
Protective Effects of Berberine on Renal Injury in Streptozotocin (STZ)-Induced Diabetic Mice

Abstract
:
1. Introduction
2. Results
2.1. The Effects of BBR on Metabolic and Biochemical Parameters in STZ-Induced Diabetic Mice
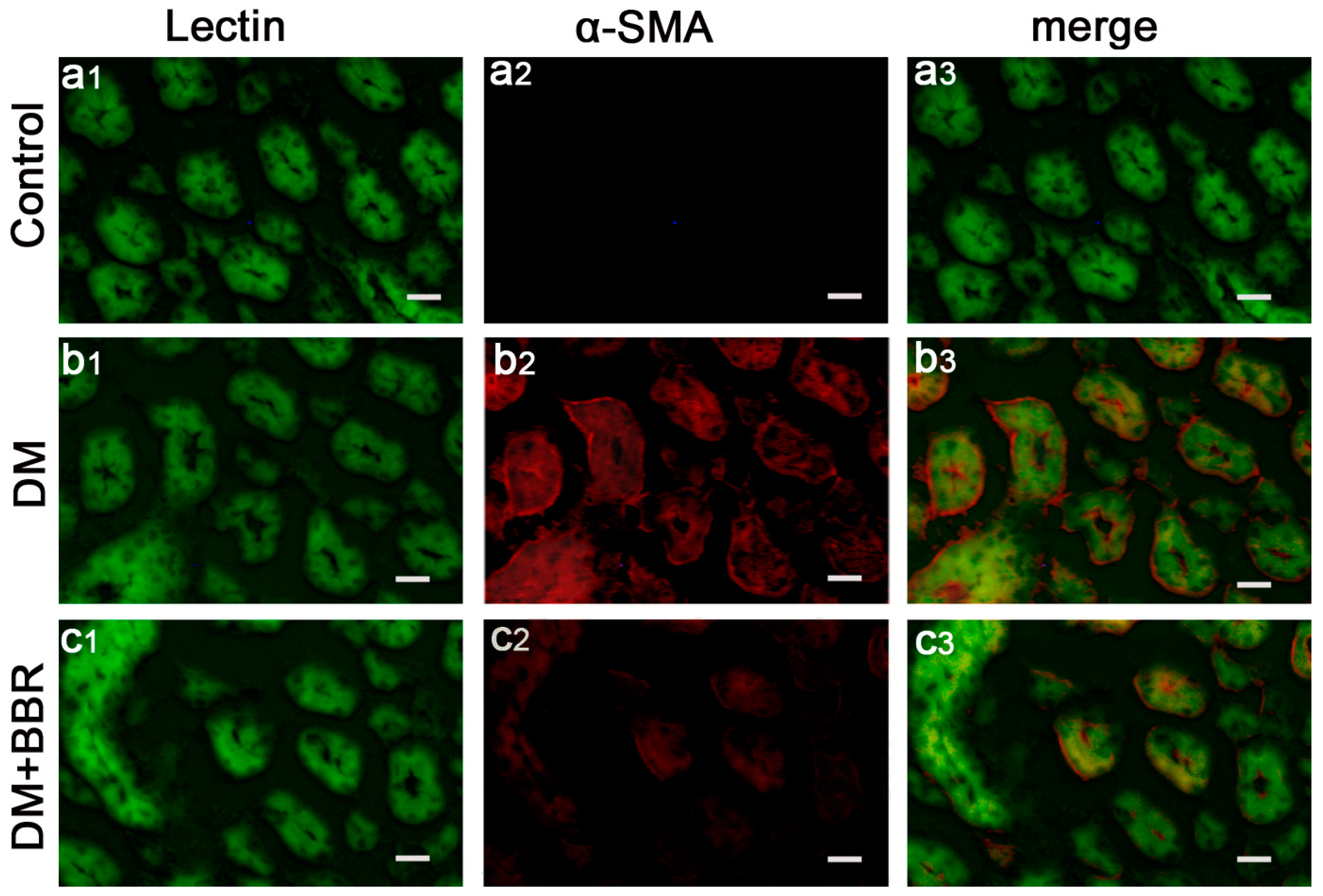
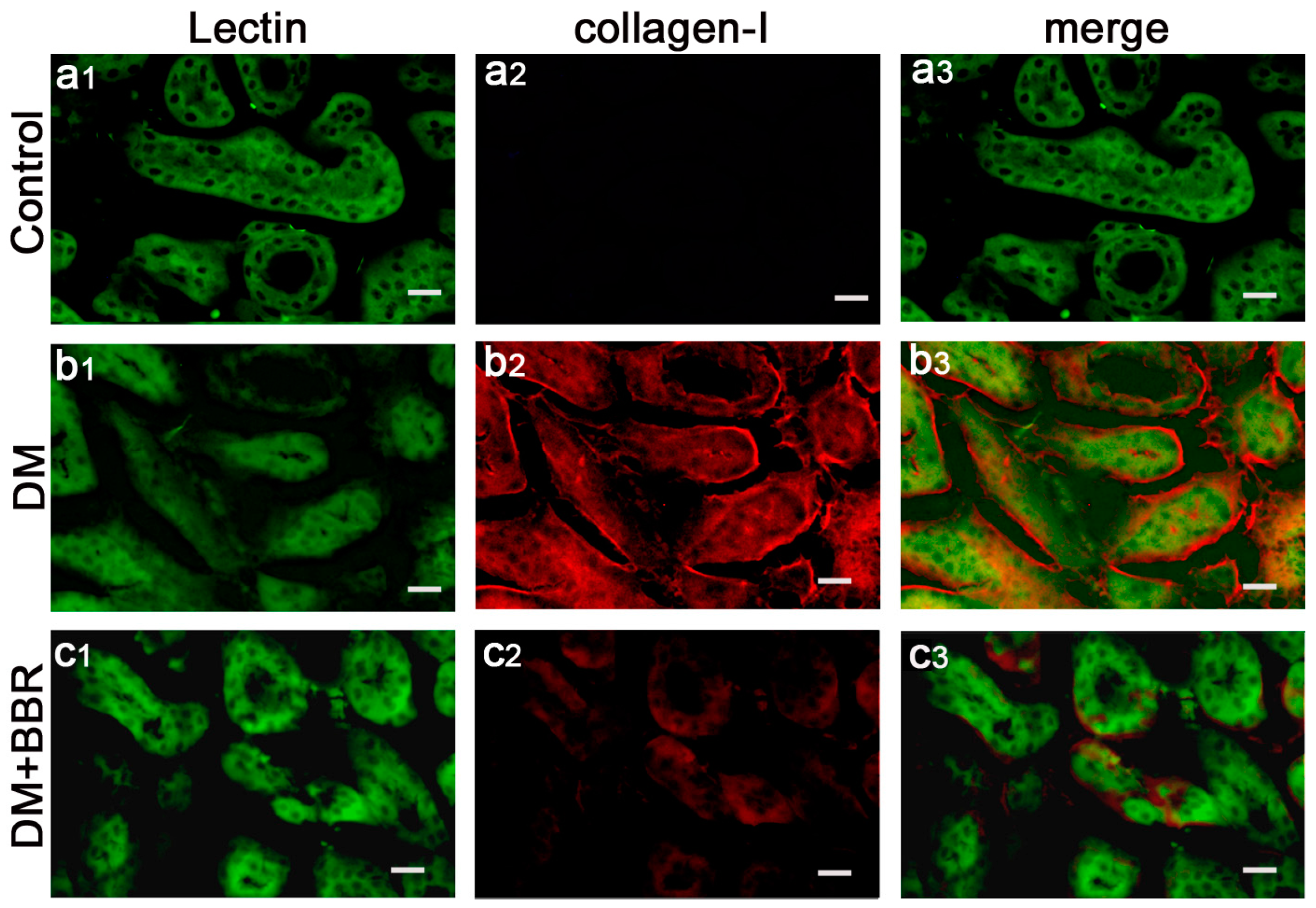
