
Intra-Peritoneal Administration of Mitochondrial DNA Provokes Acute Lung Injury and Systemic Inflammation via Toll-Like Receptor 9

 ,
,
Abstract
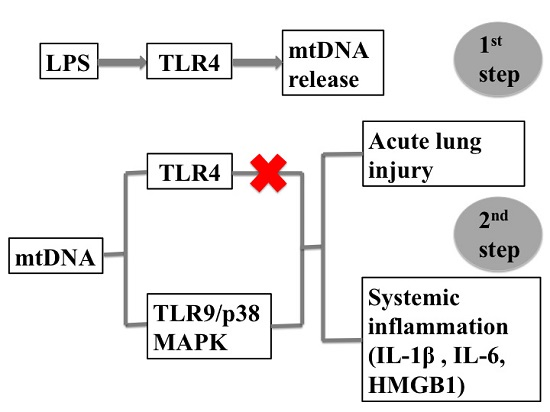
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
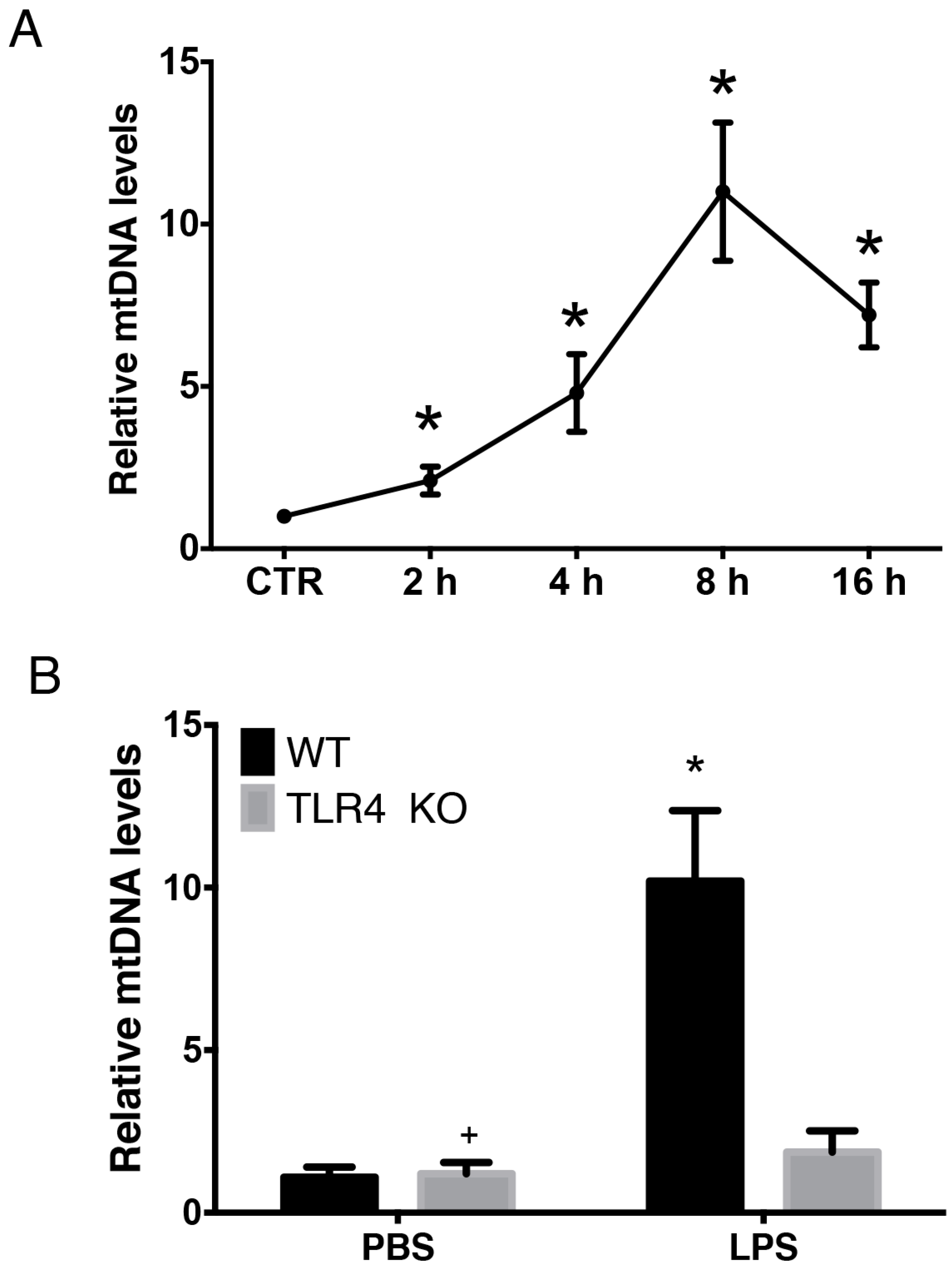
2.1. LPS-Induced Circulating Mitochondria DNA (mtDNA) is Released in a TLR4 Dependent Manner
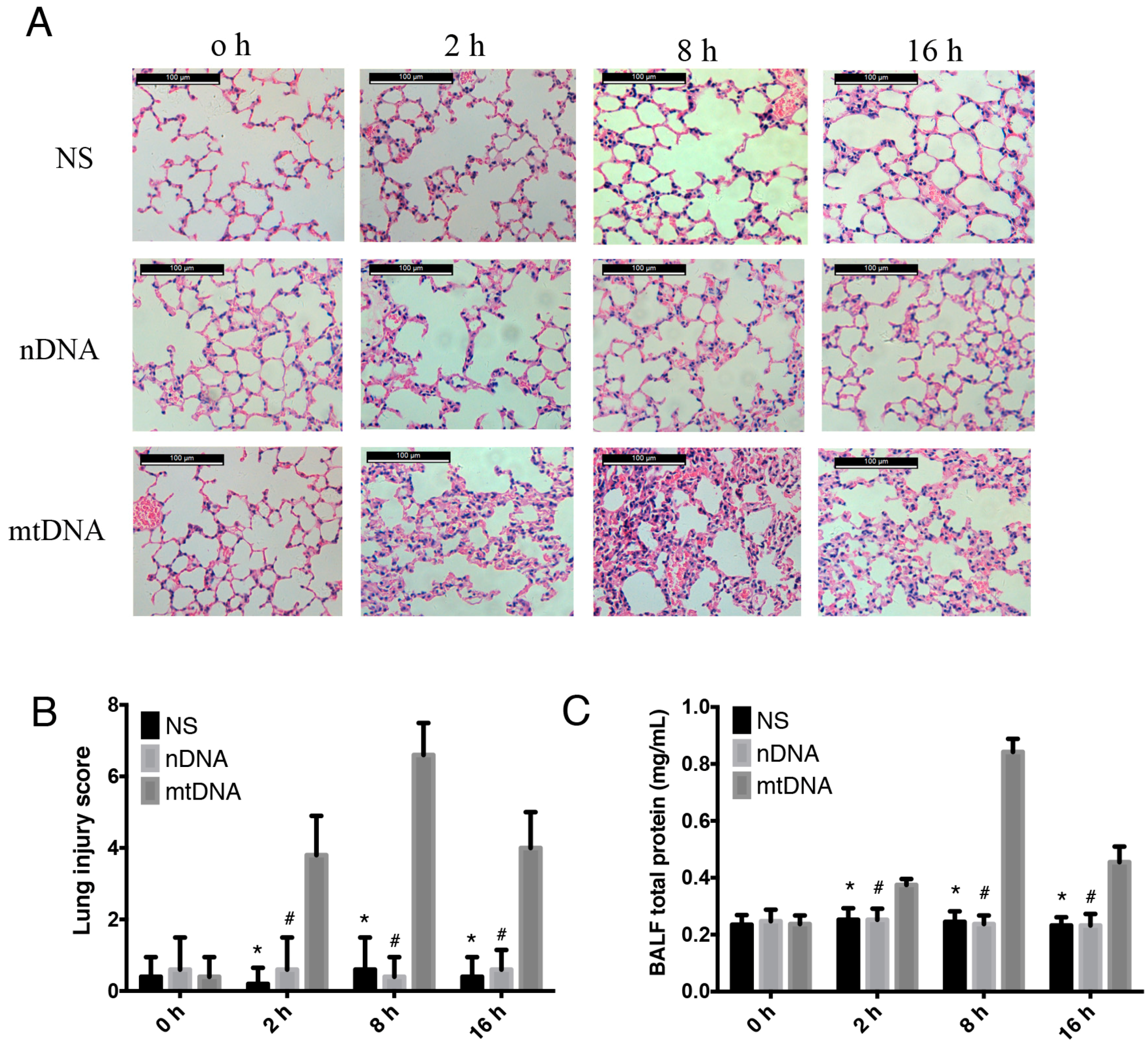
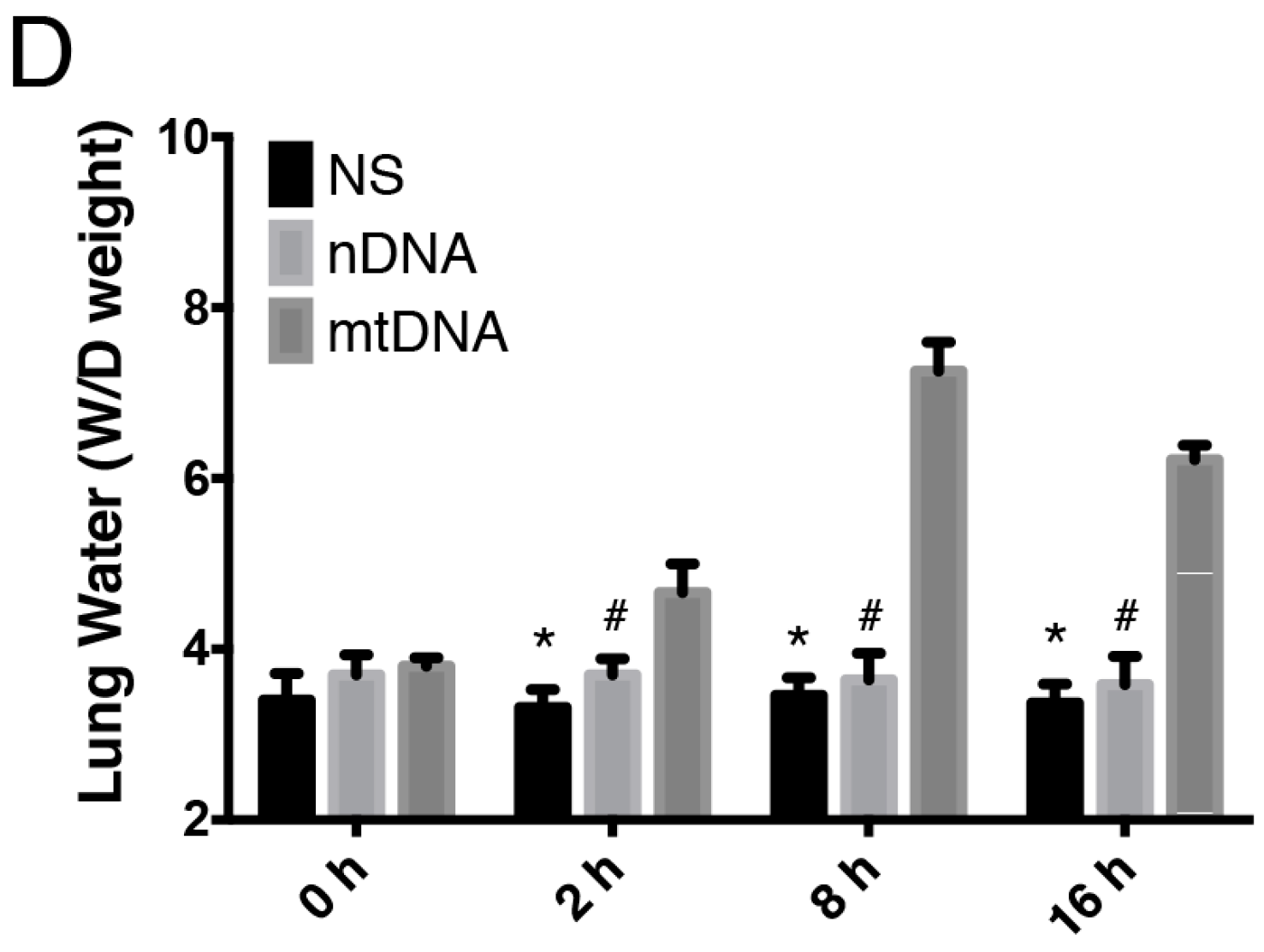
2.2. Intra-Peritoneal Administration of mtDNA Leads to Acute Lung Injury (ALI)
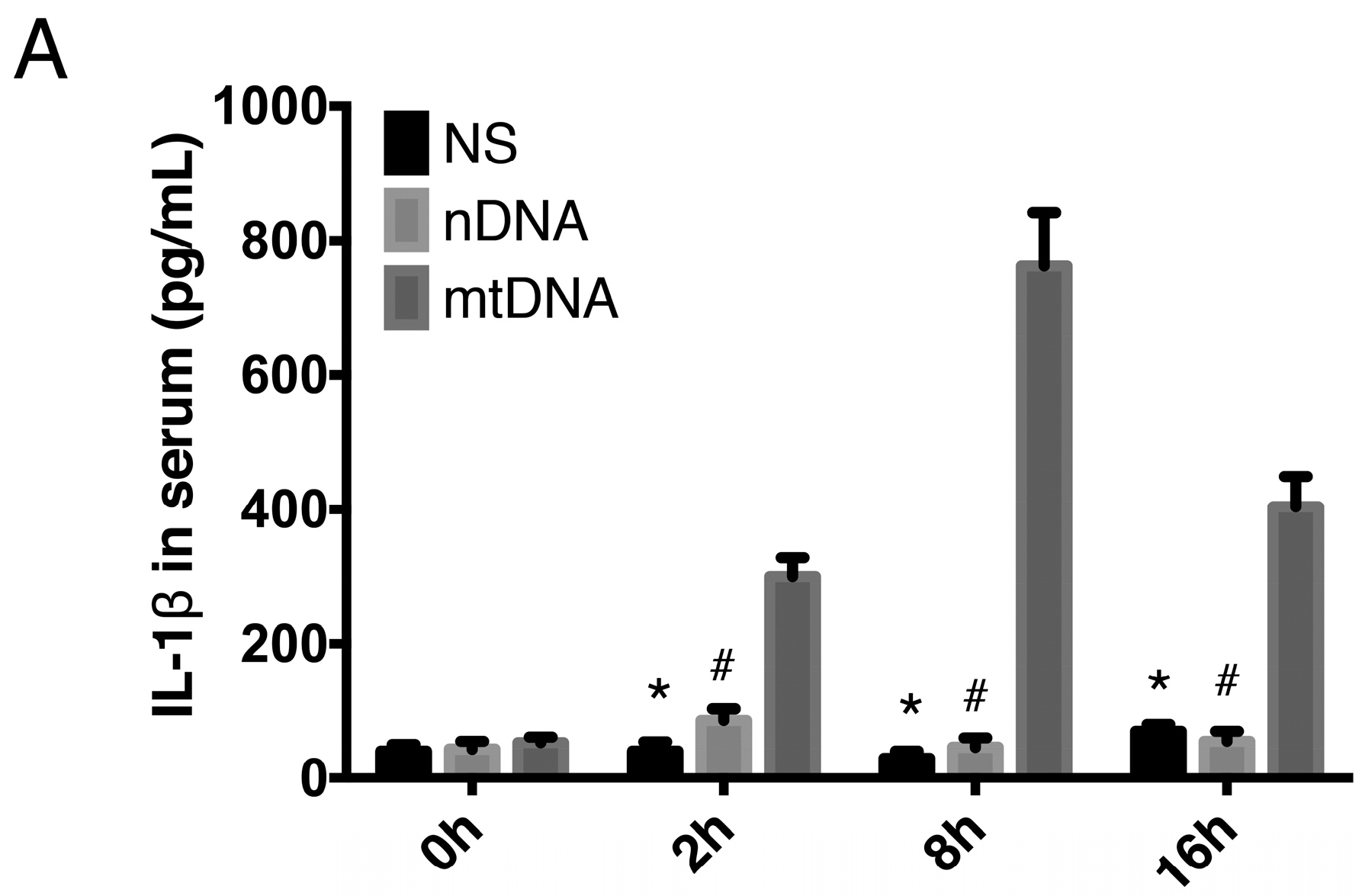
2.3. Intra-Peritoneal Administration of mtDNA Leads to Systemic Inflammation
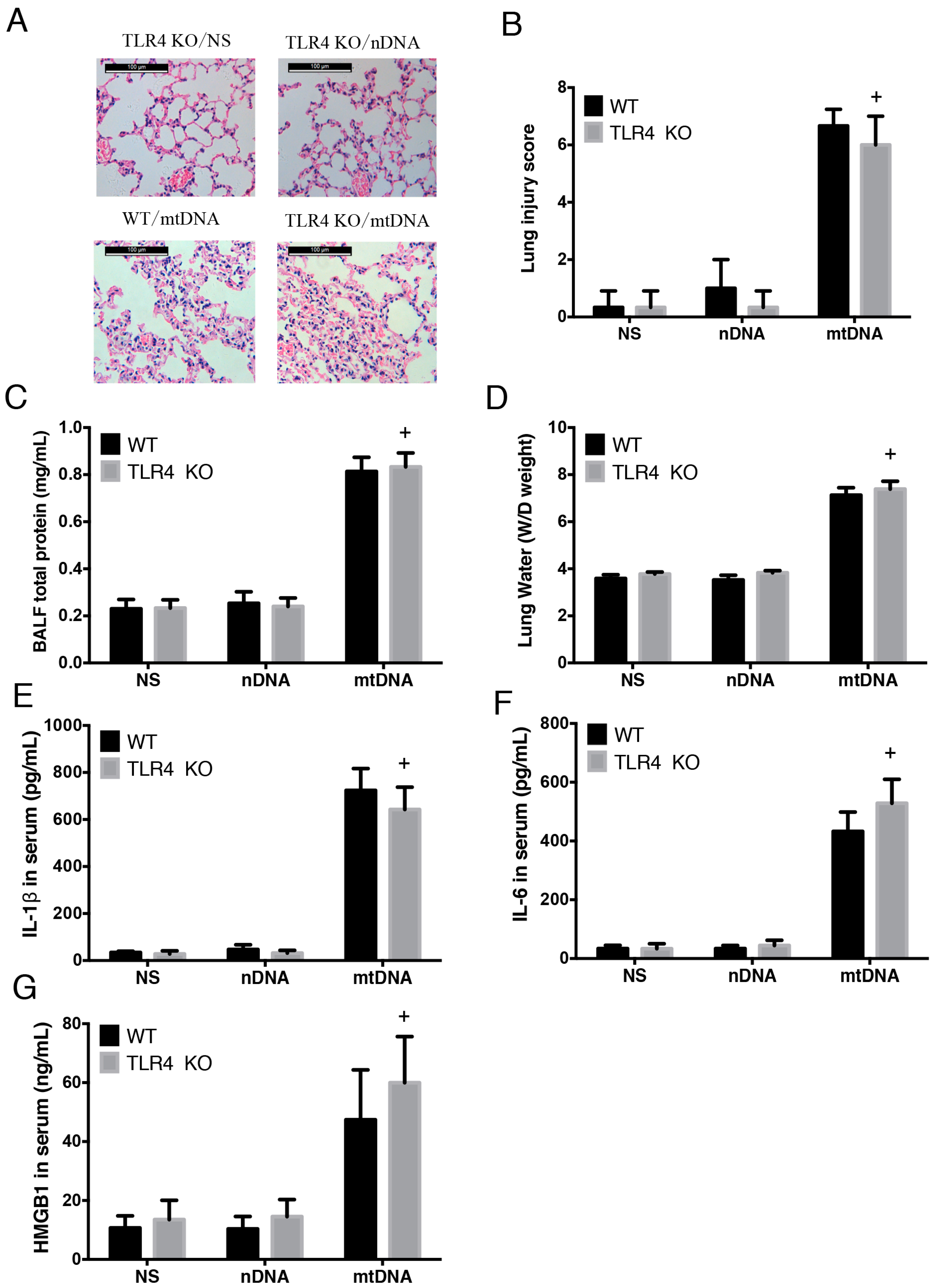
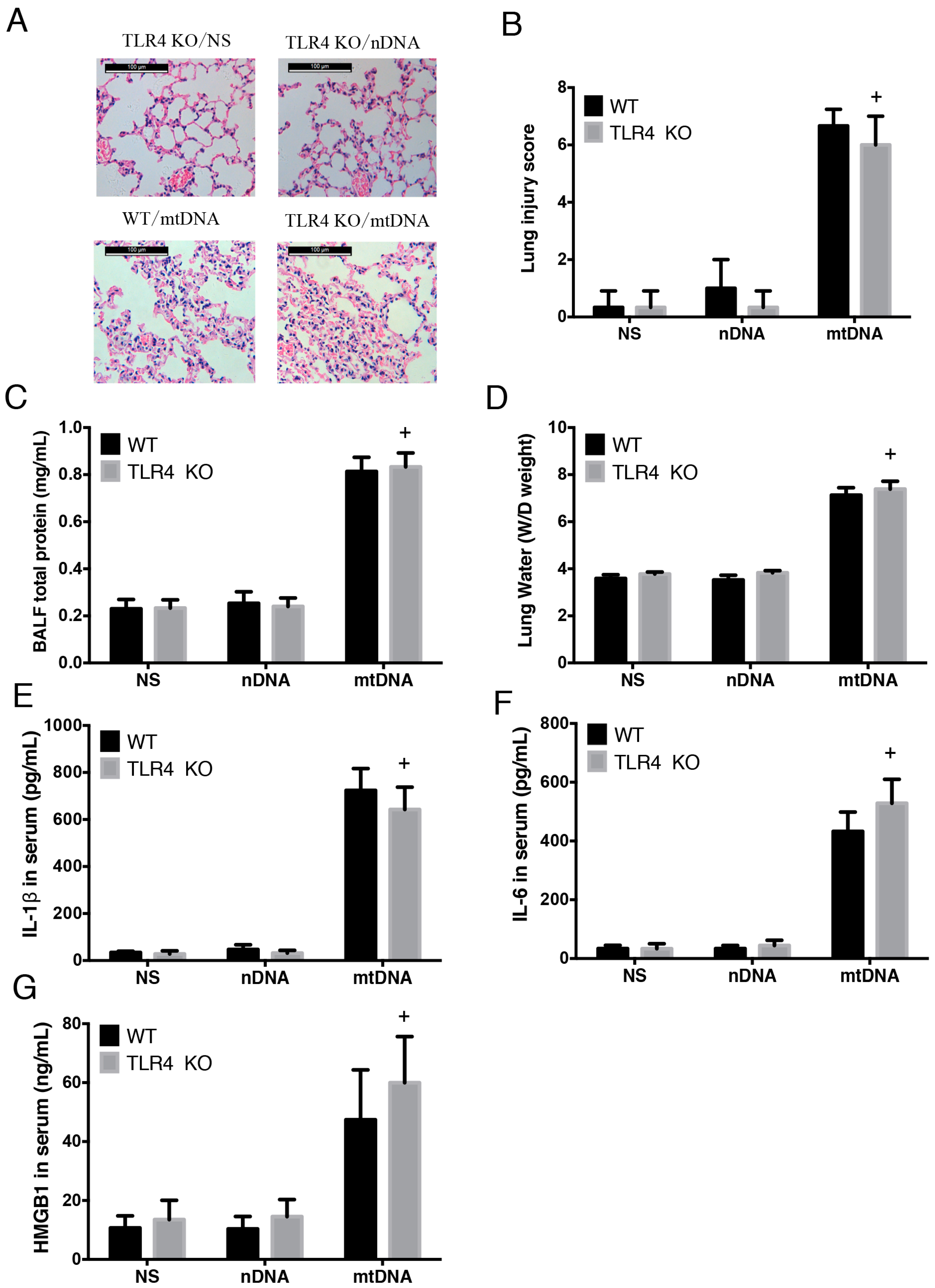
2.4. Intra-Peritoneal Administration of mtDNA Induces Acute Lung Injury and Systemic Inflammation in a TLR4-Independent Manner
2.5. Intra-Peritoneal Administration of mtDNA Induces Acute Lung Injury and Systemic Inflammation in a TLR9-Dependent Manner
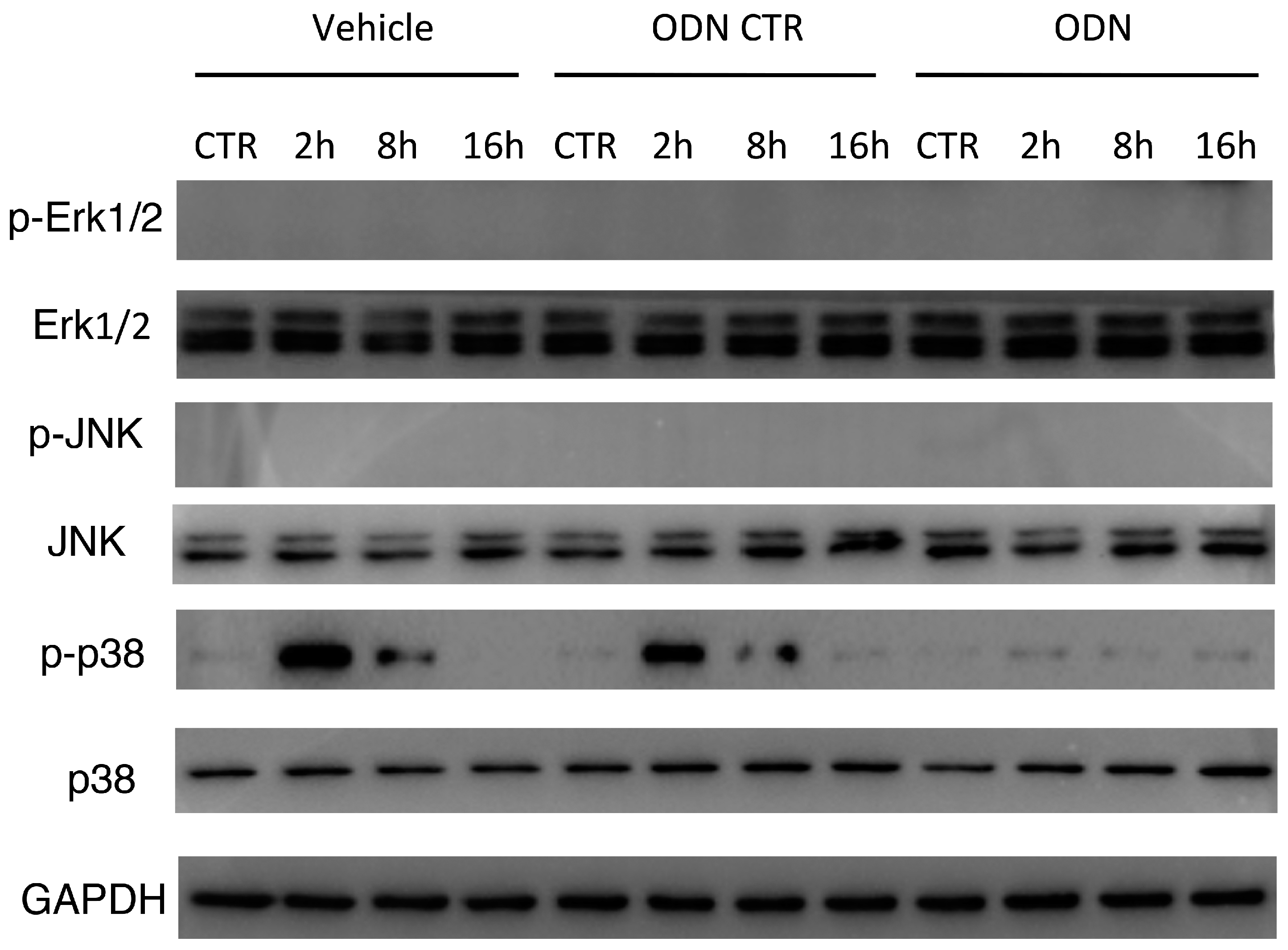
2.6. Intra-Peritoneal Administration of mtDNA Leads to p38 MAPK Activation via TLR9
3. Discussion
4. Materials and Methods
4.1. Main Reagents
4.2. Animals
4.3. In Vivo Experimental Design
4.4. Preparation of mtDNA and nDNA
4.5. Hematoxylin and Eosin Staining
4.6. Quantification of Inflammatory Cytokines
4.7. Circulating DNA Purification And Quantitative Real-Time PCR
- 18S forward, 5′-TAGAGGGACAAGTGGCGTTC-3′;
- 18S reverse, 5′-CGCTGAGCCAGTCAGTGT-3′;
- mtCOI forward, 5′-GCCCCAGATATAGCATTCCC-3′;
- mtCOI reverse, 5′-GTTCATCCTGTTCCT GCTCC-3′.
4.8. Tissue Protein Extraction and Western Blotting
