
Lactate as a Metabolite and a Regulator in the Central Nervous System


 , ,
, , 
Abstract
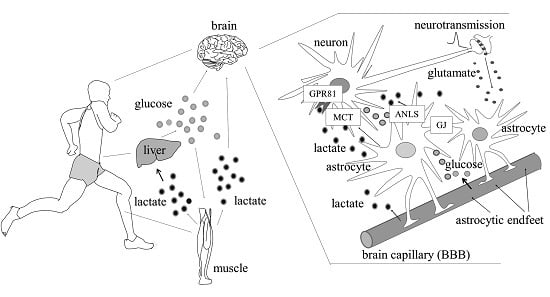
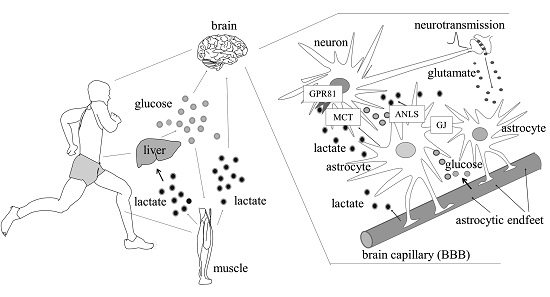
:
{kind=link}
1. Introduction
2. Exercise, Lactate Production in the Periphery and Fatigue
2.1. Exercise and Lactate Production
2.2. Fatigue
3. Lactate Uptake across the Blood-Brain Barrier and Monocarboxylate Carriers (MCTs)
3.1. Lactate Can Cross the Blood-Brain Barrier (BBB)
3.2. The Monocarboxylate Carriers (MCTs)
4. Glial Cell—Neurons Lactate Shuttle and Brain Energy Metabolism
4.1. Lactate Shuttling and Energy Metabolism
4.2. Glucose Sensing
4.3. Role of Extracellular Vesicles (EVs) in Cell-to-Cell Communications in the Brain
5. Lactate as a Substrate during Exercise and in Memory Processes
6. Lactate as a Signaling Molecule in the Brain
7. Brain Glucose Metabolism and Elevated Lactate in Pathological Conditions
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Roy, C.S.; Sherrington, C.S. On the Regulation of the Blood-supply of the Brain. J. Physiol. 1890, 11, 85–158. [Google Scholar] [PubMed]
- Fletcher, W.M.; Hopkins, F.G. Lactic acid in amphibian muscle. J. Physiol. 1907, 35, 247–309. [Google Scholar] [PubMed]
- Hill, A.V.; Long, C.N.H.; Lupton, H. Muscular exercise, lactic acid, and the supply and utilization of oxygen. Part VI. The oxygen debt and the end of exercise. Proc. R. Soc. Lond. B Biol. Sci. 1924, 97, 127–137. [Google Scholar]
- Gladden, L.B. Lactate metabolism: A new paradigm for the third millennium. J. Physiol. 2004, 558, 5–30. [Google Scholar] [PubMed]
- McIlwain, H. Metabolic Response in vitro to Electrical Stimulation of Sections of Mammalian Brain. Biochem. J. 1951, 49, 382–393. [Google Scholar] [PubMed]
- McIlwain, H. Glucose Level, Metabolism, and Response to Electrical Impulses in Cerebral Tissues from Man and Laboratory Animals. Biochem. J. 1953, 55, 618–624. [Google Scholar] [PubMed]
- Magistretti, P.J. Neuron–glia metabolic coupling and plasticity. J. Exp. Biol. 2006, 209, 2304–2311. [Google Scholar] [PubMed]
- Greenhaff, P.L.; Nevill, M.E.; Soderlund, K.; Bodin, K.; Boobis, L.H.; Williams, C.; Hultman, E. The metabolic responses of human type I and II muscle fibres during maximal treadmill sprinting. J. Physiol. 1994, 478, 149–155. [Google Scholar] [PubMed]
- Baker, J.S.; McCormick, M.C.; Robergs, R.A. Interaction among skeletal muscle metabolic energy systems during intense exercise. J. Nutr. Metab. 2010, 2010. [Google Scholar] [CrossRef]
- Crisp, A.H.; Verlengia, R.; Rocha, G.L.; da Mota, G.R.; Pellegrinotti, I.L.; Lopes, R.L. Lactate and monocarboxylate transporters (MCTS): A review of cellular aspects. J. Exerc. Physiol. Online 2015, 18, 1–13. [Google Scholar]
- Chatham, J.C. Lactate—The forgotten fuel! J. Physiol. 2002, 542. [Google Scholar] [CrossRef]
- Pérez de Heredia, F.; Wood, I.S.; Trayhurn, P. Hypoxia stimulates lactate release and modulates monocarboxylate transporter (MCT1, MCT2, and MCT4) expression in human adipocytes. Pflugers Arch. 2010, 459, 509–518. [Google Scholar] [PubMed]
- Adeva-Andany, M.; López-Ojén, M.; Funcasta-Calderón, R.; Ameneiros-Rodríguez, E.; Donapetry-García, C.; Vila-Altesor, M.; Rodríguez-Seijas, J. Comprehensive review on lactate metabolism in human health. Mitochondrion 2014, 17, 76–100. [Google Scholar] [PubMed]
- Brooks, G.A. The lactate shuttle during exercise and recovery. Med. Sci. Sports Exerc. 1986, 18, 360–368. [Google Scholar] [PubMed]
- Facey, A.; Irving, R.; Dilworth, L. Overview of Lactate Metabolism and the Implications for Athletes. Am. J. Sports Sci. Med. 2013, 1, 42–46. [Google Scholar]
- Noakes, T.D. Evidence that reduced skeletal muscle recruitment explains the lactate paradox during exercise at high altitude. J. Appl. Physiol. 2009, 106, 737–738. [Google Scholar] [PubMed]
- Amann, M.; Romer, L.M.; Subudhi, A.W.; Pegelow, D.F.; Dempsey, J.A. Severity of arterial hypoxaemia affects the relative contributions of peripheral muscle fatigue to exercise performance in healthy humans. J. Physiol. 2007, 581, 389–403. [Google Scholar] [PubMed]
- Vollestad, N.K.; Sejersted, O.M. Biochemical correlates of fatigue. A brief review. Eur. J. Appl. Physiol. Occup. Physiol. 1988, 57, 336–347. [Google Scholar] [PubMed]
- Posterino, G.S.; Dutka, T.; Lamb, G.D. l(+)-lactate does not affect twitch and tetanic responses in mechanically skinned mammalian muscle fibres. Pflügers Arch. 2001, 442, 197–203. [Google Scholar] [PubMed]
- Hall, M.M.; Rajasekaran, S.; Thomsen, T.W.; Peterson, A.R. Lactate: Friend or Foe. PM&R 2016, 8, S8–S15. [Google Scholar]
- Robergs, R.A.; Ghiasvand, F.; Parker, D. Biochemistry of exercise induced metabolic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R502–R516. [Google Scholar] [PubMed]
- Lamb, G.D.; Stephenson, D.G.; Stienen, G.J. Effects of osmolality and ionic strength on the mechanism of Ca2 release in skinned skeletal muscle fibres of the toad. J. Physiol. 1993, 464, 629–648. [Google Scholar] [PubMed]
- Allen, D.G.; Lamb, G.D.; Westerbland, H. Skeletal Muscle Fatigue: Cellular Mechanisms. Physiol. Rev. 2008, 88, 287–332. [Google Scholar] [PubMed]
- Bangsbo, J.; Madsen, K.; Kiens, B.; Richter, E.A. Effect of muscle acidity on muscle metabolism and fatigue during intense exercise in man. J. Physiol. 1996, 495, 587–596. [Google Scholar] [PubMed]
