
Toxic Effects of the Major Components of Diesel Exhaust in Human Alveolar Basal Epithelial Cells (A549)



Abstract
:1. Introduction
2. Results
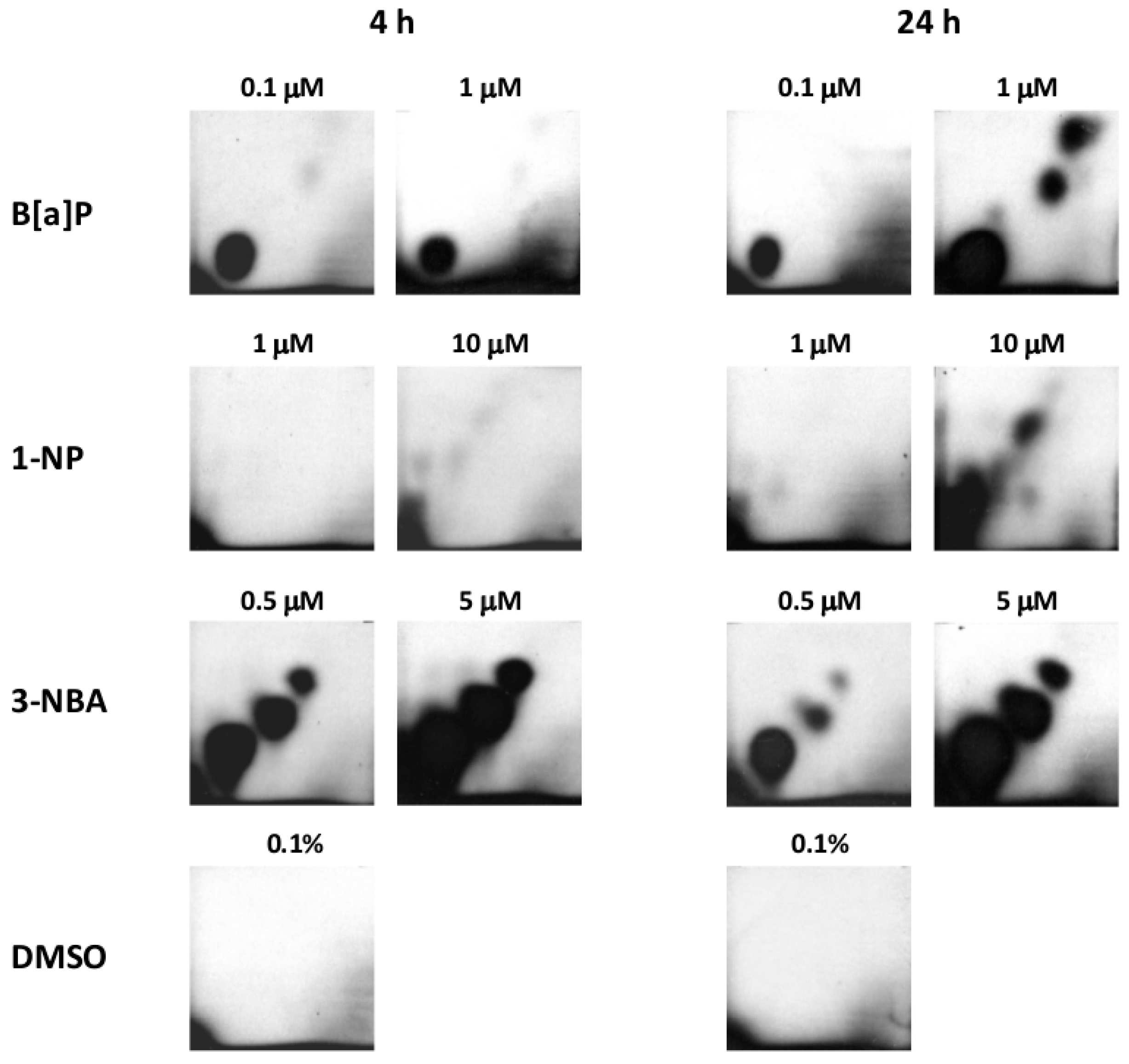
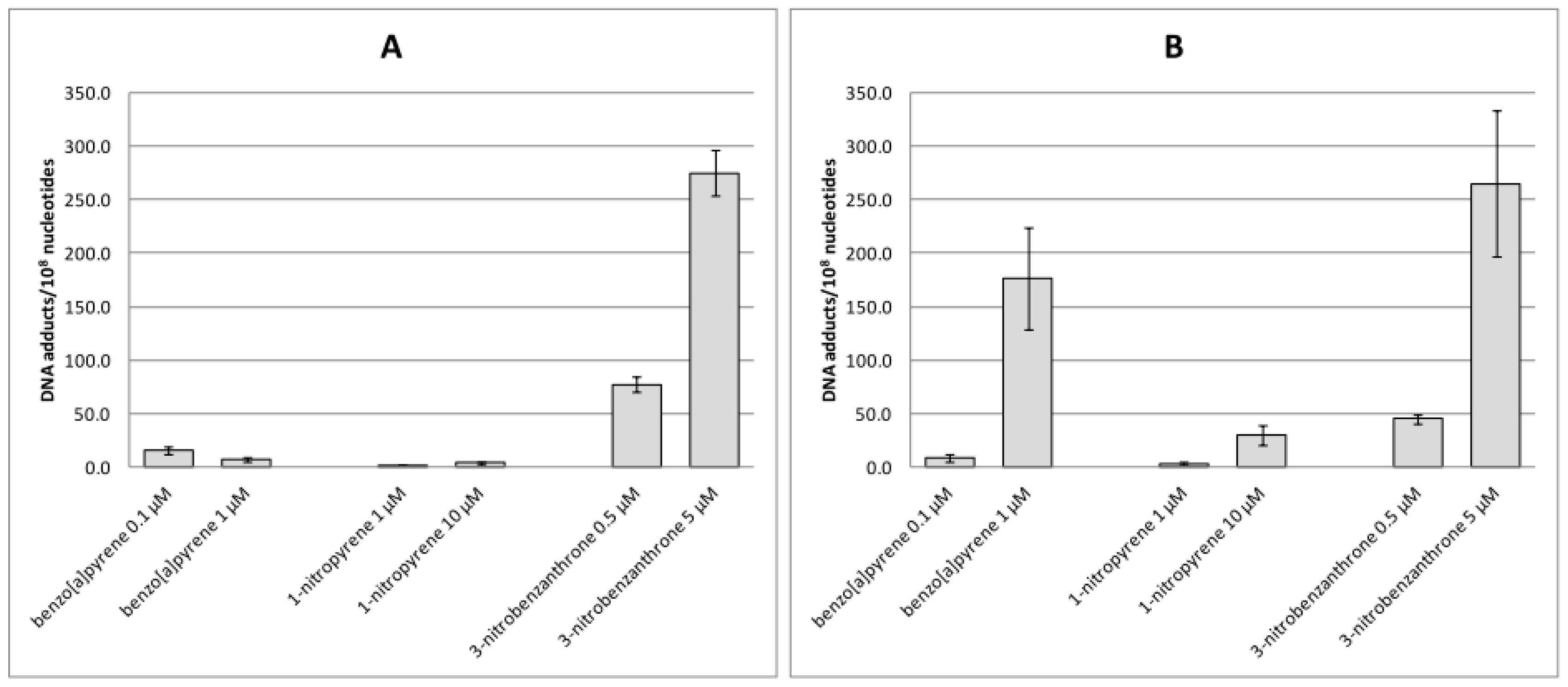
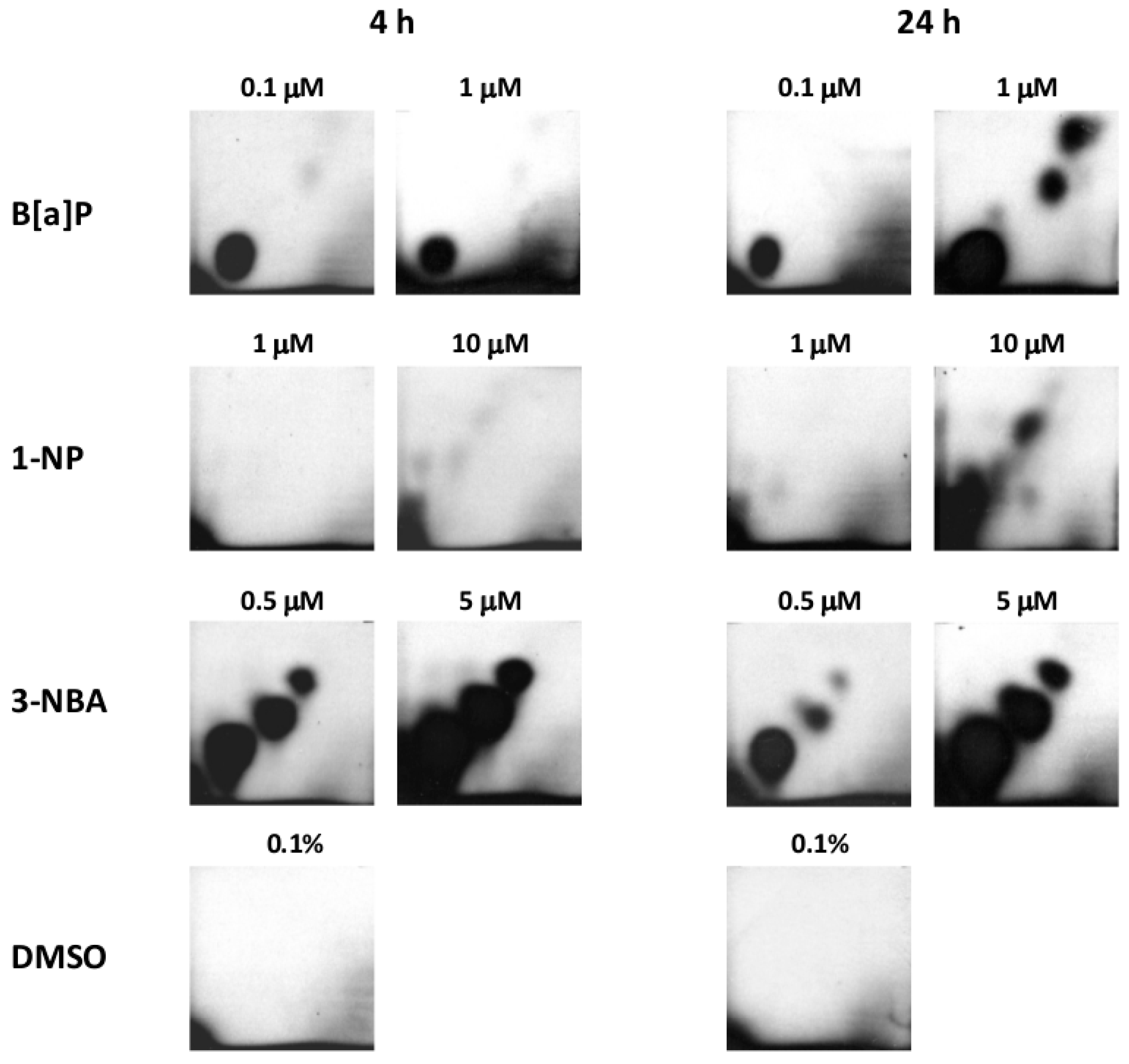
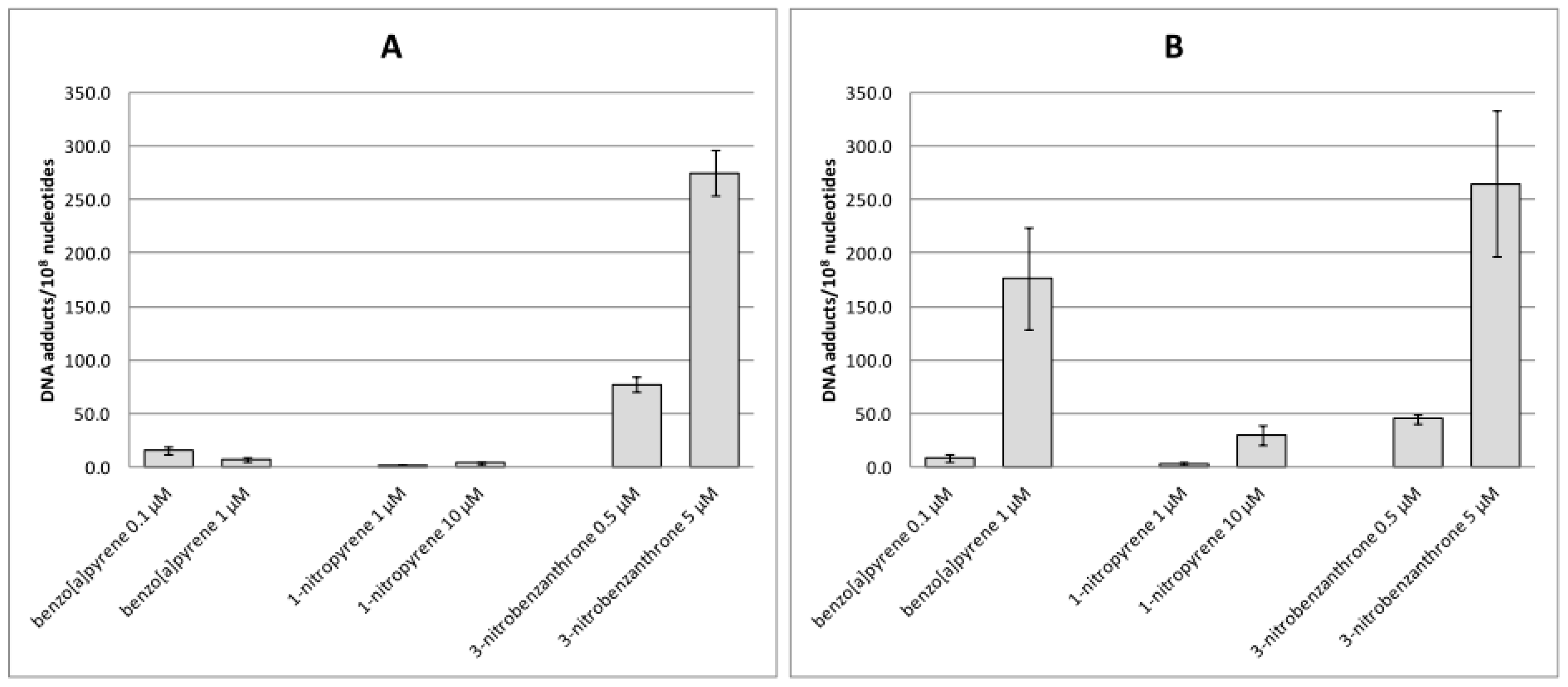
2.1. Bulky DNA Adducts
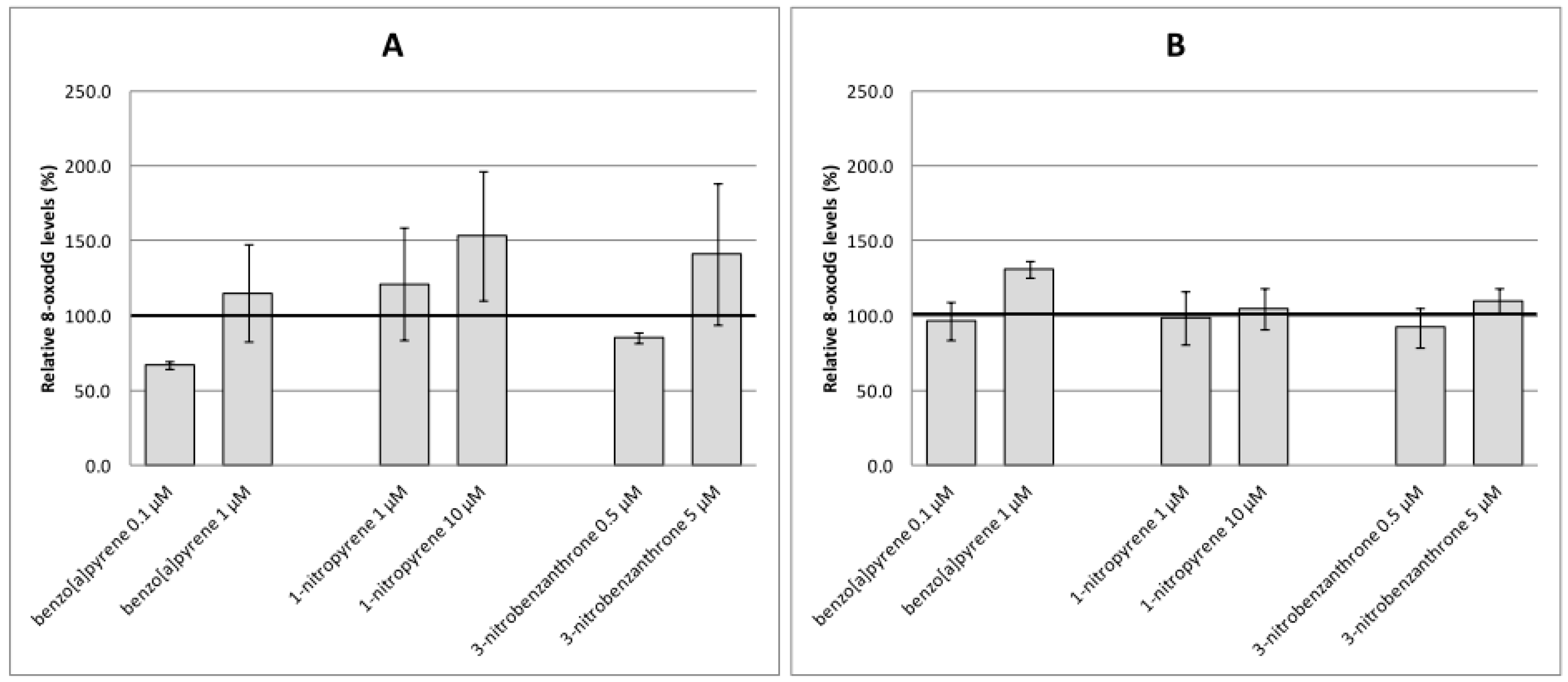
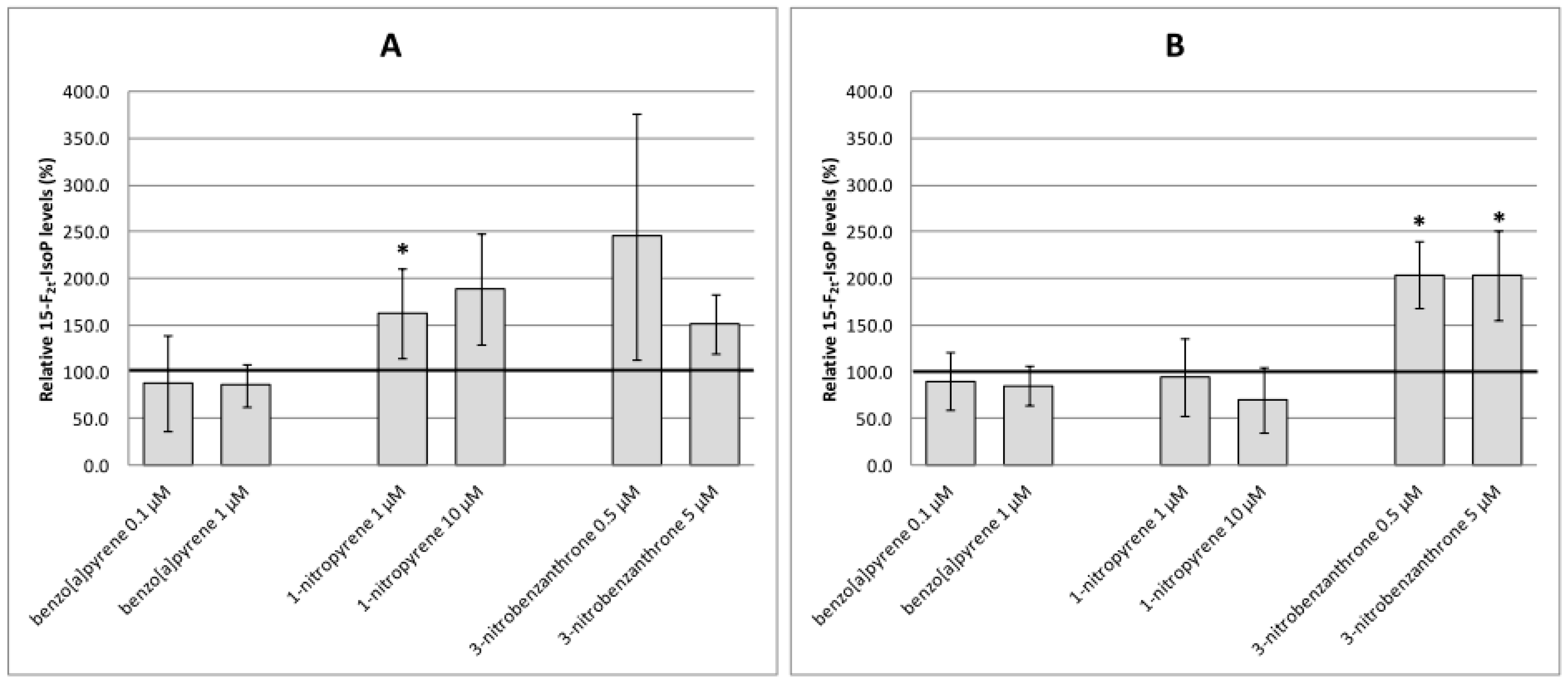
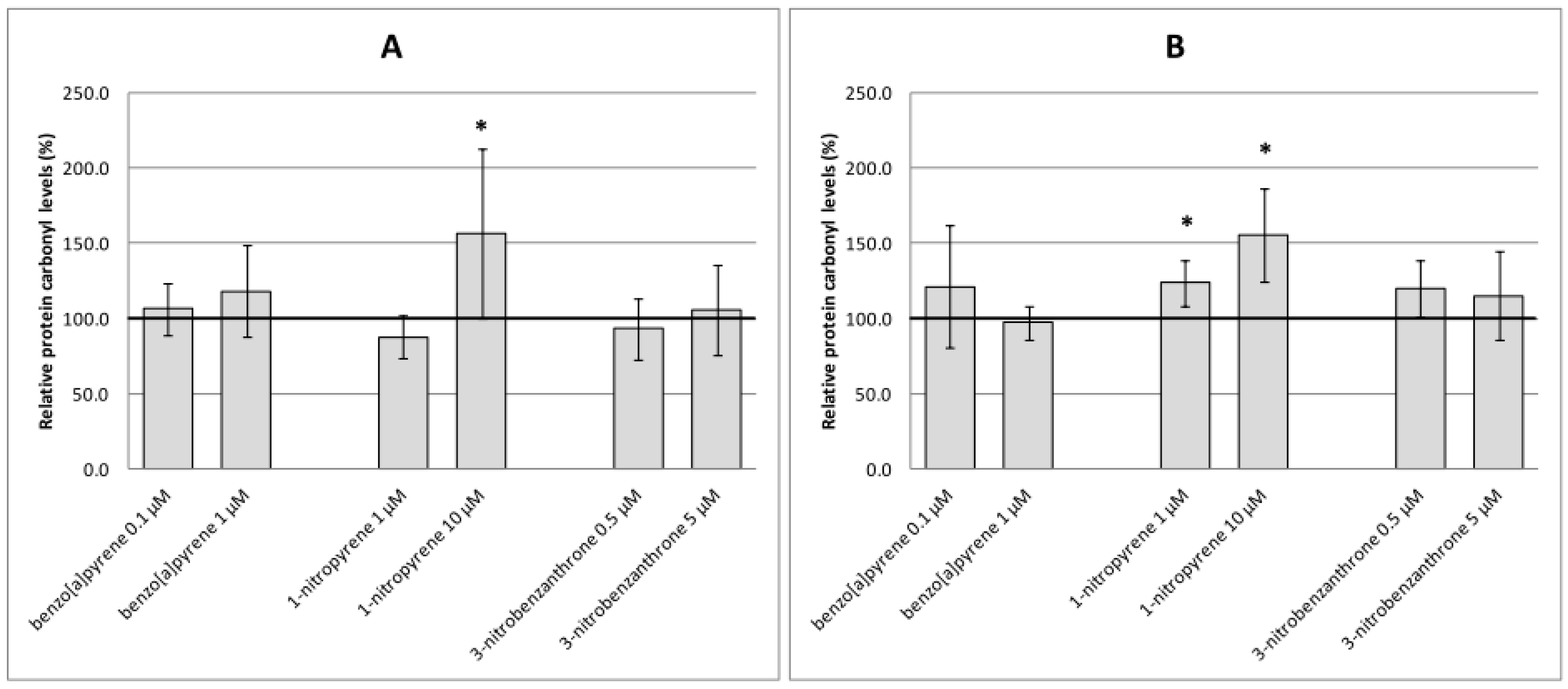
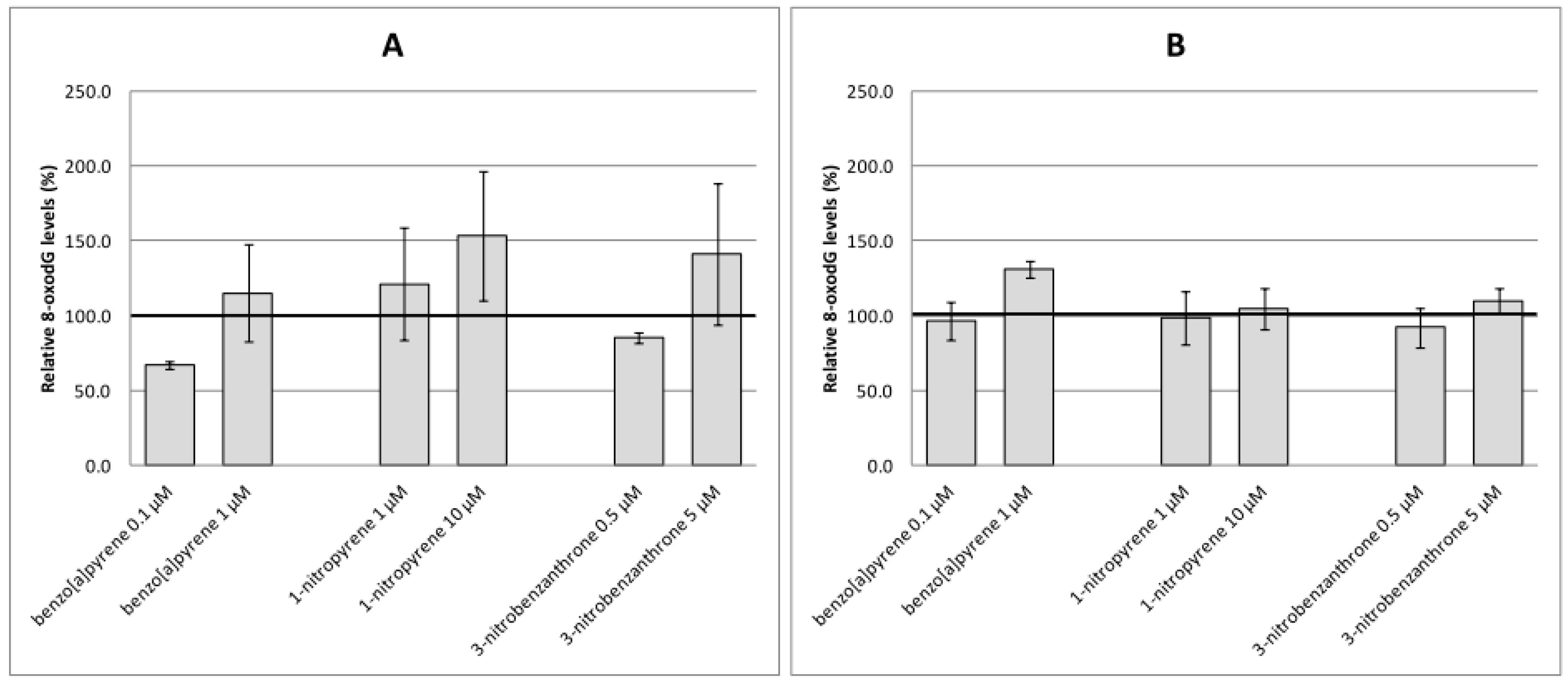
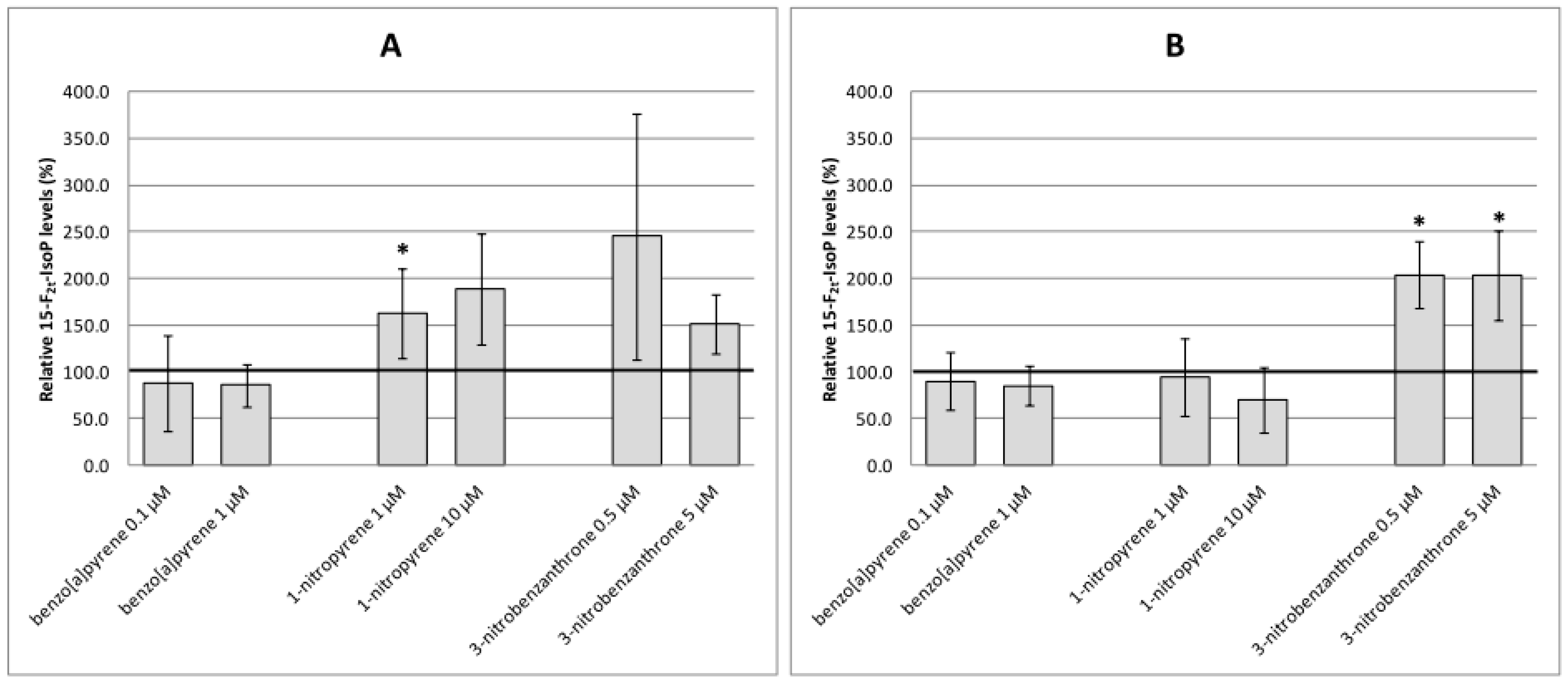
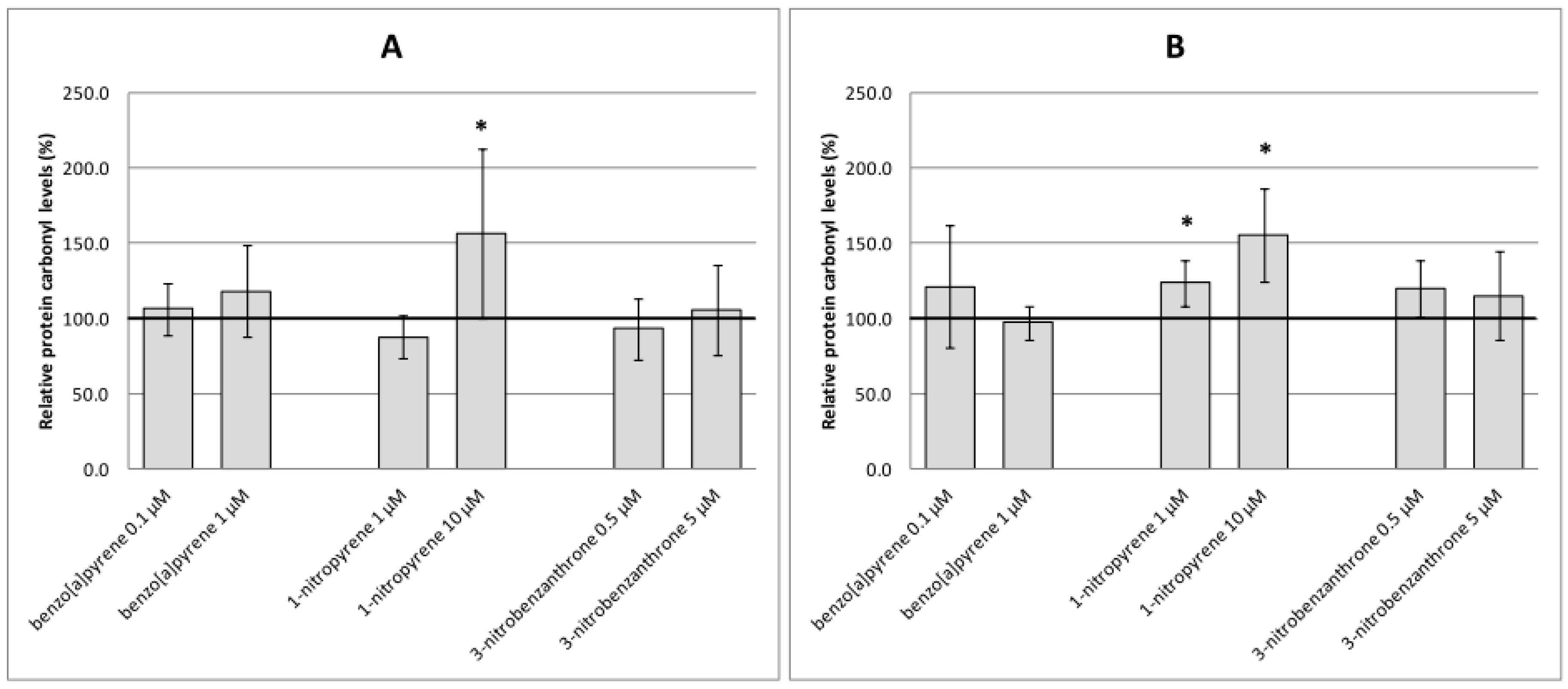
2.2. Oxidative Damage to DNA, Lipids and Proteins
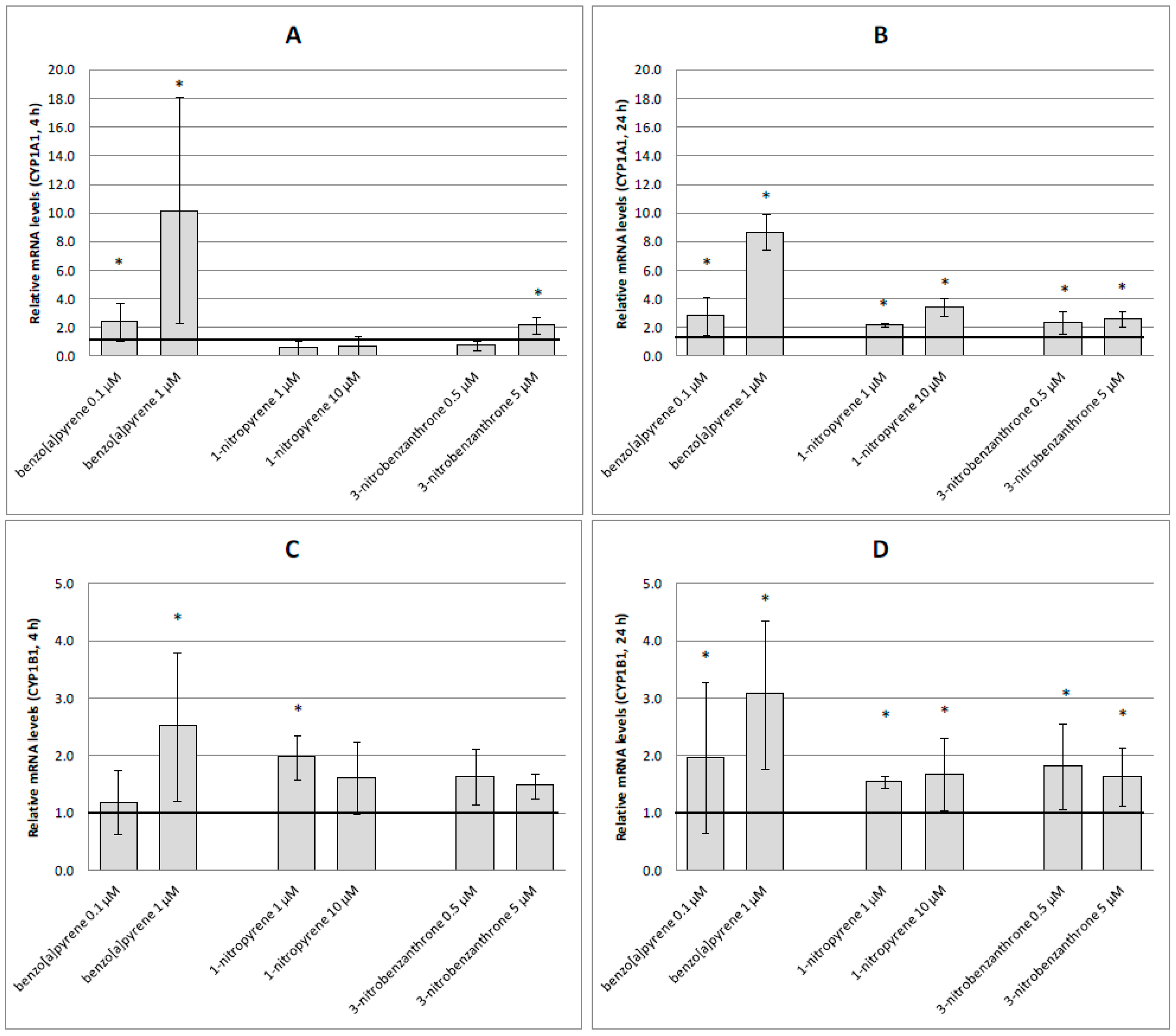
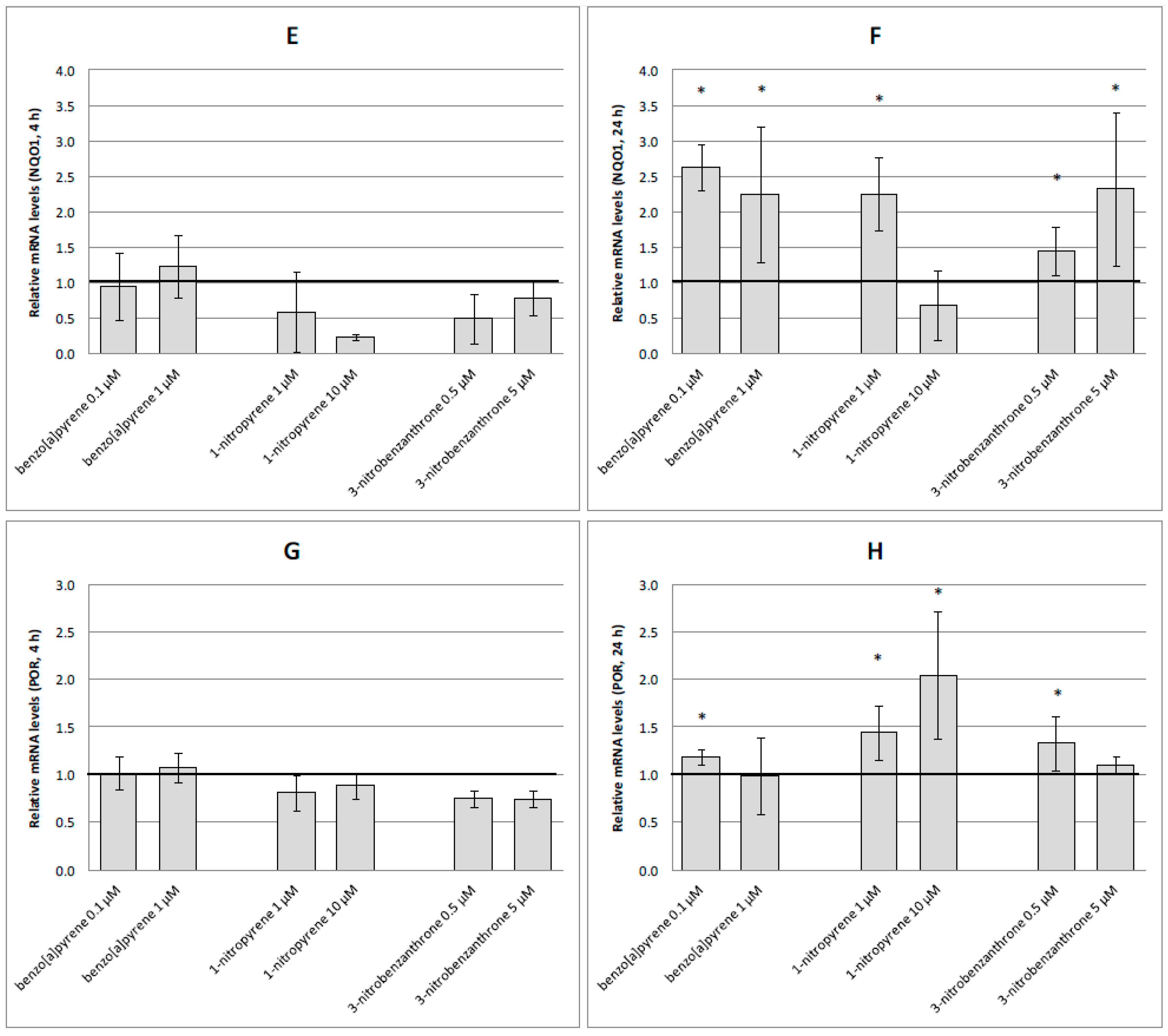
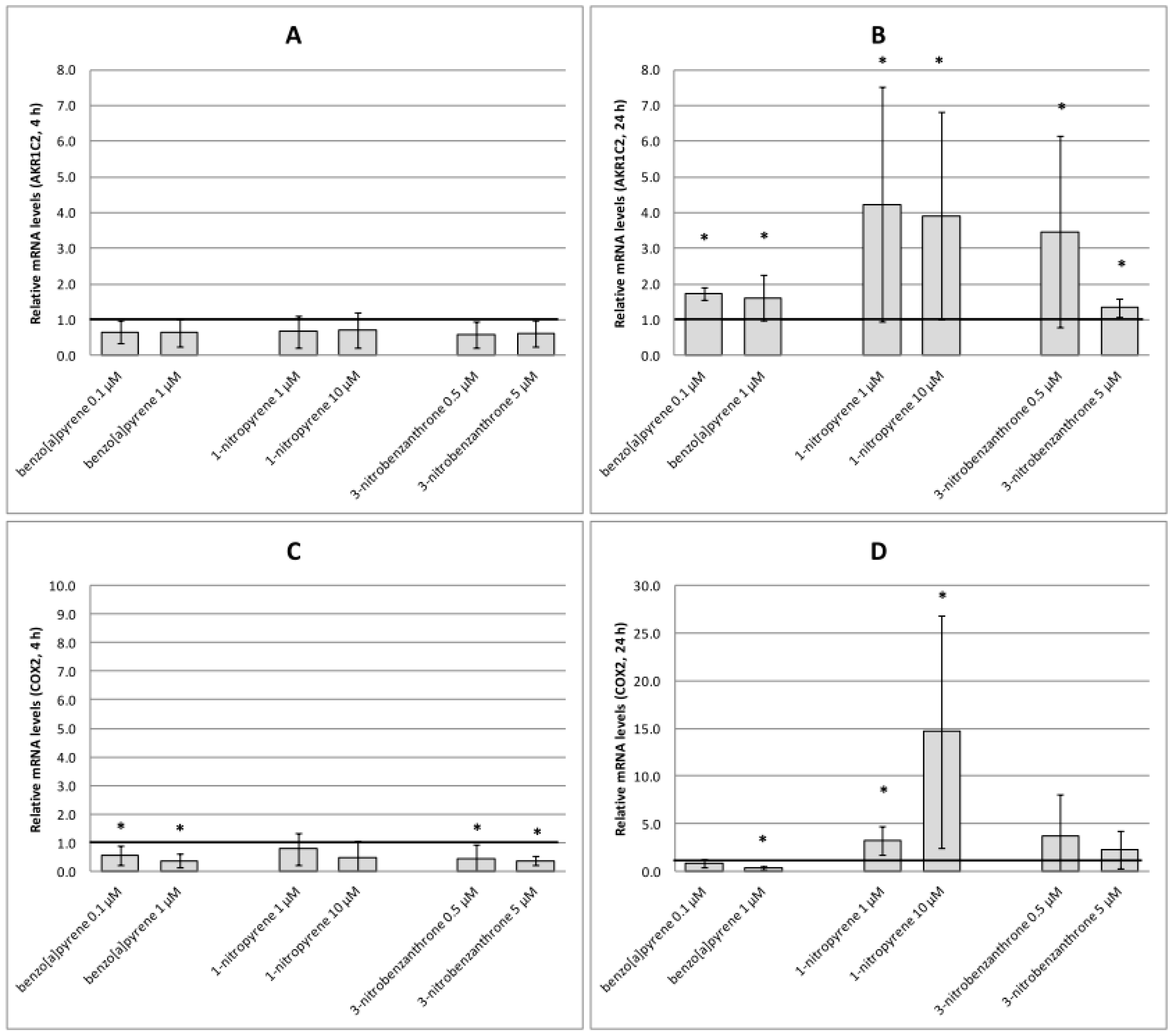
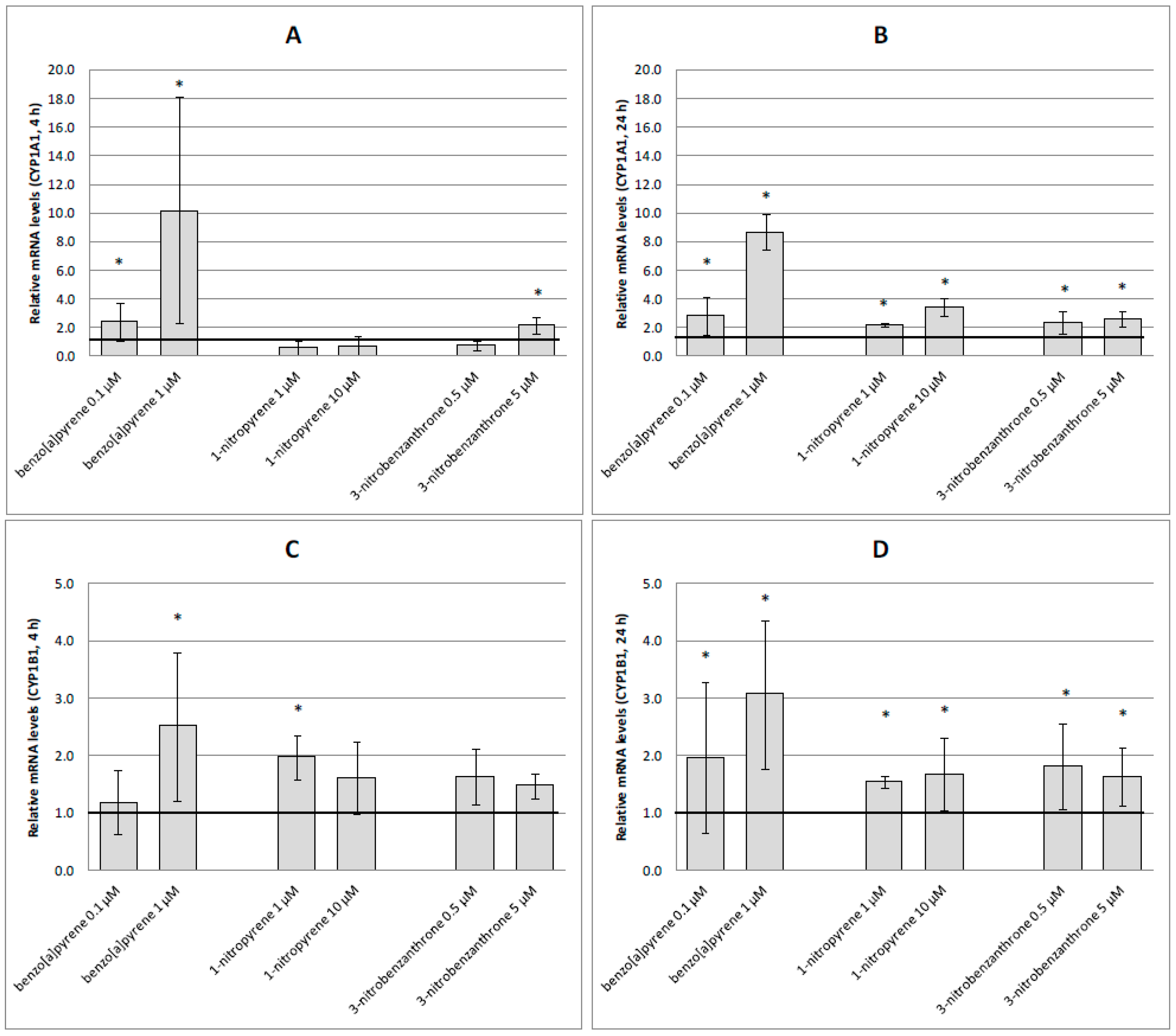
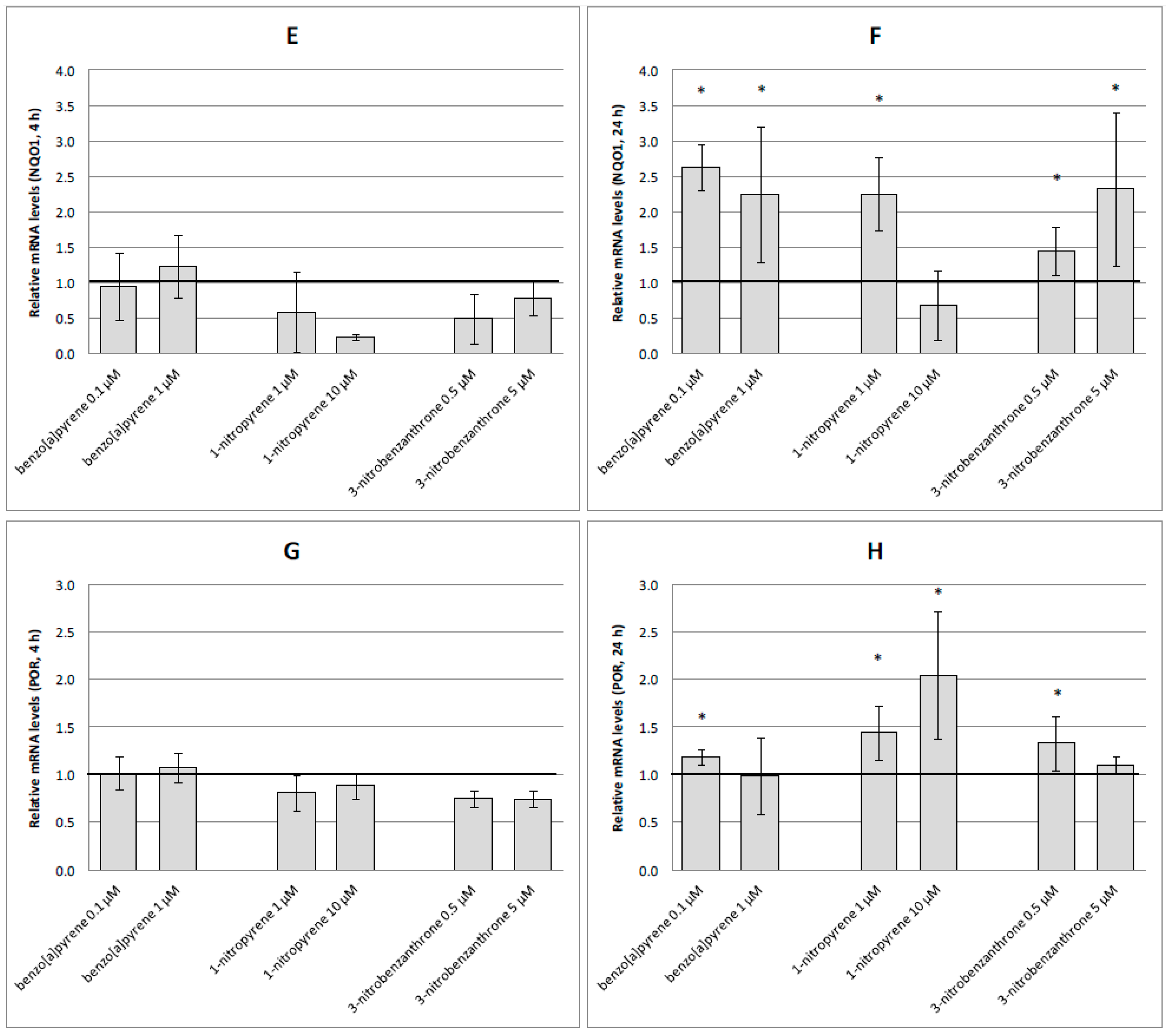
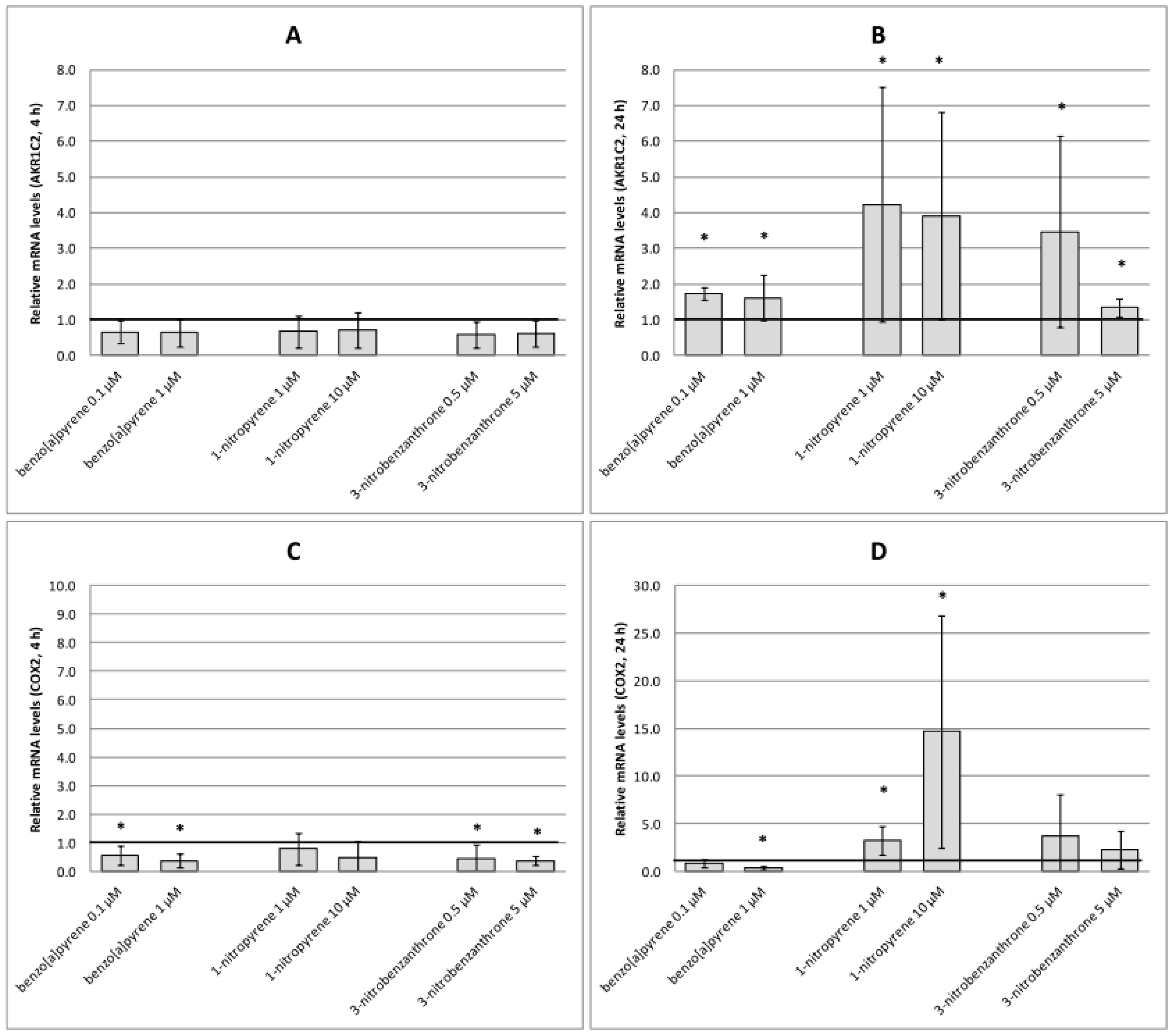
2.3. mRNA Expression of Selected Genes
3. Discussion
4. Materials and Methods
4.1. Test Compounds
4.2. Cell Cultures and Treatment
4.3. DNA Isolation and DNA Adduct Analysis
4.4. RNA Isolation and Quality Control
4.5. Analyses of mRNA Expression
4.6. Analyses of 8-oxo-7,8-Dihydro-2′-deoxyguanosine (8-oxodG)
4.7. Preparation of Cell Lysates; Analyses of Lipid Peroxidation and Protein Oxidation
4.8. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AA | Arachidonic Acid |
| AKR1C2 | Aldo-Keto Reductase |
| B[a]P | Benzo[a]pyrene |
| COX2 | Cyclooxygenase-2 |
| ER stress | Endoplasmic Reticulum Stress |
| 15-F2t-IsoP | 15-F2t-Isoprostane |
| 3-NBA | 3-nitrobenzanthrone |
| N-OH-ABA | N-hydroxy-3-aminobenzanthrone |
| 1-NP | 1-nitropyrene |
| NQO1 | NAD(P)H:QuinoneOxidoreductase |
| 8-oxodG | 8-oxo-7,8-dihydro-2′-deoxyguanosine |
| PAHs | Polycyclic Aromatic Hydrocarbons |
| POR | NAPDH: Cytochrome P450 Oxidoreductase |
| ROS | Reactive Oxygen Species |
| XO | Xanthine Oxidase |