4.9. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Angus, D.C.; Pereira, C.A.; Silva, E. Epidemiology of severe sepsis around the world. Endocr. Metab. Immune Disord. Drug Targets 2006, 6, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992, 101, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Xie, G.; Yao, S.; Wu, X.; Guo, Q.; Gu, M.; Fang, Q.; Xu, Q.; Wang, D.; Jin, Y.; et al. Epidemiology of severe sepsis in critically ill surgical patients in ten university hospitals in China. Crit. Care Med. 2007, 35, 2538–2546. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, L.C. Mitochondrial dysfunction during sepsis. Endocr. Metab. Immune Disord. Drug Targets 2010, 10, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Singer, M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 2014, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Koenitzer, J.R.; Freeman, B.A. Redox signaling in inflammation: Interactions of endogenous electrophiles and mitochondria in cardiovascular disease. Ann. N. Y. Acad. Sci. 2010, 1203, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Kazama, H.; Ricci, J.E.; Herndon, J.M.; Hoppe, G.; Green, D.R.; Ferguson, T.A. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008, 29, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial damps cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, L.; Karlsson, J.; Karlsson, A.; Rabiet, M.J.; Boulay, F.; Fu, H.; Bylund, J.; Dahlgren, C. Serum amyloid a mediates human neutrophil production of reactive oxygen species through a receptor independent of formyl peptide receptor like-1. J. Leukoc. Biol. 2008, 83, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Pullerits, R.; Bokarewa, M.; Jonsson, I.M.; Verdrengh, M.; Tarkowski, A. Extracellular