- Bruton, J.D.; Lännergren, J.; Westerblad, H. Effects of CO2-induced acidification on the fatigue resistance of single mouse muscle fibers at 28 degrees C. J. Appl. Physiol. 1998, 85, 478–483. [Google Scholar] [PubMed]
- Norman, B.; Sabina, R.L.; Jansson, E. Regulation of skeletal muscle ATP catabolism by AMPD1 genotype during sprint exercise in asymptomatic subjects. J. Appl. Physiol. 2001, 91, 258–264. [Google Scholar] [PubMed]
- Allen, D.G.; Trajanovska, S. The multiple roles of phosphate in muscle fatigue. Front. Physiol. 2012, 3, 1–8. [Google Scholar]
- Fitts, R.H. Mechanisms of muscular fatigue. In Principles of Exercise Biochemistry, 3rd ed.; Poortmans, J.R., Ed.; Karger: Basel, Switzerland, 2003; pp. 279–300. [Google Scholar]
- Westerblad, H.; Allen, D.G.; Lännergren, J. Muscle fatigue: Lactic acid or inorganic phosphate the major cause? News Physiol. Sci. 2002, 17, 17–21. [Google Scholar] [PubMed]
- Dubouchaud, H.; Butterfield, G.E.; Wolfel, E.E.; Bergman, B.C.; Brooks, G.A. Endurance training, expression, and physiology of LDH, MCT1, and MCT4 in human skeletal muscle. Am. J. Physiol. 2000, 278, E571–E579. [Google Scholar]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [PubMed]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [PubMed]
- Smith, Q.R. Transport of glutamate and other amino acids at the Blood-Brain Barrier. J. Nutr. 2000, 130, 1016S–1022S. [Google Scholar] [PubMed]
- Huber, J.D.; Egleton, R.D.; Davis, T.P. Molecular physiology and pathophysiology of tight junctions in the bloodbrain barrier. Trends Neurosci. 2001, 24, 719–725. [Google Scholar] [PubMed]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [PubMed]
- Hawkins, R.A.; Viña, J.R.; Mokashi, A.; Peterson, D.R.; O’Kane, R.; Simpson, I.A.; Dejoseph, M.R.; Rasgado-Flores, H. Synergism between the two membranes of the blood-brain barrier: Glucose and amino acid transport. Am. J. Neurosci. Res. 2013, 1, 1–25. [Google Scholar]
- Pérez-Escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497. [Google Scholar] [PubMed]
- Jones, R.S.; Morris, M.E. Monocarboxylate Transporters: Therapeutic targets and prognostic factors in disease. Clin. Pharmacol. Ther. 2016. [Google Scholar] [CrossRef]
- Bergersen, L.H. Is lactate food for neurons? Comparison of monocarboxylate transporter subtypes in brain and muscle. Neuroscience 2007, 145, 11–19. [Google Scholar] [PubMed]
- Halestrap, A.P. The monocarboxylate transporter family-structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [PubMed]
- Lauritzen, K.H.; Eid, T.; Bergersen, L.H. Monocarboxylate transporters in temporal lobe epilepsy: Roles of lactate and ketogenic diet. Brain Struct. Funct. 2013, 220, 1–12. [Google Scholar] [PubMed]
- Gallagher-Colombo, S.; Maminishkis, A.; Tate, S.; Grunwald, G.B.; Philp, N.J. Modulation of MCT3 expression during wound healing of the retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5343–5350. [Google Scholar]
- Gao, C.; Zhou, L.; Zhu, W.; Wang, H.; Wang, R.; He, Y.; Li, Z. Monocarboxylate transporter-dependent mechanism confers resistance to oxygen- and glucose-deprivation injury in astrocyte-neuron co-cultures. Neurosci. Lett. 2015, 594, 99–104. [Google Scholar] [PubMed]
- Wilson, M.C.; Meredith, D.; Fox, J.E.; Manoharan, C.; Davies, A.J.; Halestrap, A.P. Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate transporter isoforms 1 and 4: The ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J. Biol. Chem. 2005, 280, 27213–27221. [Google Scholar] [PubMed]
- Ovens, M.J.; Manoharan, C.; Wilson, M.C.; Murray, C.M.; Halestrap, A.P. The inhibition of monocarboxylate transporter 2 (MCT2) by AR-C155858 is modulated by the associated ancillary protein. Biochem. J. 2010, 431, 217–225. [Google Scholar] [PubMed]
- Wilson, M.C.; Kraus, M.; Marzban, H.; Sarna, J.R.; Wang, Y.; Hawkes, R.; Halestrap, A.P.; Beesley, P.W. The neuroplastin adhesion molecules are accessory proteins that chaperone the monocarboxylate transporter MCT2 to the neuronal cell surface. PLoS ONE 2013, 8, e78654. [Google Scholar]
- Domènech-Estévez, E.; Baloui, H.; Repond, C.; Rosafio, K.; Médard, J.J.; Tricaud, N.; Pellerin, L.; Chrast, R. Distribution of monocarboxylate transporters in the peripheral nervous system suggests putative roles in lactate shuttling and myelination. J. Neurosci. 2015, 35, 4151–4156. [Google Scholar] [PubMed]
- Giaume, C.; Koulakoff, A.; Roux, L.; Holcman, D.; Rouach, N. Astroglial networks: A step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci. 2010, 11, 87–99. [Google Scholar] [PubMed]
- Pannasch, U.; Rouach, N. Emerging role for astroglial networks in information processing: From synapse to behavior. Trends Neurosci. 2013, 36, 405–417. [Google Scholar] [PubMed]
- Leloup, C.; Allard, C.; Carneiro, L.; Fioramonti, X.; Collins, S.; Pénicaud, L. Glucose and hypothalamic astrocytes: More than a fueling role? Neuroscience 2016, 323, 110–120. [Google Scholar] [PubMed]
- Lauf, U.; Giepmans, B.N.; Lopez, P.; Braconnot, S.; Chen, S.C.; Falk, M.M. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 10446–10451. [Google Scholar] [PubMed]
- Moore, K.B.; O’Brien, J. Connexins in neurons and glia: Targets for intervention in disease and injury. Neural Regen. Res. 2015, 10, 1013–1017. [Google Scholar] [PubMed]