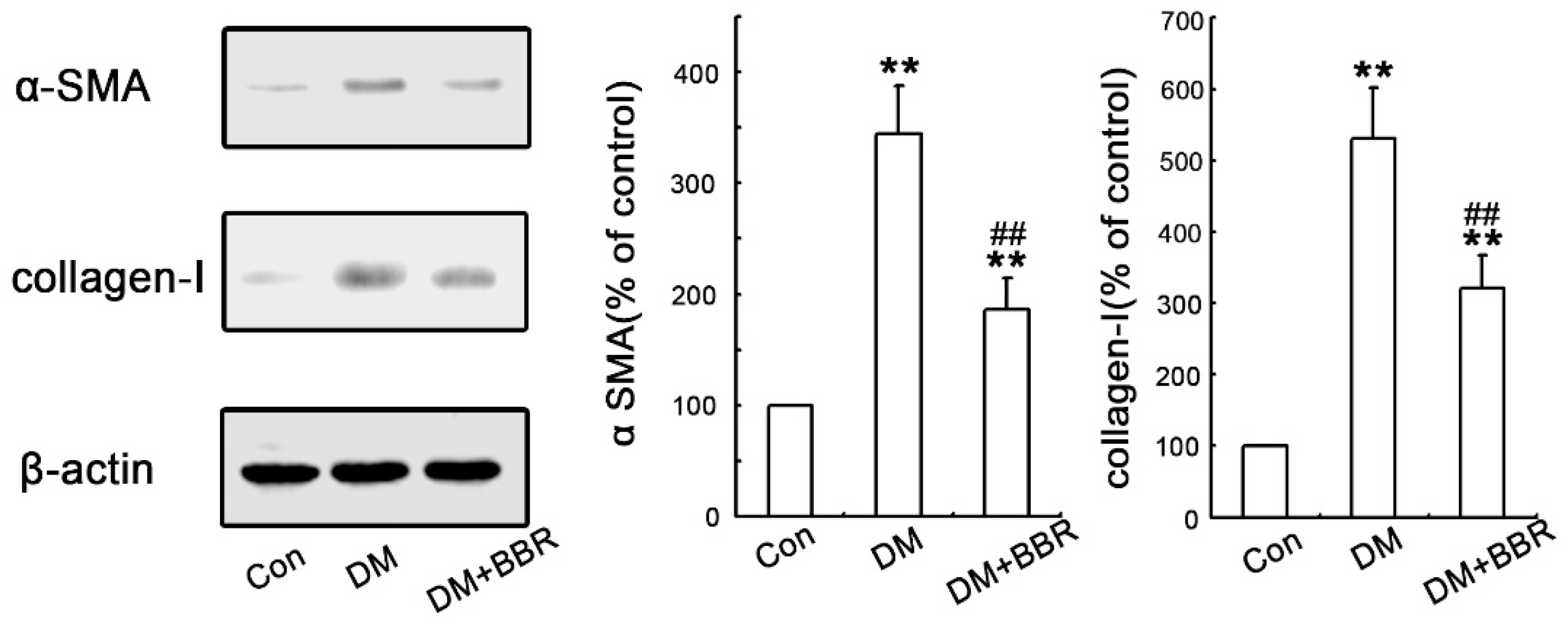
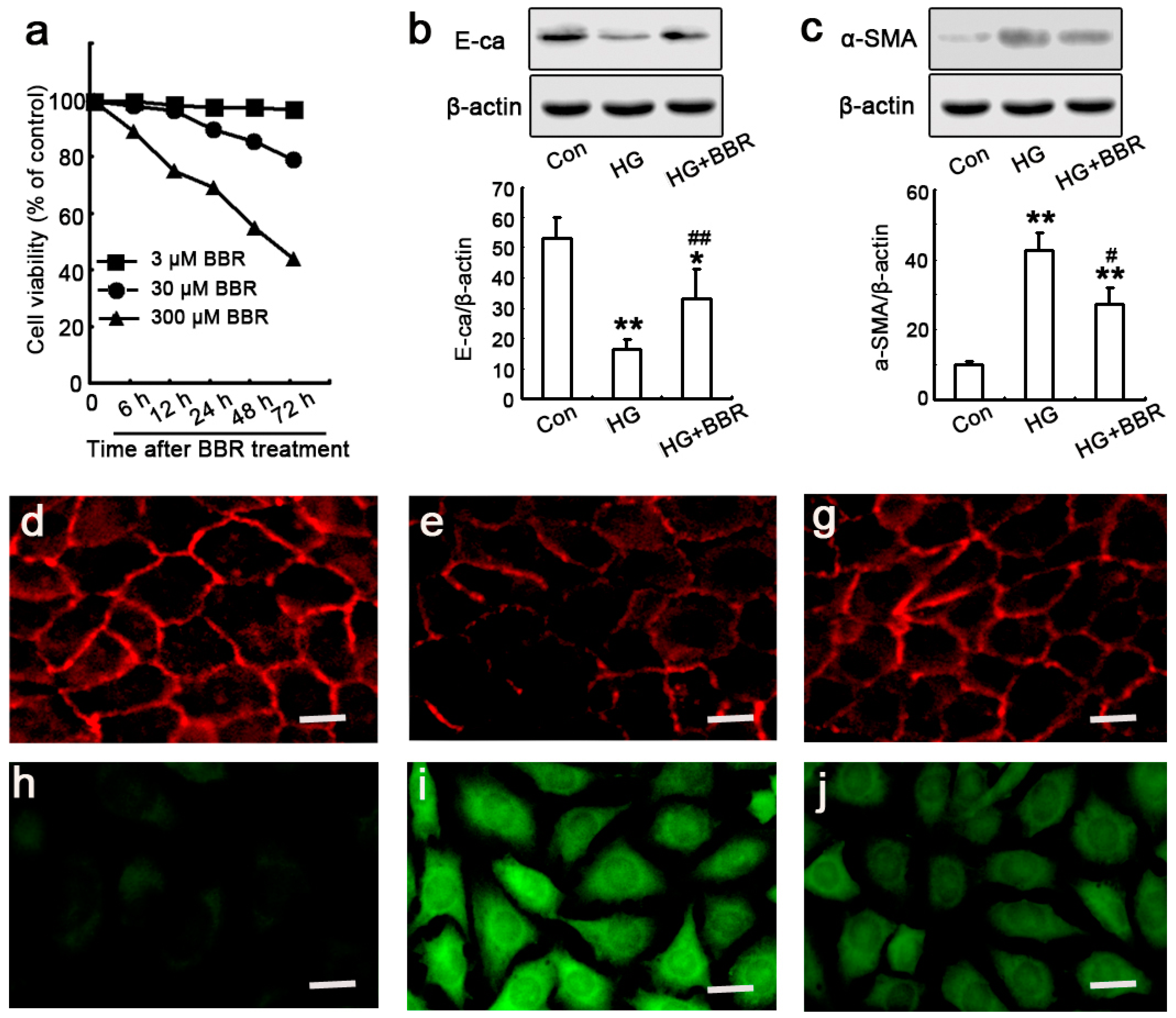
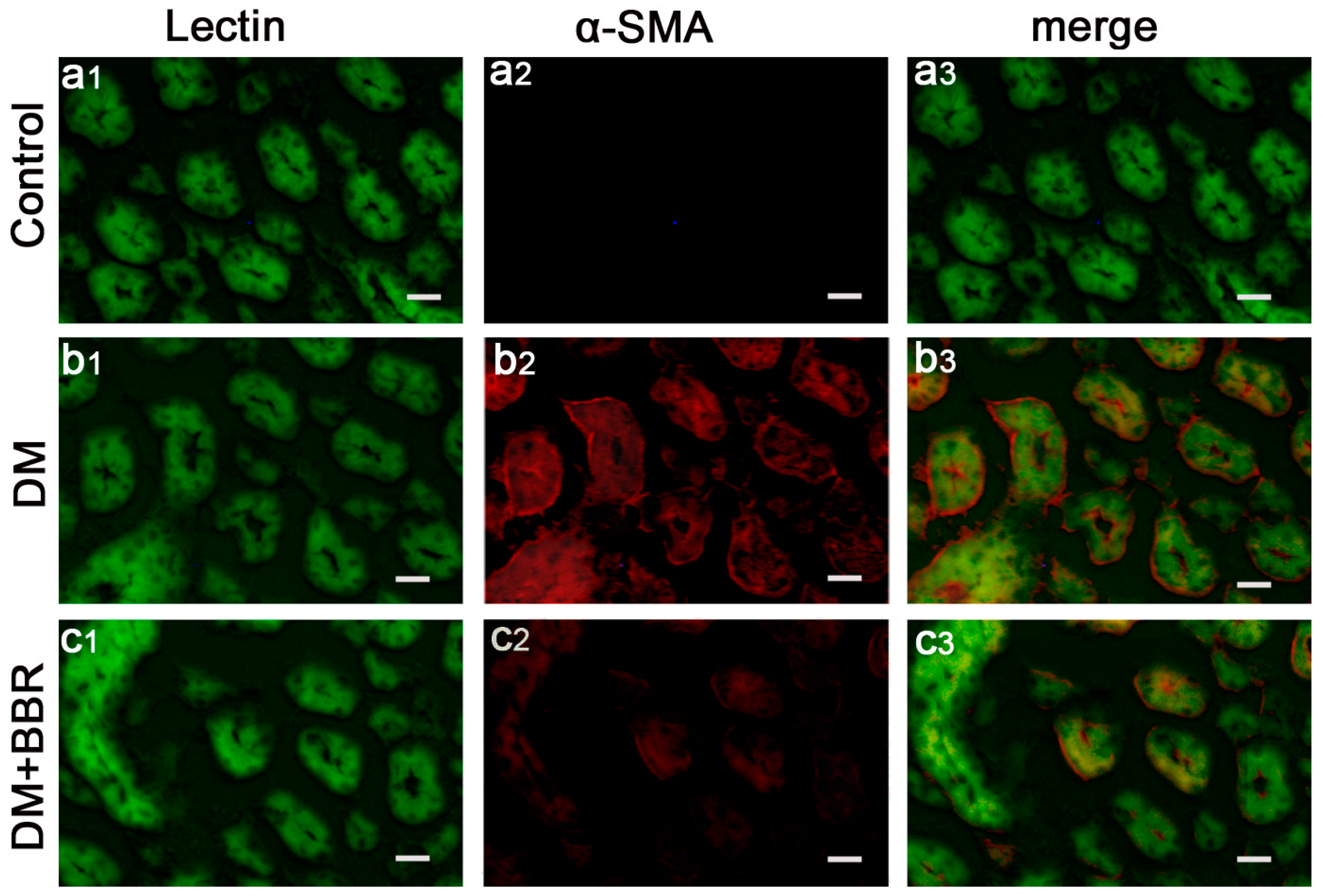