cytochrome c, a mitochondrial apoptosis-related protein, induces arthritis. Rheumatology 2005, 44, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Struck, J.; Uhlein, M.; Morgenthaler, N.G.; Furst, W.; Hoflich, C.; Bahrami, S.; Bergmann, A.; Volk, H.D.; Redl, H. Release of the mitochondrial enzyme carbamoyl phosphate synthase under septic conditions. Shock 2005, 23, 533–538. [Google Scholar] [PubMed]
- Yu, E.P.; Bennett, M.R. Mitochondrial DNA damage and atherosclerosis. Trends Endocrinol. Metab. 2014, 25, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Cossarizza, A.; Pinti, M.; Nasi, M.; Gibellini, L.; Manzini, S.; Roat, E.; De Biasi, S.; Bertoncelli, L.; Montagna, J.P.; Bisi, L.; et al. Increased plasma levels of extracellular mitochondrial DNA during HIV infection: A new role for mitochondrial damage-associated molecular patterns during inflammation. Mitochondrion 2011, 11, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.W.; Lin, T.K.; Chen, S.D.; Chang, W.N.; Wang, H.C.; Yang, T.M.; Lin, Y.J.; Jan, C.R.; Huang, C.R.; Liou, C.W.; et al. The value of serial plasma nuclear and mitochondrial DNA levels in patients with acute ischemic stroke. Clin. Chim. Acta 2011, 412, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Walko, T.D., 3rd; Bola, R.A.; Hong, J.D.; Au, A.K.; Bell, M.J.; Kochanek, P.M.; Clark, R.S.; Aneja, R.K. Cerebrospinal fluid mitochondrial DNA: A novel damp in pediatric traumatic brain injury. Shock 2014, 41, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Helbling, D.; Buchaklian, A.; Wang, J.; Wong, L.J.; Dimmock, D. Reduced mitochondrial DNA content and heterozygous nuclear gene mutations in patients with acute liver failure. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.T.; Hsiao, S.Y.; Tsai, T.C.; Su, C.M.; Chang, W.N.; Huang, C.R.; Wang, H.C.; Lin, W.C.; Chang, H.W.; Lin, Y.J.; et al. Plasma nuclear and mitochondrial DNA levels as predictors of outcome in severe sepsis patients in the emergency room. J. Transl. Med. 2012, 10, 130. [Google Scholar] [CrossRef] [PubMed]
- Sursal, T.; Stearns-Kurosawa, D.J.; Itagaki, K.; Oh, S.Y.; Sun, S.; Kurosawa, S.; Hauser, C.J. Plasma bacterial and mitochondrial DNA distinguish bacterial sepsis from sterile systemic inflammatory response syndrome and quantify inflammatory tissue injury in nonhuman primates. Shock 2013, 39, 55–62. [Google Scholar] [PubMed]
- MacKenzie, E.J. Epidemiology of injuries: Current trends and future challenges. Epidemiol. Rev. 2000, 22, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gaspar, M.; Santolaria, F.; Jarque-Lopez, A.; Gonzalez-Reimers, E.; Milena, A.; de la Vega, M.J.; Rodriguez-Rodriguez, E.; Gomez-Sirvent, J.L. Prognostic value of cytokines in SIRS general medical patients. Cytokine 2001, 15, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.; Burn, D.J.; Gordon, C.; Swan, C.; Chinnery, P.F.; Baudouin, S.V. Fall in circulating mononuclear cell mitochondrial DNA content in human sepsis. Intensive Care Med. 2010, 36, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, K.; Kox, M.; Scheffer, G.J.; Pickkers, P. Plasma nuclear and mitochondrial DNA levels, and markers of inflammation, shock, and organ damage in patients with septic shock. Shock 2015, 45, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Kyung, S.Y.; Rogers, A.J.; Gazourian, L.; Youn, S.; Massaro, A.F.; Quintana, C.; Osorio, J.C.; Wang, Z.; Zhao, Y.; et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: Derivation and validation. PLoS Med. 2013, 10, e1001577. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sursal, T.; Adibnia, Y.; Zhao, C.; Zheng, Y.; Li, H.; Otterbein, L.E.; Hauser, C.J.; Itagaki, K. Mitochondrial damps increase endothelial permeability through neutrophil dependent and independent pathways. PLoS ONE 2013, 8, e59989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Wu, G.; Yao, Y.; Zeng, J.; Shi, D.; Lv, T.; Luo, L.; Song, Y. Intratracheal administration of mitochondrial DNA directly provokes lung inflammation through the TLR9-p38 MAPK pathway. Free Radic. Biol. Med. 2015, 83, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Unuma, K.; Aki, T.; Funakoshi, T.; Hashimoto, K.; Uemura, K. Extrusion of mitochondrial contents from lipopolysaccharide-stimulated cells: Involvement of autophagy. Autophagy 2015, 11, 1520–1536. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, R.; Li, J.; Zhang, L.; Ding, G.; Luo, P.; He, S.; Dong, Y.; Jiang, W.; Lu, Y.; et al. Antimalarial artesunate protects sepsis model mice against heat-killed Escherichia coli challenge by decreasing TLR4, TLR9 mRNA expressions and transcription factor NF-κB activation. Int. Immunopharmacol. 2008, 8, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Fan, J. Pattern recognition receptor-dependent mechanisms of acute lung injury. Mol. Med. 2010, 16, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.M.; Dellinger, R.P.; Townsend, S.R.; Linde-Zwirble, W.T.; Marshall, J.C.; Bion, J.; Schorr, C.; Artigas, A.; Ramsay, G.; Beale, R.; et