- Bosone, C.; Andreu, A.; Echevarria, D. GAP junctional communication in brain secondary organizers. Dev. Growth Differ. 2016, 58, 446–455. [Google Scholar] [PubMed]
- Genoud, C.; Houades, V.; Kraftsik, R.; Welker, E.; Giaume, C. Proximity of excitatory synapses and astroglial gap junctions in layer IV of the mouse barrel cortex. Neuroscience 2015, 291, 241–249. [Google Scholar] [PubMed]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [PubMed]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.; Charles, A.C. Intercellular calcium signaling in astrocytes via ATP release through connexinhemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [PubMed]
- Stehberg, J.; Moraga-Amaro, R.; Salazar, C.; Becerra, A.; Echeverría, C.; Orellana, J.A.; Bultynck, G.; Ponsaerts, R.; Leybaert, L.; Simon, F.; et al. Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 2012, 26, 3649–3657. [Google Scholar] [PubMed]
- Li, X.; Zhao, H.; Tan, X.; Kostrzewa, R.M.; Du, G.; Chen, Y.; Zhu, J.; Miao, Z.; Yu, H.; Kong, J.; et al. Inhibition of connexion43 improves functional recovery after ischemic brain injury in neonatal rats. Glia 2015, 63, 1553–1567. [Google Scholar] [PubMed]
- Matsui, T.; Soya, S.; Okamoto, M.; Ichitani, Y.; Kawanaka, K.; Soya, H. Brain glycogen decreases during prolonged exercise. J. Physiol. 2011, 589, 3383–3393. [Google Scholar] [PubMed]
- Chambers, T.W.; Daly, T.P.; Hockley, A.; Brown, A.M. Contribution of glycogen in supporting axon conduction in the peripheral and central nervous systems: The role of lactate. Front. Neurosci. 2014, 8, 1–6. [Google Scholar]
- Dienel, G.A. The metabolic trinity, glucose-glycogen-lactate, links astrocytes and neurons in brain energetics, signaling, memory, and gene expression. Neurosci. Lett. 2015. [Google Scholar] [CrossRef]
- Dienel, G.A.; Cruz, N.F. Contributions of glycogen to astrocytic energetics during brain activation. Metab. Brain Dis. 2015, 30, 281–298. [Google Scholar] [PubMed]
- Matsui, T.; Soya, S.; Okamoto, M.; Ichitani, Y.; Kawanaka, K.; Soya, H. Brain Glycogen Decreases during Intense Exercise without Hypoglycemia: The Possible Involvement of Serotonin. Neurochem. Res. 2015, 40, 1333–1340. [Google Scholar] [PubMed]
- Evans, R.D.; Brown, A.M.; Ransom, B.R. Glycogen function in adult central and peripheral nerves. J. Neurosci. Res. 2013, 91, 1044–1049. [Google Scholar] [PubMed]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, R.; Magistretti, P.J.; Pellerin, L. Trans-inhibition of glutamate transport prevents excitatory amino acid-induced glycolysis in astrocytes. Brain Res. 1999, 850, 39–46. [Google Scholar] [CrossRef]
- Voutsinos-Porche, B.; Bonvento, G.; Tanaka, K.; Steiner, P.; Welker, E.; Chatton, J.Y.; Magistretti, P.J.; Pellerin, L. Glial glutamate transporters mediate a functional metabolic crosstalk between neurons and astrocytes in the mouse developing cortex. Neuron 2003, 37, 275–286. [Google Scholar] [CrossRef]
- Chatton, J.Y.; Magistretti, P.J.; Barros, L.F. Sodium signaling and astrocyte energy metabolism. Glia 2016. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Pellerin, L. Astrocytes Couple Synaptic Activity to Glucose Utilization in the Brain. News Physiol. Sci. 1999, 14, 177–182. [Google Scholar] [PubMed]
- Schurr, A.; West, C.A.; Rigor, B.M. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 1988, 240, 1326–1328. [Google Scholar] [CrossRef] [PubMed]
- Schurr, A.; Payne, R.S.; Miller, J.J.; Rigor, B.M. Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: An in vitro study. Brain Res. 1997, 744, 105–111. [Google Scholar] [CrossRef]
- Schurr, A.; Payne, R.S.; Miller, J.J.; Rigor, B.M. Glia are the main source of lactate utilized by neurons for recovery of function posthypoxia. Brain Res. 1997, 774, 221–224. [Google Scholar] [CrossRef]
- Schurr, A. Cerebral glycolysis: A century of persistent misunderstanding and misconception. Front. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Ashford, C.A.; Holmes, E.G. Contributions to the study of brain metabolism: Role of phosphates in lactic acid production. Biochem. J. 1929, 23, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.G. Oxidations in central and peripheral nervous tissue. Biochem. J. 1930, 24, 914–925. [Google Scholar] [CrossRef]
- Holmes, E.G.; Ashford, C.A. Lactic acid oxidation in brain with reference to the “Meyerhof cycle”. Biochem. J. 1930, 24, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Ashford, C.A.; Holmes, E.G. Further observations on the oxidation of lactic acid by brain tissue. Biochem. J. 1931, 25, 2028–2049. [Google Scholar] [CrossRef] [PubMed]
- Quastel, J.H.; Wheatley, A.H. Oxidations by the brain. Biochem. J. 1932, 26, 725–744. [Google Scholar] [CrossRef] [PubMed]
- Wender, R.; Brown, A.M.; Fern, R.; Swanson, R.A.; Farrell, K.; Ransom, B.R. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 2000, 20, 6804–6810. [Google Scholar] [PubMed]
- Brown, A.M.; Wender, R.; Ransom, B.R. Metabolic substrates other than glucose support axon function in central white matter. J. Neurosci. Res. 2001, 66, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L. Lactate as a pivotal element in neuron-glia metabolic cooperation. Neurochem. Int. 2003, 43, 331–338. [Google Scholar] [CrossRef]