References
- Loomis, D.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Baan, R.; Mattock, H.; Straif, K. The carcinogenicity of outdoor air pollution. Lancet Oncol. 2013, 14, 1262–1263. [Google Scholar] [CrossRef]
- IARC. Diesel and gasoline engine exhausts and some nitroarenes. In IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans; IARC Publications: Lyon, France, 2013; Volume 105, p. 467. [Google Scholar]
- Bamford, H.A.; Bezabeh, D.Z.; Schantz, S.; Wise, S.A.; Baker, J.E. Determination and comparison of nitrated-polycyclic aromatic hydrocarbons measured in air and diesel particulate reference materials. Chemosphere 2003, 50, 575–587. [Google Scholar] [CrossRef]
- IARC. Chemical agents and related occupations. In IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans; IARC Publications: Lyon, France, 2012; Volume 100, pp. 137–138. [Google Scholar]
- Penning, T.M.; Burczynski, M.E.; Hung, C.F.; McCoull, K.D.; Palackal, N.T.; Tsuruda, L.S. Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: Generation of reactive and redox active o-quinones. Chem. Res. Toxicol. 1999, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Toriba, A.; Kitaoka, H.; Dills, R.L.; Mizukami, S.; Tanabe, K.; Takeuchi, N.; Ueno, M.; Kameda, T.; Tang, N.; Hayakawa, K.; et al. Identification and quantification of 1-nitropyrene metabolites in human urine as a proposed biomarker for exposure to diesel exhaust. Chem. Res. Toxicol. 2007, 20, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Silvers, K.J.; Couch, L.H.; Rorke, E.A.; Howard, P.C. Role of nitroreductases but not cytochromes P450 in the metabolic activation of 1-nitropyrene in the HepG2 human hepatoblastoma cell line. Biochem. Pharmacol. 1997, 54, 927–936. [Google Scholar] [CrossRef]
- WHO. Selected nitro and nitrooxy-polycyclic-aromatic hydrocarbons. In Environ. Health Criteria; World Health Organization: Geneva, Switzerland, 2003; Volume 229, pp. 125–140. [Google Scholar]