al. The surviving sepsis campaign: Results of an international guideline-based performance improvement program targeting severe sepsis. Crit. Care Med. 2010, 38, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Japiassu, A.M.; Santiago, A.P.; d’Avila, J.C.; Garcia-Souza, L.F.; Galina, A.; Castro Faria-Neto, H.C.; Bozza, F.A.; Oliveira, M.F. Bioenergetic failure of human peripheral blood monocytes in patients with septic shock is mediated by reduced F1FO adenosine-5′-triphosphate synthase activity. Crit. Care Med. 2011, 39, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Brealey, D.; Singer, M. Mitochondrial dysfunction in sepsis. Curr. Infect. Dis. Rep. 2003, 5, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Chamorro-Vina, C.; Ruiz, J.R.; Santana-Sosa, E.; Gonzalez Vicent, M.; Madero, L.; Perez, M.; Fleck, S.J.; Perez, A.; Ramirez, M.; Lucia, A. Exercise during hematopoietic stem cell transplant hospitalization in children. Med. Sci. Sports Exerc. 2010, 42, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.A.; Rovere-Querini, P. The mitochondrion—A trojan horse that kicks off inflammation? N. Engl. J. Med. 2010, 362, 2132–2134. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, S.; Kudo, D.; Yamada, M.; Miyagawa, N.; Furukawa, H.; Kushimoto, S. Plasma mitochondrial DNA levels in patients with trauma and severe sepsis: Time course and the association with clinical status. J. Crit. Care 2013, 28, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, C. Origins. On the origin of eukaryotes. Science 2009, 325, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. Cytoplasmic DNA innate immune