- Pellerin, L.; Bouzier-Sore, A.K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P.J. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Cruz, N.F. Nutrition during brain activation: Does cell-to-cell lactate shuttling contribute significantly to sweet and sour food for thought? Neurochem. Int. 2004, 45, 321–351. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; BaltanTekkök, S.; Ransom, B.R. Energy transfer from astrocytes to axons: The role of CNS glycogen. Neurochem. Int. 2004, 45, 529–536. [Google Scholar] [PubMed]
- Fillenz, M. The role of lactate in brain metabolism. Neurochem. Int. 2005, 47, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cereb. Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A.; Coles, J.A. Cellular pathways of energy metabolism in the brain: Is glucose used by neurons or astrocytes? Glia 2007, 55, 1238–1250. [Google Scholar] [CrossRef] [PubMed]
- Tsacopoulos, M.; Magistretti, P.J. Metabolic coupling between glia and neurons. J. Neurosci. 1996, 16, 877–885. [Google Scholar] [PubMed]
- Magistretti, P.J.; Pellerin, L. Cellular bases of brain energy metabolism and their relevance to functional brain imaging: Evidence for a prominent role of astrocytes. Cereb. Cortex 1996, 6, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Pellerin, L. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A.; Wittendorp-Rechenmann, E.; Lam, C.D. Selective uptake of [14C]2-deoxyglucose by neurons and astrocytes: High-resolution microautoradiographic imaging by cellular 14C-trajectography combined with immunohistochemistry. J. Cereb. Blood Flow Metab. 2004, 24, 1004–1014. [Google Scholar] [CrossRef]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the central nervous system. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, K.J.; Du, J.; Sloat, S.R.; Contreras, L.; Linton, J.D.; Turner, S.J.; Sadilek, M.; Satrústegui, J.; Hurley, J.B. Pyruvate kinase and aspartate-glutamate carrier distributions reveal key metabolic links between neurons and glia in retina. Proc. Natl. Acad. Sci. USA 2014, 111, 15579–15584. [Google Scholar] [CrossRef] [PubMed]
- Zielke, H.R.; Zielke, C.L.; Baab, P.J. Oxidation of 14C-labeled compounds perfused by microdialysis in the brains of free-moving rats. J. Neurosci. Res. 2007, 85, 3145–3149. [Google Scholar] [CrossRef] [PubMed]
- Zielke, H.R.; Zielke, C.L.; Baab, P.J. Direct measurement of oxidative metabolism in the living brain by microdialysis: A review. J. Neurochem. 2009, 109 (Suppl. 1), 24–29. [Google Scholar] [CrossRef] [PubMed]
- Caesar, K.; Hashemi, P.; Douhou, A.; Bonvento, G.; Boutelle, M.G.; Walls, A.B.; Lauritzen, M. Glutamate receptor-dependent increments in lactate, glucose and oxygen metabolism evoked in rat cerebellum in vivo. J. Physiol. 2008, 586, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Contreras, L.; Satrústegui, J. Calcium signaling in brain mitochondria: Interplay of malate aspartate NADH shuttle and calcium uniporter/mitochondrial dehydrogenase pathways. J. Biol. Chem. 2009, 284, 7091–7099. [Google Scholar] [CrossRef] [PubMed]
- Ivannikov, M.V.; Sugimori, M.; Llinás, R.R. Calcium clearance and its energy requirements in cerebellar neurons. Cell Calcium 2010, 47, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Obel, L.F.; Walls, A.B.; Schousboe, A.; Faek, S.A.; Jajo, F.S.; Waagepetersen, H.S. Novel model of neuronal bioenergetics: Postsynaptic utilization of glucose but not lactate correlates positively with Ca2+ signalling in cultured mouse glutamatergic neurons. ASN Neuro 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Astrocytic energetics during excitatory neurotransmission: What are contributions of glutamate oxidation and glycolysis? Neurochem. Int. 2013, 63, 244–258. [Google Scholar] [PubMed]
- An, J.; Haile, W.B.; Wu, F.; Torre, E.; Yepes, M. Tissue-type plasminogen activator mediates neuroglial coupling in the central nervous system. Neuroscience 2014, 257, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Newman, L.A.; Korol, D.L.; Gold, P.E. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS ONE 2011, 6, e28427. [Google Scholar] [CrossRef] [PubMed]
- Sotelo-Hitschfeld, T.; Niemeyer, M.I.; Mächler, P.; Ruminot, I.; Lerchundi, R.; Wyss, M.T.; Stobart, J.; Fernández-Moncada, I.; Valdebenito, R.; Garrido-Gerter, P.; et al. Channel-mediated lactate release by K⁺-stimulated astrocytes. J. Neurosci. 2015, 35, 4168–4178. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I.; Malkov, A.E.; Waseem, T.; Mukhtarov, M.; Buldakova, S.; Gubkina, O.; Zilberter, M.; Zilberter, Y. Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J. Cereb. Blood Flow Metab. 2014, 34, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Lai, J.C.; Chowdhury, G.M.; Hyder, F.; Rothman, D.L.; Shulman, R.G.; Behar, K.L. Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc. Natl. Acad. Sci. USA 2014, 111, 5385–5390. [Google Scholar] [CrossRef] [PubMed]