- Andersson, H.; Piras, E.; Demma, J.; Hellman, B.; Brittebo, E. Low levels of the air pollutant 1-nitropyrene induce DNA damage, increased levels of reactive oxygen species and endoplasmic reticulum stress in human endothelial cells. Toxicology 2009, 262, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, S.; Murata, M.; Fukuhara, K.; Miyata, N.; Kawanishi, S. Oxidative DNA damage by a metabolite of carcinogenic 1-nitropyrene. Biochem. Biophys. Res. Commun. 2001, 280, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M. 3-Nitrobenzanthrone, a potential human cancer hazard in diesel exhaust and urban air pollution: A review of the evidence. Mutagenesis 2005, 20, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Cole, K.J.; Phillips, D.H. Activation of 3-nitrobenzanthrone and its metabolites to DNA-damaging species in human B lymphoblastoid MCL-5 cells. Mutagenesis 2004, 19, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Tezuka, T.; Ohnishi, S.; Takamura-Enya, T.; Hisamatsu, Y.; Kawanishi, S. Carcinogenic 3-nitrobenzanthrone induces oxidative damage to isolated and cellular DNA. Free Radic. Biol. Med. 2006, 40, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Oya, E.; Ovrevik, J.; Arlt, V.M.; Nagy, E.; Phillips, D.H.; Holme, J.A. DNA damage and DNA damage response in human bronchial epithelial BEAS-2B cells following exposure to 2-nitrobenzanthrone and 3-nitrobenzanthrone: Role in apoptosis. Mutagenesis 2011, 26, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Oster, C.G.; Mayer, M.M.; Avery, M.L.; Audus, K.L. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp. Cell Res. 1998, 243, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.; Seidel, A.; Borlak, J. The environmental carcinogen 3-nitrobenzanthrone and its main metabolite 3-aminobenzanthrone enhance formation of reactive oxygen intermediates in human A549 lung epithelial cells. Toxicol. Appl. Pharmacol. 