pathways. Immunol. Rev. 2011, 243, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Shao, B.; He, Z.; Ye, T.; Luo, M.; Sang, Y.; Liang, X.; Wang, W.; Luo, S.; Yang, S.; et al. Cationic nanocarriers induce cell necrosis through impairment of Na+/K+-ATPase and cause subsequent inflammatory response. Cell Res. 2015, 25, 237–253. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Wang, G.L.; Li, J. Complete F-type mitochondrial genome of Chinese freshwater mussels Lamprotula gottschei. Mitochondrial DNA 2016, 27, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [PubMed]
- Julian, M.W.; Shao, G.; Vangundy, Z.C.; Papenfuss, T.L.; Crouser, E.D. Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFα release via RAGE- and TLR9-responsive plasmacytoid dendritic cells. PLoS ONE 2013, 8, e72354. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Liu, Z.; Liu, J.; Ren, J.X.; Sun, T.S. Mitochondrial DNA induces inflammation and increases TLR9/NF κB expression in lung tissue. Int. J. Mol. Med. 2014, 33, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Dings, R.P.; Nesmelova, I.; Griffioen, A.W.; Mayo, K.H. Discovery and development of anti-angiogenic peptides: A structural link. Angiogenesis 2003, 6, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, S.H.; Lasbury, M.E.; Tschang, D.; Liao, C.P.; Durant, P.J.; Lee, C.H. Toll-like receptor 2 mediates alveolar macrophage response to pneumocystis murina. Infect. Immun. 