- Ciofi, P.; Garret, M.; Lapirot, O.; Lafon, P.; Loyens, A.; Prévot, V.; Levine, J.E. Brain-endocrine interactions: A microvascular route in the mediobasal hypothalamus. Endocrinology 2009, 150, 5509–5519. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Anand, B.K.; Chhina, G.S.; Singh, B. Effect of glucose on the activity of hypothalamic “feeding centers”. Science 1962, 138, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Oomura, Y.; Ono, T.; Ooyama, H.; Wayner, M.J. Glucose and osmosensitiveneuronesof the rat hypothalamus. Nature 1969, 222, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Allard, C.; Carneiro, L.; Grall, S.; Cline, B.H.; Fioramonti, X.; Chrétien, C.; Baba-Aissa, F.; Giaume, C.; Pénicaud, L.; Leloup, C. Hypothalamic astroglialconnexins are required for brain glucose sensing-induced insulin secretion. J. Cereb. Blood Flow Metab. 2014, 34, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Lynch, R.M.; Tompkins, L.S.; Brooks, H.L.; Dunn-Meynell, A.A.; Levin, B.E. Localization of glucokinase gene expression in the rat brain. Diabetes 2000, 49, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Gramm, C.F.; Scorrano, L.; Zhang, C.Y.; Krauss, S.; Ranger, A.M.; Datta, S.R.; Greenberg, M.E.; Licklider, L.J.; Lowell, B.B.; et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 2003, 424, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Leloup, C.; Arluison, M.; Lepetit, N.; Cartier, N.; Marfaing-Jallat, P.; Ferré, P.; Pénicaud, L. Glucose transporter 2 (GLUT 2): Expression in specific brain nuclei. Brain Res. 1994, 638, 221–226. [Google Scholar] [CrossRef]
- Arluison, M.; Quignon, M.; Thorens, B.; Leloup, C.; Penicaud, L. Immunocytochemical localization of the glucose transporter 2 (GLUT2) in the adult rat brain. II. Electron microscopic study. J. Chem. Neuroanat. 2004, 28, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Routh, V.H. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 2005, 54, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Venner, A.; Karnani, M.M.; Gonzalez, J.A.; Jensen, L.T.; Fugger, L.; Burdakov, D. Orexin neurons as conditional glucosensors: Paradoxical regulation of sugar sensing by intracellular fuels. J. Physiol. 2011, 589, 5701–5708. [Google Scholar] [CrossRef] [PubMed]
- Schiera, G.; Proia, P.; Alberti, C.; Mineo, M.; Savettieri, G.; di Liegro, I. Neurons produce FGF2 and VEGF and secrete them at least in part by shedding extracellular vesicles. J. Cell. Mol. Med. 2007, 11, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Proia, P.; Schiera, G.; Mineo, M.; Ingrassia, A.M.; Santoro, G.; Savettieri, G.; di Liegro, I. Astrocytes shed extracellular vesicles that contain fibroblast growth factor-2 and vascular endothelial growth factor. Int. J. Mol. Med. 2008, 21, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.R.; Robinson, M.B.; Gifondorwa, D.J.; Tytell, M.; Milligan, C.E. Regulation of heat shock protein 70 release in astrocytes: Role of signaling kinases. Dev. Neurobiol. 2007, 67, 1815–1829. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cesca, F.; Loers, G.; Schweizer, M.; Buck, F.; Benfenati, F.; Schachner, M.; Kleene, R. Synapsin I is an oligomannose-carrying glycoprotein, acts as an oligomannose-binding lectin, and promotes neurite outgrowth and neuronal survival when released via glia-derived exosomes. J. Neurosci. 2011, 31, 7275–7290. [Google Scholar] [CrossRef] [PubMed]
- Lo Cicero, A.; Majkowska, I.; Nagase, H.; di Liegro, I.; Troeberg, L. Microvesicles shed by oligodendroglioma cells and rheumatoid synovial fibroblasts contain aggrecanase activity. Matrix Biol. 2012, 31, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Schiera, G.; di Liegro, C.M.; Saladino, P.; Pitti, R.; Savettieri, G.; Proia, P.; di Liegro, I. Oligodendroglioma cells synthesize the differentiation-specific linker histone H1 and release it into the extracellular environment through shed vesicles. Int. J. Oncol. 2013, 43, 1771–1776. [Google Scholar] [PubMed]
- Schiera, G.; di Liegro, C.M.; di Liegro, I. Extracellular Membrane Vesicles as Vehicles for Brain Cell-to-Cell Interactions in Physiological as well as Pathological Conditions. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Lachenal, G.; Pernet-Gallay, K.; Chivet, M.; Hemming, F.J.; Belly, A.; Bodon, G.; Blot, B.; Haase, G.; Goldberg, Y.; Sadoul, R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell. Neurosci. 2011, 46, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Chivet, M.; Hemming, F.; Pernet-Gallay, K.; Fraboulet, S.; Sadoul, R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Krämer-Albers, E.M. Extracellular vesicles as mediators of neuron-glia communication. Front. Cell Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.N. Exercise, cognitive function, and aging. Adv. Physiol. Educ. 2015, 39, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Laitman, B.M.; John, G.R. Understanding how exercise promotes cognitive integrity in the aging brain. PLoS Biol. 2015, 13, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Colcombe, S.J.; Erickson, K.I.; Raz, N.; Webb, A.G.; Cohen, N.J.; McAuley, E.; Kramer, A.F. Aerobic fitness reduces brain tissue loss in aging humans. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.F.; Chen, P.C.; Calkins, M.J.; Wu, S.Y.; Kuo, Y.M. Exercise counteracts aging-related memory impairment: A potential role for the astrocytic metabolic shuttle. Front. Aging Neurosci. 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Ide, K.; Schmalbruch, I.K.; Quistorff, B.; Horn, A.; Secher, N.H. Lactate, glucose and O2 uptake in human brain during recovery from maximal exercise. J. Physiol. 2000, 522, 159–164. [Google Scholar] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [PubMed]