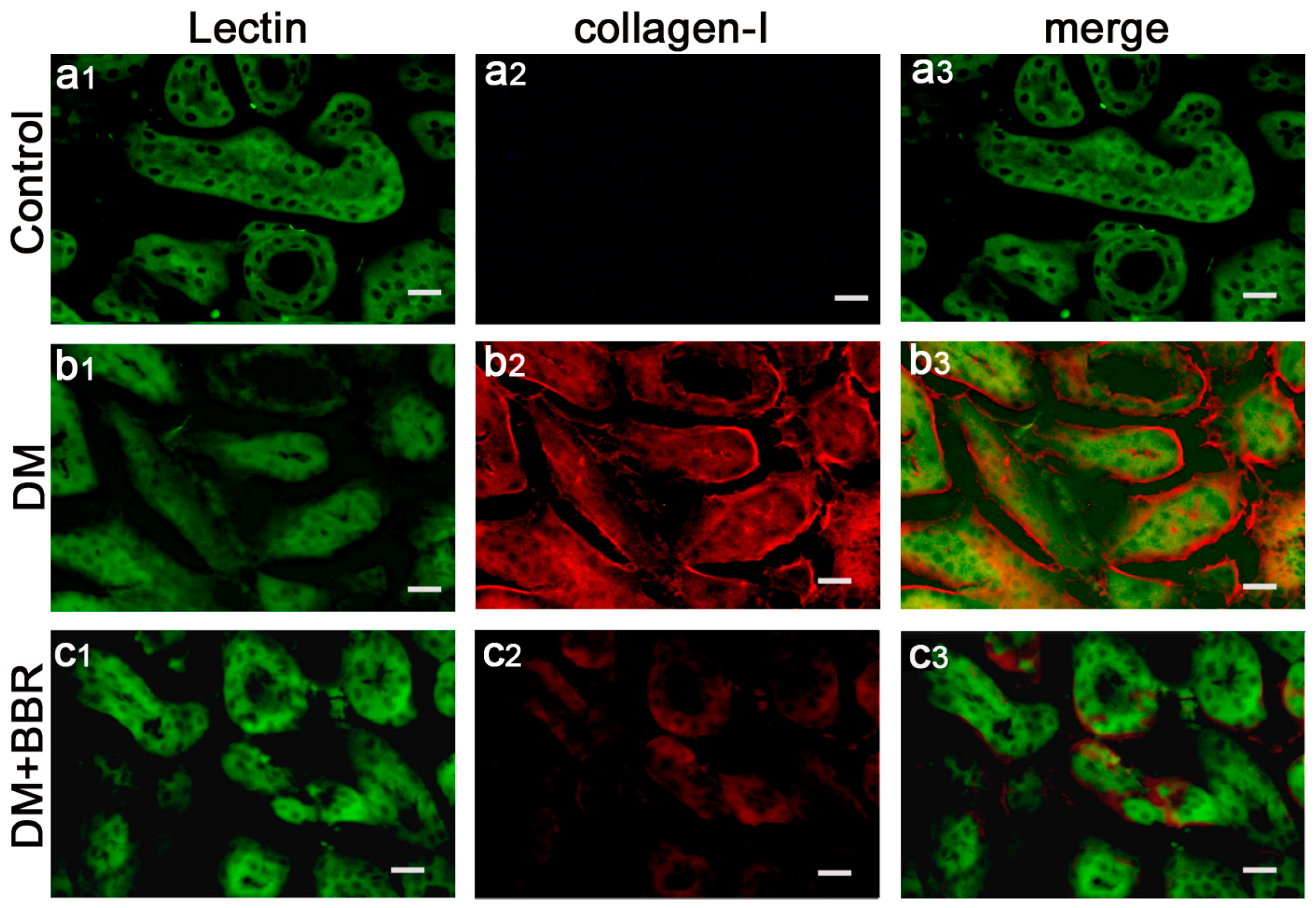
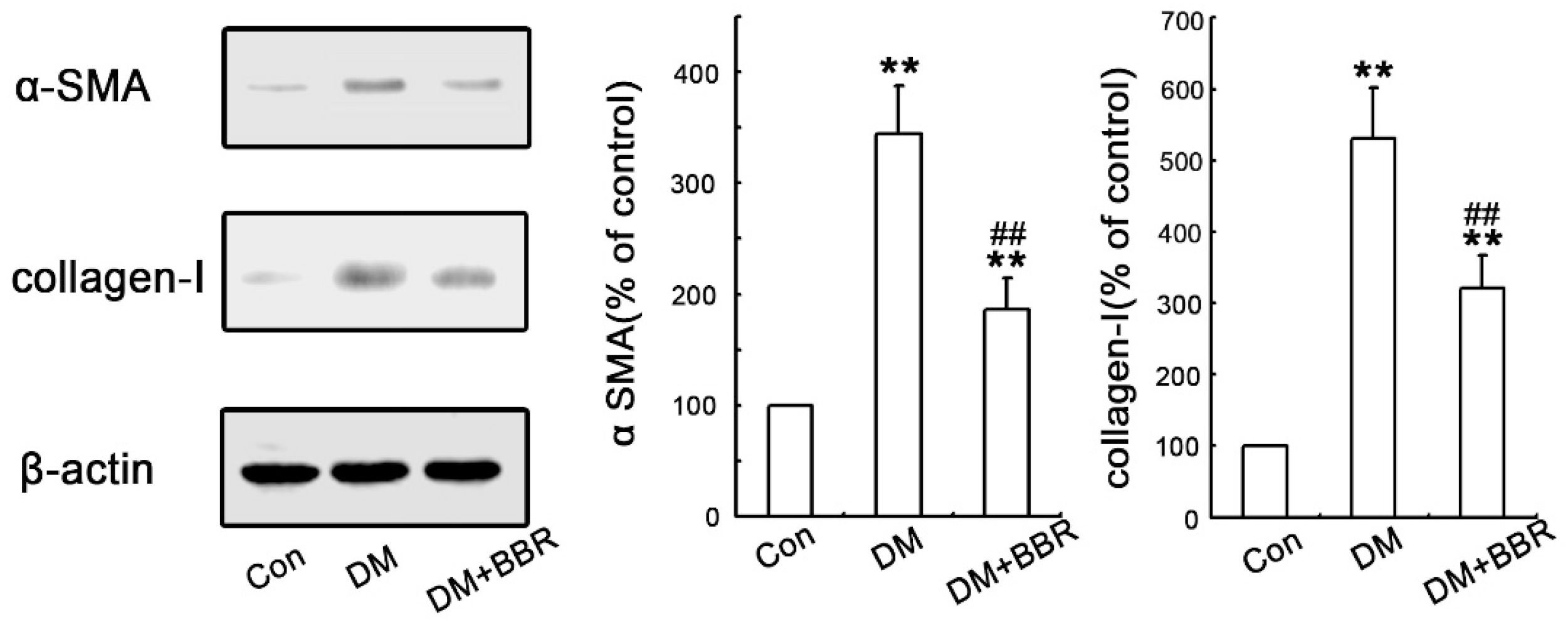