2007, 221, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Genies, C.; Maitre, A.; Lefebvre, E.; Jullien, A.; Chopard-Lallier, M.; Douki, T. The extreme variety of genotoxic response to benzo[a]pyrene in three different human cell lines from three different organs. PLoS ONE 2013, 8, e78356. [Google Scholar] [CrossRef]
- Libalova, H.; Krckova, S.; Uhlirova, K.; Milcova, A.; Schmuczerova, J.; Ciganek, M.; Klema, J.; Machala, M.; Sram, R.J.; Topinka, J. Genotoxicity but not the AhR-mediated activity of PAHs is inhibited by other components of complex mixtures of ambient air pollutants. Toxicol. Lett. 2014, 225, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Sato, H.; Nishimura, N.; Takahashi, S.; Itoh, K.; Yamamoto, M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol. Appl. Pharmacol. 2001, 173, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Hashimoto, A.H.; Amanuma, K.; Matsumoto, M.; Hiyoshi, K.; Takano, H.; Masumura, K.; Itoh, K.; Nohmi, T.; Yamamoto, M. Enhanced spontaneous and benzo(a)pyrene-induced mutations in the lung of Nrf2-deficient gpt delta mice. Cancer Res. 2007, 67, 5643–5648. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Hatanaka, N.; Kizu, R.; Hayakawa, K.; Shimada, N.; Guengerich, F.P.; Nakajima, M.; Yokoi, T. Bioactivation of diesel exhaust particle extracts and their major nitrated polycyclic aromatic hydrocarbon components, 1-nitropyrene and dinitropyrenes, by human cytochromes P450 1A1, 1A2, and 1B1. Mutat. Res. 2000, 472, 129–138. [Google Scholar] [CrossRef]