- Matsui, T.; Ishikawa, T.; Ito, H.; Okamoto, M.; Inoue, K.; Lee, M.C.; Fujikawa, T.; Ichitani, Y.; Kawanaka, K.; Soya, H. Brain glycogen supercompensation following exhaustive exercise. J. Physiol. 2012, 590, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.E. Role of Glycogenolysis in Memory and Learning: Regulation by Noradrenaline, Serotonin and ATP. Front. Integr. Neurosci. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Steinman, M.Q.; Gao, V.; Alberini, C.M. The role of lactate-mediated etabolic coupling between astrocytes and neurons in long-term memory formation. Front. Integr. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Cotman, C.W.; Berchtold, N.C.; Christie, L.A. Exercise builds brain health: Key roles of growth factor cascades and inflammation. Trends Neurosci. 2007, 30, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Vivar, C.; Potter, M.C.; van Praag, H. All about running: Synaptic plasticity, growth factors and adult hippocampal neurogenesis. Curr. Top. Behav. Neurosci. 2013, 15, 189–210. [Google Scholar] [PubMed]
- Skriver, K.; Roig, M.; Lundbye-Jensen, J.; Pingel, J.; Helge, J.W.; Kiens, B.; Nielsen, J.B. Acute exercise improves motor memory: Exploring potential biomarkers. Neurobiol. Learn. Mem. 2014, 116, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.S.; Li, W.P.; Yao, Z.B.; Zhou, X.F. Deprivation of endogenous brain derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain Res. 1999, 835, 259–265. [Google Scholar] [CrossRef]
- Cirulli, F.; Berry, A.; Chiarotti, F.; Alleva, E. Intrahippocampal administration of BDNF in adult rats affects short-term behavioral plasticity in the Morris water maze and performance in the elevated plus-maze. Hippocampus 2004, 14, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Piepmeier, A.T.; Etnier, J.L. Brain-derived neurotrophic factor (BDNF) as a potential mechanism of the effects of acute exercise on cognitive performance. J. Sport Health Sci. 2015, 4, 14–23. [Google Scholar] [CrossRef]
- Gomez-Pinilla, F.; Vaynman, S.; Ying, Z. Brain-derived neurotrophic factor functions as a metabotrophin to mediate the effects of exercise on cognition. Eur. J. Neurosci. 2008, 28, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Ferris, L.T.; Williams, J.S.; Shen, C.L. The effect of acute exercise on serum brain-derived neurotrophic factor levels and cognitive function. Med. Sci. Sports Exerc. 2007, 9, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Seifert, T.; Brassard, P.; Wissenberg, M.; Rasmussen, P.; Nordby, P.; Stallknecht, B.; Adser, H.; Jacobsen, A.H.; Pilegaard, H.; Nielsen, H.B.; et al. Endurance training enhances BDNF release fron the human brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R372–R377. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Okamoto, M.; Shibato, J.; Lee, M.C.; Matsui, T.; Rakwal, R.; Soya, H. Long-Term Mild, rather than Intense, Exercise Enhances Adult Hippocampal Neurogenesis and Greatly Changes the Transcriptomic Profile of the Hippocampus. PLoS ONE 2015, 10, e0128720. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.J.; Brasel, J.; Hintz, R.L.; Mohan, S.; Cooper, D.M. Effect of brief low- and high-intensity exercise on circulating insulin-like growth factor (IGF) I, II and IGF-binding protein 3 and its proteolysis in young healthy men. J. Clin. Endocrinol. Metab. 1996, 81, 3492–3497. [Google Scholar]
- Carro, E.; Nuñez, A.; Busiguina, S.; Torres-Aleman, I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J. Neurosci. 2000, 20, 2926–2933. [Google Scholar] [PubMed]
- Lista, I.; Sorrentino, G. Biological mechanisms of physical activity in preventing cognitive decline. Cell. Mol. Neurobiol. 2010, 30, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Byth, L.A. Ca2+- and CaMKII-mediated processes in early LTP. Ann. Neurosci. 2014, 21, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Ataei, N.; Sabzghabaee, A.M.; Movahedian, A. Calcium/Calmodulin-dependent Protein Kinase II is a Ubiquitous Molecule in Human Long-term Memory Synaptic Plasticity: A Systematic Review. Int. J. Prev. Med. 2015, 6. [Google Scholar] [CrossRef]
- Slemmon, J.R.; Morgan, J.I.; Fullerton, S.M.; Danho, W.; Hilbush, B.S.; Wengenack, T.M. Camstatins are peptide antagonists of calmodulin based upon a conserved structural motif in PEP-19, neurogranin, and neuromodulin. J. Biol. Chem. 1996, 271, 15911–15917. [Google Scholar] [CrossRef] [PubMed]
- Slemmon, J.R.; Feng, B.; Erhardt, J.A. Small proteins that modulate calmodulin-dependent signal transduction: Effects of PEP-19, neuromodulin, and neurogranin on enzyme activation and cellular homeostasis. Mol. Neurobiol. 2000, 22, 99–113. [Google Scholar] [CrossRef]
- Morgan, M.A.; Morgan, J.I. Pcp4l1 contains an auto-inhibitory element that prevents its IQ motif from binding to calmodulin. J. Neurochem. 2012, 121, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xiong, L.W.; El Ayadi, A.; Boehning, D.; Putkey, J.A. The calmodulin regulator protein, PEP-19, sensitizes ATP-induced Ca2+ release. J. Biol. Chem. 2013, 288, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Di Liegro, C.M.; Schiera, G.; di Liegro, I. Regulation of RNA transport, localization and translation in the nervous system of mammals. Int. J. Mol. Med. 2014, 33, 747–762. [Google Scholar] [PubMed]
