.14 | 4.41 ± 0.15 |
| 0.1 | 5.64 ± 0.45 | 1.78 ± 0.35 | 1.59 ± 0.13 | 4.64 ± 0.23 | |
| 1.0 | 6.01 ± 0.62 | 1.94 ± 0.58 | 1.46 ± 0.09 | 4.22 ± 0.19 | |
| 5.0 | 5.27 ± 0.33 | 1.61 ± 0.22 | 1.32 ± 0.07 | 3.17 ± 0.27 a |
| Sex | PFOS (mg/kg/day) | CD8+ (Cells × 107) | DP (Cells × 105) | DN (Cells × 107) | CD4+ (Cells × 107) |
|---|---|---|---|---|---|
| 4 weeks of age | |||||
| Male | 0.0 | 5.14 ± 0.18 | 12.55 ± 0.71 | 7.44 ± 0.42 | 1.78 ± 0.09 |
| 0.1 | 4.93 ± 0.22 | 11.92 ± 0.76 | 8.65 ± 0.31 | 1.84 ± 0.12 | |
| 1.0 | 4.66 ± 0.37 | 13.08 ± 0.82 | 6.46 ± 0.72 | 1.51 ± 0.11 | |
| 5.0 | 4.43 ± 0.29 | 12.14 ± 0.63 | 5.77 ± 0.50 a | 1.37 ± 0.06 a | |
| Female | 0.0 | 3.59 ± 0.34 | 10.79 ± 0.94 | 6.98 ± 0.68 | 1.58 ± 0.12 |
| 0.1 | 3.67 ± 0.51 | 10.84 ± 0.57 | 7.15 ± 0.40 | 1.75 ± 0.09 | |
| 1.0 | 4.21 ± 0.47 | 10.56 ± 0.43 | 6.43 ± 0.51 | 1.61 ± 0.13 | |
| 5.0 | 3.45 ± 0.26 | 10.61 ± 0.30 | 6.09 ± 0.32 | 1.39 ± 0.14 | |
| 8 weeks of age | |||||
| Male | 0.0 | 4.01 ± 0.35 | 9.11 ± 1.03 | 5.16 ± 0.54 | 1.87 ± 0.08 |
| 0.1 | 4.37 ± 0.42 | 9.25 ± 0.67 | 6.05 ± 0.67 | 1.71 ± 0.07 | |
| 1.0 | 3.52 ± 0.37 | 8.66 ± 1.24 | 5.13 ± 0.39 | 1.60 ± 0.14 | |
| 5.0 | 3.71 ± 0.41 | 8.30 ± 1.38 | 4.95 ± 0.44 | 1.53 ± 0.10 a | |
| Female | 0.0 | 3.64 ± 0.43 | 8.79 ± 0.75 | 4.65 ± 0.31 | 1.66 ± 0.11 |
| 0.1 | 3.51 ± 0.57 | 8.65 ± 0.69 | 5.03 ± 0.28 | 1.58 ± 0.16 | |
| 1.0 | 3.79 ± 0.36 | 7.72 ± 0.58 | 4.50 ± 0.24 | 1.69 ± 0.10 | |
| 5.0 | 3.22 ± 0.40 | 7.93 ± 1.25 | 5.49 ± 0.48 | 1.47 ± 0.08 |
| Sex | PFOS | IL-2 | IFNγ | IL-4 | IL-10 |
|---|---|---|---|---|---|
| 4 weeks of age | |||||
| Male | 0.0 | 54.02 ± 4.20 | 24.67 ± 2.53 | 11.87 ± 2.02 | 53.21 ± 4.32 |
| 0.1 | 51.89 ± 5.21 | 22.38 ± 2.05 | 13.95 ± 1.35 | 57.75 ± 3.97 | |
| 1.0 | 47.08 ± 3.14 | 20.03 ± 1.70 | 15.81 ± 1.92 | 54.68 ± 4.21 | |
| 5.0 | 38.49 ± 3.68 a | 19.63 ± 1.53 | 17.78 ± 1.23 a | 60.61 ± 3.64 | |
| Female | 0.0 | 49.65 ± 4.01 | 22.07 ± 2.28 | 13.55 ± 1.85 | 49.45 ± 4.31 |
| 0.1 | 46.50 ± 3.07 | 24.90 ± 2.14 | 15.61 ± 1.09 | 53.96 ± 5.53 | |
| 1.0 | 41.45 ± 3.98 | 20.47 ± 1.79 | 17.59 ± 1.37 | 56.07 ± 4.78 | |