- Ovrevik, J.; Arlt, V.M.; Oya, E.; Nagy, E.; Mollerup, S.; Phillips, D.H.; Lag, M.; Holme, J.A. Differential effects of nitro-PAHs and amino-PAHs on cytokine and chemokine responses in human bronchial epithelial BEAS-2B cells. Toxicol. Appl. Pharmacol. 2010, 242, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Iwanari, M.; Nakajima, M.; Kizu, R.; Hayakawa, K.; Yokoi, T. Induction of CYP1A1, CYP1A2, and CYP1B1 mRNAs by nitropolycyclic aromatic hydrocarbons in various human tissue-derived cells: Chemical-, cytochrome P450 isoform-, and cell-specific differences. Arch. Toxicol. 2002, 76, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Johansson, C.; Zeisig, M.; Moller, L. Oxidative stress and DNA damage caused by the urban air pollutant 3-NBA and its isomer 2-NBA in human lung cells analyzed with three independent methods. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 827, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Adachi, S.; Takamura-Enya, T.; Zeisig, M.; Moller, L. DNA adduct formation and oxidative stress from the carcinogenic urban air pollutant 3-nitrobenzanthrone and its isomer 2-nitrobenzanthrone, in vitro and in vivo. Mutagenesis 2007, 22, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Dracinska, H.; Mizerovska, J.; Frei, E.; Schmeiser, H.H.; Hudecek, J.; Hodek, P.; Phillips, D.H.; Arlt, V.M. The environmental pollutant and carcinogen 3-nitrobenzanthrone induces cytochrome P450 1A1 and NAD(P)H:quinoneoxidoreductase in rat lung and kidney, thereby enhancing its own genotoxicity. Toxicology 2008, 247, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Mizerovska, J.; Dracinska, H.; Arlt, V.M.; Schmeiser, H.H.; Frei, E.; Stiborova, M. Oxidation of 3-aminobenzanthrone, a human metabolite of carcinogenic environmental pollutant 3-nitrobenzanthrone, by cytochromes P450—Similarity between human and rat enzymes. Neuro Endocrinol. Lett. 2009, 30, 52–59. [Google Scholar] [PubMed]