- Saladino, P.; di Liegro, C.M.; Proia, P.; Sala, A.; Schiera, G.; Lo Cicero, A.; di Liegro, I. RNA-binding activity of the rat calmodulin-binding PEP-19 protein and of the long PEP-19 isoform. Int. J. Mol. Med. 2012, 29, 141–145. [Google Scholar] [PubMed]
- Dalsgaard, M.K.; Quistorff, B.; Danielsen, E.R.; Selner, C.; Vogelsang, T.; Secher, N.H. A reduced cerebral metabolic ratio in exercise reflects metabolism and not accumulation of lactate within the human brain. J. Physiol. 2004, 554, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Van Hall, G.; Strømstad, M.; Rasmussen, P.; Jans, O.; Zaar, M.; Gam, C.; Quistorff, B.; Secher, N.H.; Nielsen, H.B. Blood lactate is an important energy source for the human brain. J. Cereb. Blood Flow Metab. 2009, 29, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Coco, M.; Alagona, G.; Rapisarda, G.; Costanzo, E.; Calogero, R.A.; Perciavalle, V. Elevated blood lactate is associated with increased motor cortex excitability. Somatosens. Mot. Res. 2010, 27, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, T.; Schulte, S.; Sperlich, B.; Achtzehn, S.; Fricke, H.; Strüder, H.K. Lactate infusion at rest increases BDNF blood concentration in humans. Neurosci. Lett. 2011, 488, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ruchti, E.; Petit, J.M.; Jourdain, P.; Grenningloh, G.; Allaman, I.; Magistretti, P.J. Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc. Natl. Acad. Sci. USA 2014, 111, 12228–12233. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, L.; Puyal, J.; Chatton, J.Y. Lactate modulates the activity of primary cortical neurons through a receptor-mediated pathway. PLoS ONE 2013, 8, e71721. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, K.H.; Morland, C.; Puchades, M.; Holm-Hansen, S.; Hagelin, E.M.; Lauritzen, F.; Attramadal, H.; Storm-Mathisen, J.; Gjedde, A.; Bergersen, L.H. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex. 2014, 24, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Morland, C.; Lauritzen, K.H.; Puchades, M.; Holm-Hansen, S.; Andersson, K.; Gjedde, A.; Attramadal, H.; Storm-Mathisen, J.; Bergersen, L.H. The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: Expression and action in brain. J. Neurosci. Res. 2015, 93, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Barros, L.F. Metabolic signaling by lactate in the brain. Trends Neurosci. 2013, 36, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Philp, A.; Macdonald, A.L.; Watt, P.W. Lactate-a signal coordinating cell and systemic function. J. Exp. Biol. 2005, 208, 4561–4575. [Google Scholar] [CrossRef] [PubMed]
- Mosienko, V.; Teschemacher, A.G.; Kasparov, S. Is L-lactate a novel signaling molecule in the brain? J. Cereb. Blood Flow. Metab. 2015, 35, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.R.; Choi, H.B.; Rungta, R.L.; Ellis-Davies, G.C.; MacVicar, B.A. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 2008, 456, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Rinholm, J.E.; Hamilton, N.B.; Kessaris, N.; Richardson, W.D.; Bergersen, L.H.; Attwell, D. Regulation of oligodendrocyte development and myelination by glucose and lactate. J. Neurosci. 2011, 31, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Arbeláez, A.M.; Cryer, P.E. Lactate and the mechanism of hypoglycemia-associated autonomic failure in diabetes. Diabetes 2013, 62, 3999–4001. [Google Scholar] [CrossRef] [PubMed]
- Maran, A.; Cranston, I.; Lomas, J.; Macdonald, I.; Amiel, S.A. Protection by lactate of cerebral function during hypoglycemia. Lancet 1994, 343, 16–20. [Google Scholar] [CrossRef]
- Veneman, T.; Mitrakou, A.; Mokan, M.; Cryer, P.; Gerich, J. Effect of hyperketonemia and hyperlacticacidemia on symptoms, cognitive dysfunction, and counterregulatory hormone responses during hypoglycemia in normal humans. Diabetes 1994, 43, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Boumezbeur, F.; Peterson, K.F.; Cline, G.W.; Mason, G.F.; Behar, K.L.; Shulman, G.I.; Rothman, D.L. The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13C nuclear magnetic resonance spectroscopy. J. Neurosci. 2010, 30, 13983–13991. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.I.; Jiang, L.; Herman, P.; Zhao, C.; Sanganahalli, B.G.; Mason, G.F.; Hyder, F.; Rothman, D.L.; Sherwin, R.S.; Behar, K.L. Lactate preserves neuronal metabolism and function following antecedent recurrent hypoglycemia. J. Clin. Investig. 2013, 123, 1988–1998. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A.; Martin, N.A. Cerebral metabolism following traumatic brain injury: New discoveries with implications for treatment. Front. Neurosci. 2015, 8, 408. [Google Scholar] [CrossRef]