2006, 74, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Pan, P.; Su, X.; Zhang, L.; Qin, Q.; Tan, H.; Huang, L.; Li, Y. Interferon regulatory factor-1 mediates alveolar macrophage pyroptosis during LPS-induced acute lung injury in mice. Shock 2016, 46, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Lyu, J.; Liu, S.; Huang, J.; Liu, C.; Xiang, D.; Xie, M.; Zeng, Q. Silencing of uncoupling protein 2 by small interfering RNA aggravates mitochondrial dysfunction in cardiomyocytes under septic conditions. Int. J. Mol. Med. 2015, 35, 1525–1536. [Google Scholar] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Deng, S.; Zhao, S.; Ai, Y.; Zhang, L.; Pan, P.; Su, X.; Tan, H.; Wu, D. Intra-Peritoneal Administration of Mitochondrial DNA Provokes Acute Lung Injury and Systemic Inflammation via Toll-Like Receptor 9. Int. J. Mol. Sci. 2016, 17, 1425. https://doi.org/10.3390/ijms17091425
Zhang L, Deng S, Zhao S, Ai Y, Zhang L, Pan P, Su X, Tan H, Wu D. Intra-Peritoneal Administration of Mitochondrial DNA Provokes Acute Lung Injury and Systemic Inflammation via Toll-Like Receptor 9. International Journal of Molecular Sciences. 2016; 17(9):1425. https://doi.org/10.3390/ijms17091425
Chicago/Turabian StyleZhang, Lemeng, Songyun Deng, Shuangping Zhao, Yuhang Ai, Lina Zhang, Pinhua Pan, Xiaoli Su, Hongyi Tan, and Dongdong Wu. 2016. "Intra-Peritoneal Administration of Mitochondrial DNA Provokes Acute Lung Injury and Systemic Inflammation via Toll-Like Receptor 9" International Journal of Molecular Sciences 17, no. 9: 1425. https://doi.org/10.3390/ijms17091425
APA StyleZhang, L., Deng, S., Zhao, S., Ai, Y., Zhang, L., Pan, P., Su, X., Tan, H., & Wu, D. (2016). Intra-Peritoneal Administration of Mitochondrial DNA Provokes Acute Lung Injury and Systemic Inflammation via Toll-Like Receptor 9. International Journal of Molecular Sciences, 17(9), 1425. https://doi.org/10.3390/ijms17091425