| 5.0 | 39.33 ± 2.99 | 18.03 ± 2.30 | 19.46 ± 1.93 a | 62.40 ± 4.99 | |
| 8 weeks of age | |||||
| Male | 0.0 | 34.35 ± 3.92 | 26.87 ± 2.99 | 15.32 ± 1.88 | 39.42 ± 4.61 |
| 0.1 | 31.76 ± 4.11 | 25.63 ± 3.74 | 16.71 ± 2.54 | 40.65 ± 5.62 | |
| 1.0 | 38.22 ± 3.83 | 23.68 ± 4.20 | 19.50 ± 1.93 | 42.98 ± 3.71 | |
| 5.0 | 32.42 ± 5.80 | 24.85 ± 3.52 | 21.79 ± 2.06 a | 46.93 ± 4.98 | |
| Female | 0.0 | 36.16 ± 5.84 | 25.40 ± 3.58 | 19.78 ± 3.20 | 36.19 ± 3.36 |
| 0.1 | 35.68 ± 3.70 | 24.39 ± 4.24 | 20.04 ± 2.78 | 38.47 ± 5.44 | |
| 1.0 | 37.27 ± 4.48 | 23.88 ± 3.82 | 21.72 ± 3.43 | 42.80 ± 3.28 | |
| 5.0 | 34.89 ± 4.53 | 21.92 ± 4.03 | 25.69 ± 2.56 | 41.34 ± 4.60 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, S.-Q.; Chen, Z.-X.; Kong, M.-L.; Xie, Y.-Q.; Zhou, Y.; Qin, X.-D.; Paul, G.; Zeng, X.-W.; Dong, G.-H. Testosterone-Mediated Endocrine Function and TH1/TH2 Cytokine Balance after Prenatal Exposure to Perfluorooctane Sulfonate: By Sex Status. Int. J. Mol. Sci. 2016, 17, 1509. https://doi.org/10.3390/ijms17091509
Zhong S-Q, Chen Z-X, Kong M-L, Xie Y-Q, Zhou Y, Qin X-D, Paul G, Zeng X-W, Dong G-H. Testosterone-Mediated Endocrine Function and TH1/TH2 Cytokine Balance after Prenatal Exposure to Perfluorooctane Sulfonate: By Sex Status. International Journal of Molecular Sciences. 2016; 17(9):1509. https://doi.org/10.3390/ijms17091509
Chicago/Turabian StyleZhong, Shou-Qiang, Zan-Xiong Chen, Min-Li Kong, Yan-Qi Xie, Yang Zhou, Xiao-Di Qin, Gunther Paul, Xiao-Wen Zeng, and Guang-Hui Dong. 2016. "Testosterone-Mediated Endocrine Function and TH1/TH2 Cytokine Balance after Prenatal Exposure to Perfluorooctane Sulfonate: By Sex Status" International Journal of Molecular Sciences 17, no. 9: 1509. https://doi.org/10.3390/ijms17091509
APA StyleZhong, S.-Q., Chen, Z.-X., Kong, M.-L., Xie, Y.-Q., Zhou, Y., Qin, X.-D., Paul, G., Zeng, X.-W., & Dong, G.-H. (2016). Testosterone-Mediated Endocrine Function and TH1/TH2 Cytokine Balance after Prenatal Exposure to Perfluorooctane Sulfonate: By Sex Status. International Journal of Molecular Sciences, 17(9), 1509. https://doi.org/10.3390/ijms17091509