- Kim, Y.D.; Ko, Y.J.; Kawamoto, T.; Kim, H. The effects of 1-nitropyrene on oxidative DNA damage and expression of DNA repair enzymes. J. Occup. Health 2005, 47, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Hanzalova, K.; Rossner, P., Jr.; Sram, R.J. Oxidative damage induced by carcinogenic polycyclic aromatic hydrocarbons and organic extracts from urban air particulate matter. Mutat. Res. 2010, 696, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Bolck, B.; Ibrahim, M.; Steinritz, D.; Morguet, C.; Duhr, S.; Suhr, F.; Lu-Hesselmann, J.; Bloch, W. Detection of key enzymes, free radical reaction products and activated signaling molecules as biomarkers of cell damage induced by benzo[a]pyrene in human keratinocytes. Toxicol. in Vitro 2014, 28, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Fullove, T.P.; Yu, H. DNA damage and repair of human skin keratinocytes concurrently exposed to pyrene derivatives and UVA light. Toxicol. Res. Camb. 2013, 2, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Persson, H.L.; Nilsson, K.J.; Brunk, U.T. Novel cellular defenses against iron and oxidation: Ferritin and autophagocytosis preserve lysosomal stability in airway epithelium. Redox. Rep. 2001, 6, 57–63. [Google Scholar] [CrossRef] [PubMed]
- De Mejia, E.G.; Ramirez-Mares, M.V. Leaf extract from Ardisiacompressa protects against 1-nitropyrene-induced cytotoxicity and its antioxidant defense disruption in cultured rat hepatocytes. Toxicology 2002, 179, 151–162. [Google Scholar] [CrossRef]
- Landvik, N.E.; Gorria, M.; Arlt, V.M.; Asare, N.; Solhaug, A.; Lagadic-Gossmann, D.; Holme, J.A. Effects of nitrated-polycyclic aromatic hydrocarbons and diesel exhaust particle extracts on cell signalling related to apoptosis: Possible implications for their mutagenic and carcinogenic effects. Toxicology 2007, 231, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Fullove, T.P.; Johnson, B.; Yu, H. Structure-dependent lipid peroxidation by photoirradiation of pyrene and its