2.2. BBR Can Supress the Fibrosis in Diabetic Kidneys by Inhibiting EMT
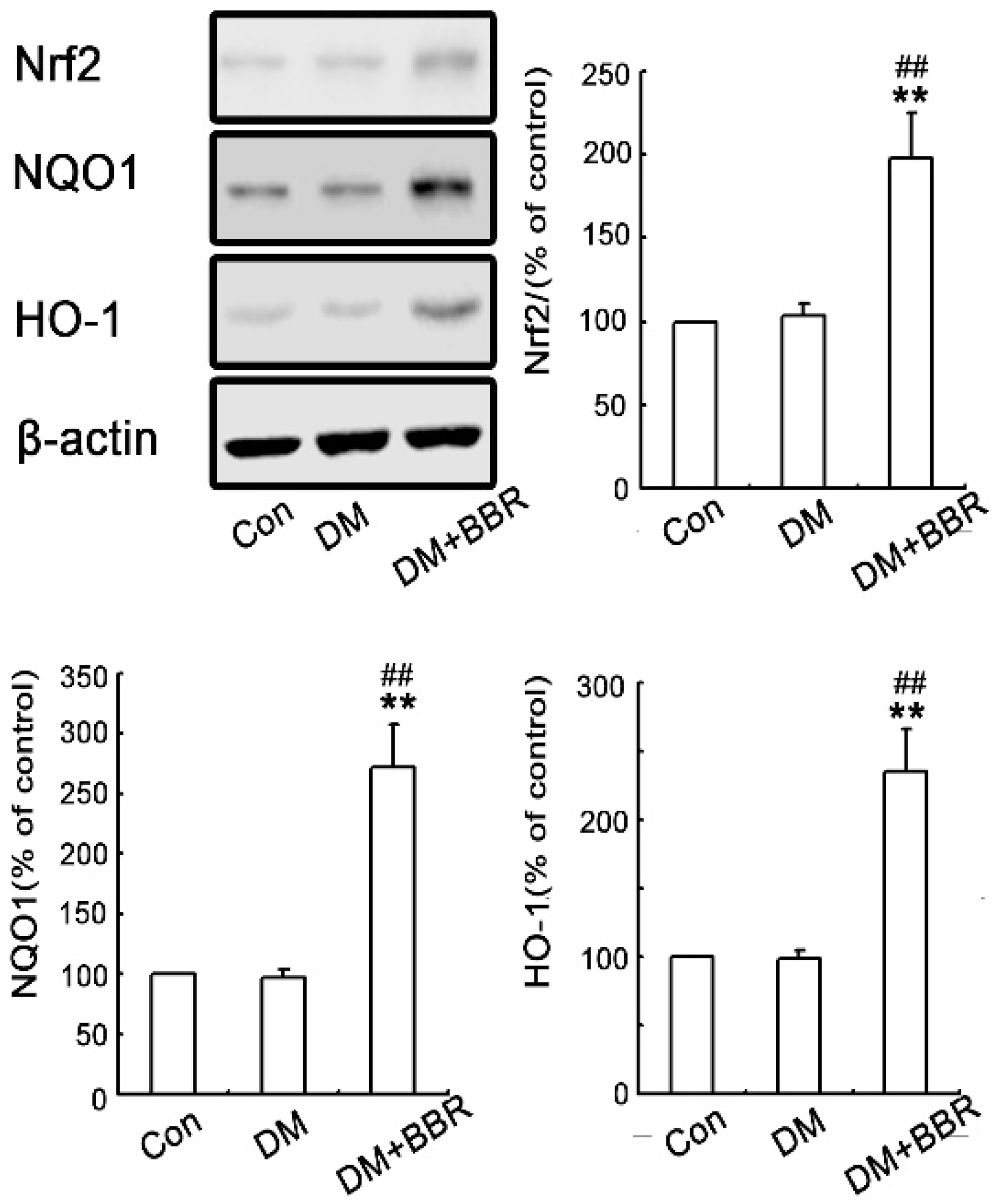