- Jalloh, I.; Carpenter, K.L.; Grice, P.; Howe, D.J.; Mason, A.; Gallagher, C.N.; Helmy, A.; Murphy, M.P.; Menon, D.K.; Carpenter, T.A.; et al. Glycolysis and the pentose phosphate pathway after human traumatic brain injury: Microdialysis studies using 1,2-13C2 glucose. J. Cereb. Blood Flow Metab. 2015, 35, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.C.; Martin, N.A.; Horning, M.A.; McArthur, D.L.; Hovda, DA.; Vespa, P.; Brooks, G.A. Lactate: Brain fuel in human traumatic brain injury: A comparison with normal healthy control subjects. J. Neurotrauma 2015, 32, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.N.; Carpenter, K.L.H.; Grice, P.; Howe, D.J.; Mason, A.; Timofeev, I.; Menon, D.K.; Kirkpatrick, P.J.; Pickard, J.D.; Sutherland, G.R.; et al. The human brain utilizes lactate via the tricarboxylic acid cycle: A 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain 2009, 132, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.L.; Jalloh, I.; Hutchinson, P.J. Glycolysis and the significance of lactate in traumatic brain injury. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Bouzat, P.; Sala, N.; Suys, T.; Zerfauth, J.B.; Marques-Vidal, P.; Feihl, F.; Bloch, J.; Messerer, M.; Levivier, M.; Meuli, R.; et al. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med. 2014, 40, 412–421. [Google Scholar] [CrossRef]
- Lama, S.; Auer, R.N.; Tyson, R.; Gallagher, C.N.; Tomanek, B.; Sutherland, G.R. Lactate storm marks cerebral metabolism following brain trauma. J. Biol. Chem. 2014, 289, 20200–20208. [Google Scholar] [CrossRef] [PubMed]
- Ros, J.; Pecinska, N.; Alessandri, B.; Landolt, H.; Fillenz, M. Lactate reduces glutamate-induced neurotoxicity in rat cortex. J. Neurosci. Res. 2001, 66, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Berthet, C.; Castillo, X.; Magistretti, P.J.; Hirt, L. New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: Extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc. Dis. 2012, 34, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Castillo, X.; Rosafio, K.; Wyss, M.T.; Drandarov, K.; Buck, A.; Pellerin, L.; Weber, B.; Hirt, L. A probable dual mode of action for both l- and d-lactate neuroprotection in cerebral ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Detka, J.; Kurek, A.; Kucharczyk, M.; Głombik, K.; Basta-Kaim, A.; Kubera, M.; Lasoń, W.; Budziszewska, B. Brain glucose metabolism in an animal model of depression. Neuroscience 2015, 295, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Bosoi, C.R.; Zwingmann, C.; Marin, H.; Parent-Robitaille, C.; Huynh, J.; Tremblay, M.; Rose, C.F. Increased brain lactate is central to the development of brain edema in rats with chronic liver disease. J. Hepatol. 2014, 60, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Bosoi, C.R.; Rose, C.F. Elevated cerebral lactate: Implications in the pathogenesis of hepatic encephalopathy. Metab. Brain Dis. 2014, 29, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Redjems-Bennani, N.; Jeandel, C.; Lefebvre, E.; Blain, H.; Vidailhet, M.; Guéant, J.L. Abnormal substrate levels that depend upon mitochondrial function in cerebrospinal fluid from Alzheimer patients. Gerontology 1998, 44, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Xu, G.; Weigel-Van Aken, K.A.K. Lactic Acid Induces Aberrant Amyloid Precursor Protein Processing by Promoting Its Interaction with Endoplasmic Reticulum Chaperone Proteins. PLoS ONE 2010, 5, e13820. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Junior, R.S.; Cevada, T.; Oliveira, B.R.; Lattari, E.; Portugal, E.M.; Carvalho, A.; Deslandes, A.C. We need to move more: Neurobiological hypotheses of physical exercise as a treatment for Parkinson’s disease. Med. Hypotheses 2015, 85, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Herbst, E.A.F.; Holloway, G.P. Exercise training normalizes mitochondrial respiratory capacity within the striatum of R6/1 model of Huntington’s Disease. Neuroscience 2015, 303, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Wens, I.; Dalgas, U.; Vandennabeele, F.; Verboven, K.; Hansen, D.; Deckx, N.; Cools, N.; Eijnde, B.O. High intensity aerobic and resistance exercise can improve glucose tolerance in persons with Multiple Sclerosis: A randomized controlled trial. Am. J. Phys. Med. Rehabil. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Venkatraghavan, L.; Mariappan, R.; Ebinu, J.; Meng, Y.; Khan, O.; Tung, T.; Reyhani, S.; Bernstein, M.; Zadeh, G. Serum lactate as a potential biomarker of non-glial brain tumors. J. Clin. Neurosci. 2015, 22, 1625–1627. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Proia, P.; Di Liegro, C.M.; Schiera, G.; Fricano, A.; Di Liegro, I. Lactate as a Metabolite and a Regulator in the Central Nervous System. Int. J. Mol. Sci. 2016, 17, 1450. https://doi.org/10.3390/ijms17091450
Proia P, Di Liegro CM, Schiera G, Fricano A, Di Liegro I. Lactate as a Metabolite and a Regulator in the Central Nervous System. International Journal of Molecular Sciences. 2016; 17(9):1450. https://doi.org/10.3390/ijms17091450
Chicago/Turabian StyleProia, Patrizia, Carlo Maria Di Liegro, Gabriella Schiera, Anna Fricano, and Italia Di Liegro. 2016. "Lactate as a Metabolite and a Regulator in the Central Nervous System" International Journal of Molecular Sciences 17, no. 9: 1450. https://doi.org/10.3390/ijms17091450
APA StyleProia, P., Di Liegro, C. M., Schiera, G., Fricano, A., & Di Liegro, I. (2016). Lactate as a Metabolite and a Regulator in the Central Nervous System. International Journal of Molecular Sciences, 17(9), 1450. https://doi.org/10.3390/ijms17091450