mono-substituted derivatives. J. Environ. Sci. Health Toxic Hazard. Subst. Environ. Eng. 2013, 48, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wahl, L.M. Oxidative stress augments the production of matrix metalloproteinase-1, cyclooxygenase-2, and prostaglandin E2 through enhancement of NF-kappa B activity in lipopolysaccharide-activated human primary monocytes. J. Immunol. 2005, 175, 5423–5429. [Google Scholar] [CrossRef] [PubMed]
- Morrow, C.S.; Chiu, J.; Cowan, K.H. Posttranscriptional control of glutathione S-transferase pi gene expression in human breast cancer cells. J. Biol. Chem. 1992, 267, 10544–10550. [Google Scholar] [PubMed]
- Gupta, R.C. Enhanced Sensitivity of 32P-Postlabeling Analysis of Aromatic Carcinogen: DNA Adducts. Cancer Res. 1985, 45, 5656–5662. [Google Scholar] [PubMed]
- Phillips, D.H.; Castegnaro, M. Standardization and validation of DNA adduct postlabelling methods: Report of interlaboratory trials and production of recommended protocols. Mutagenesis 1999, 14, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Randerath, E.; Avitts, T.A.; Reddy, M.V.; Miller, R.H.; Everson, R.B.; Randerath, K. Comparative 32P-postlabeling analysis of cigarette smoke-induced DNA damage in human tissues and mouse skin. Cancer Res. 1986, 46, 5869–5877. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Compound | Concentration (μM) | Cytotoxicity (%) | |
|---|---|---|---|
| 4 h | 24 h | ||
| B[a]P | 0.1 | ND | ND |
| 1.0 | ND | ND | |
| 1-NP | 1.0 | ND | ND |
| 10 | ND | 0.6 | |
| 3-NBA | 0.5 | ND | ND |
| 5.0 | ND | 2.0 | |
| Gene | RefSeq ID | Primer Sequence | |
|---|---|---|---|
| CYP1A1 | NM_000499 | Forward | CACCATCCCCCACAGCAC |
| Reverse | TTACAAAGACACAACGCCCC | ||
| CYP1B1 | NM_000104 | Forward | GCTTTTTCTCTTCATCTCCATC |
| Reverse | TTCATTTTCGCAGGCTCATTTG | ||
| ALDH3A1 | NM_001135168 | Forward | TCTTGGCTCTTGCCGTTC |