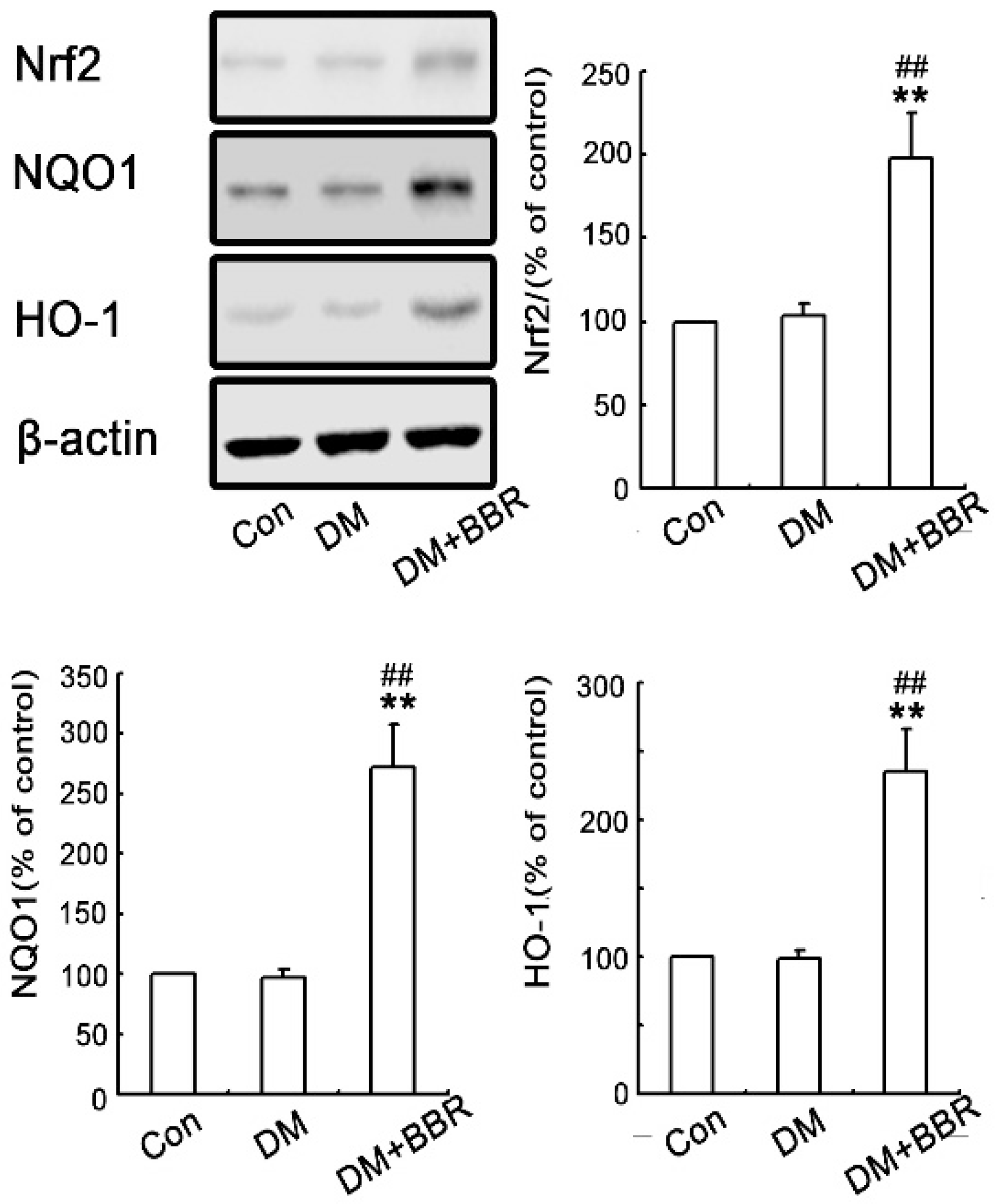
2.3. BBR Enhanced Activation of Nrf2/HO-1 Signaling in