| Reverse | CGCTGATCTTGCTCATGG | ||
| COX2 | NM_000963 | Forward | CTTCACGCATCAGTTTTTCAAG |
| Reverse | TCACCGTAAATATGATTTAAGTCCAC | ||
| AKR1C2 | NM_001135241 | Forward | AGCACCTCCACCTTCTCTCTC |
| Reverse | AGGTGAAGGGGAAGTAAGCAT | ||
| NQO1 | NM_000903 | Forward | ATGTATGACAAAGGACCCTTCC |
| Reverse | TCCCTTGCAGAGAGTACATGG | ||
| POR | NM_000941 | Forward | GAGACCCCACCGACAATG |
| Reverse | TGGCATTGAAGTGCTCGTAG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossner, P.; Strapacova, S.; Stolcpartova, J.; Schmuczerova, J.; Milcova, A.; Neca, J.; Vlkova, V.; Brzicova, T.; Machala, M.; Topinka, J. Toxic Effects of the Major Components of Diesel Exhaust in Human Alveolar Basal Epithelial Cells (A549). Int. J. Mol. Sci. 2016, 17, 1393. https://doi.org/10.3390/ijms17091393
Rossner P, Strapacova S, Stolcpartova J, Schmuczerova J, Milcova A, Neca J, Vlkova V, Brzicova T, Machala M, Topinka J. Toxic Effects of the Major Components of Diesel Exhaust in Human Alveolar Basal Epithelial Cells (A549). International Journal of Molecular Sciences. 2016; 17(9):1393. https://doi.org/10.3390/ijms17091393
Chicago/Turabian StyleRossner, Pavel, Simona Strapacova, Jitka Stolcpartova, Jana Schmuczerova, Alena Milcova, Jiri Neca, Veronika Vlkova, Tana Brzicova, Miroslav Machala, and Jan Topinka. 2016. "Toxic Effects of the Major Components of Diesel Exhaust in Human Alveolar Basal Epithelial Cells (A549)" International Journal of Molecular Sciences 17, no. 9: 1393. https://doi.org/10.3390/ijms17091393
APA StyleRossner, P., Strapacova, S., Stolcpartova, J., Schmuczerova, J., Milcova, A., Neca, J., Vlkova, V., Brzicova, T., Machala, M., & Topinka, J. (2016). Toxic Effects of the Major Components of Diesel Exhaust in Human Alveolar Basal Epithelial Cells (A549). International Journal of Molecular Sciences, 17(9), 1393. https://doi.org/10.3390/ijms17091393