Diabetic Mice Kidneys
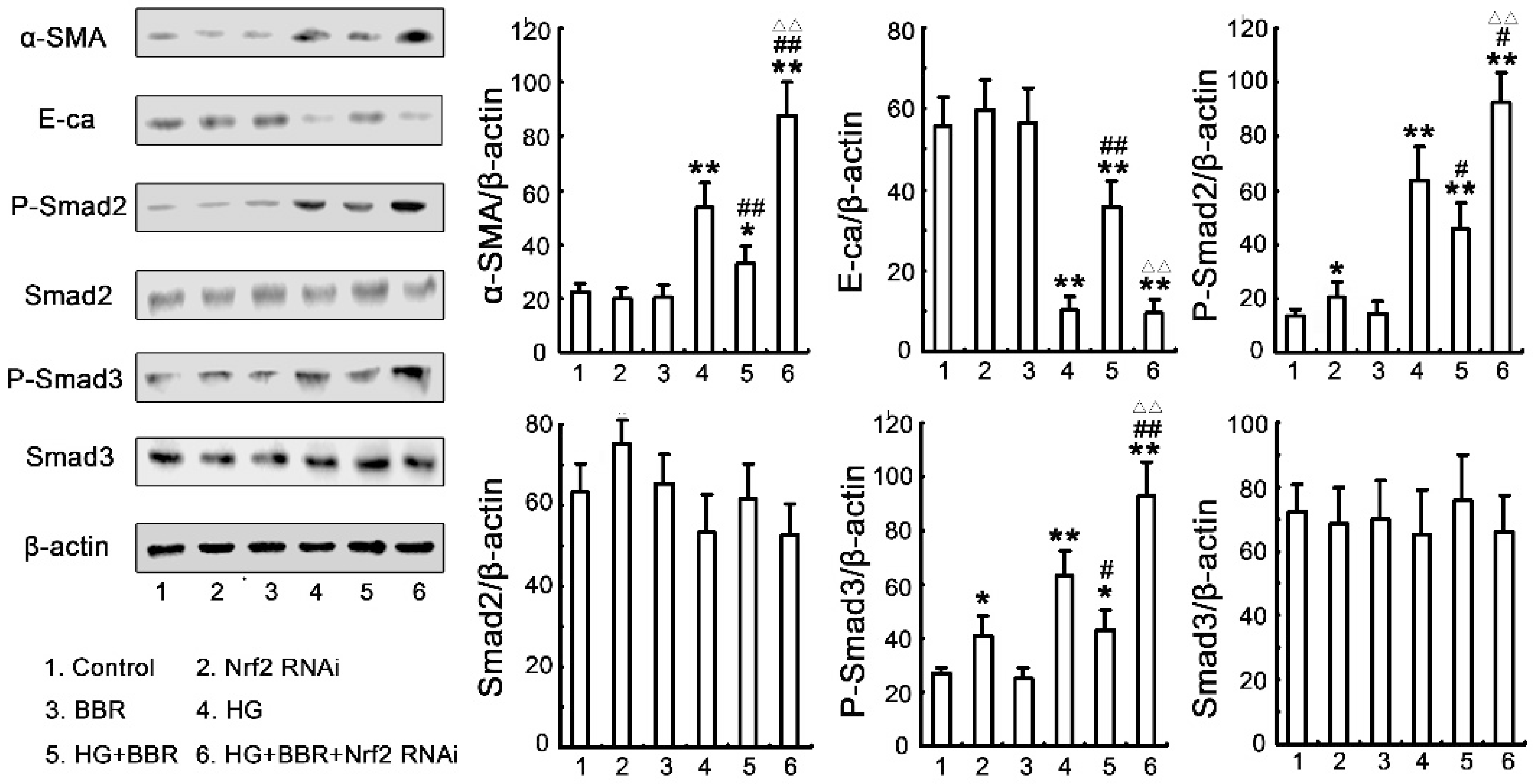
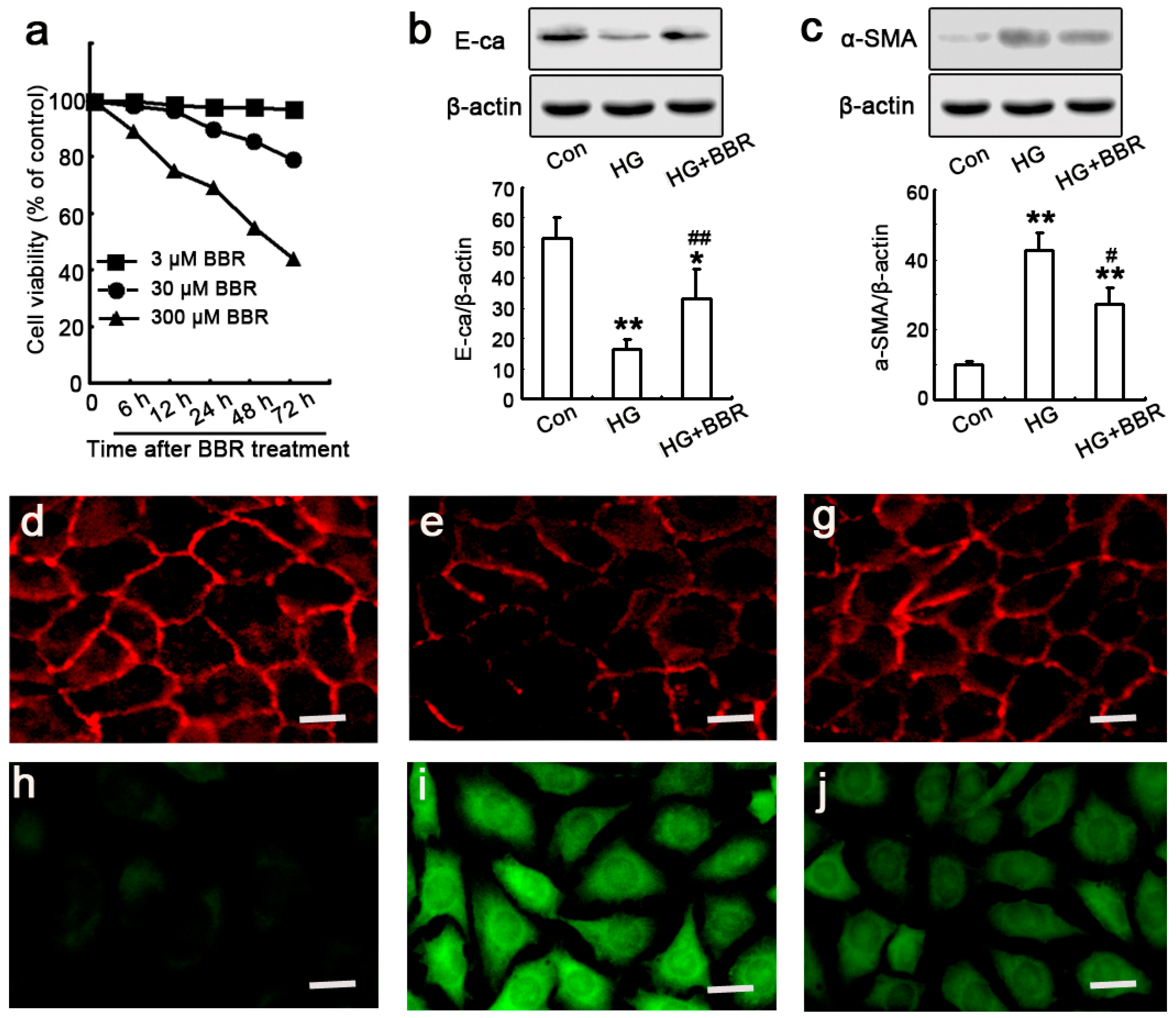
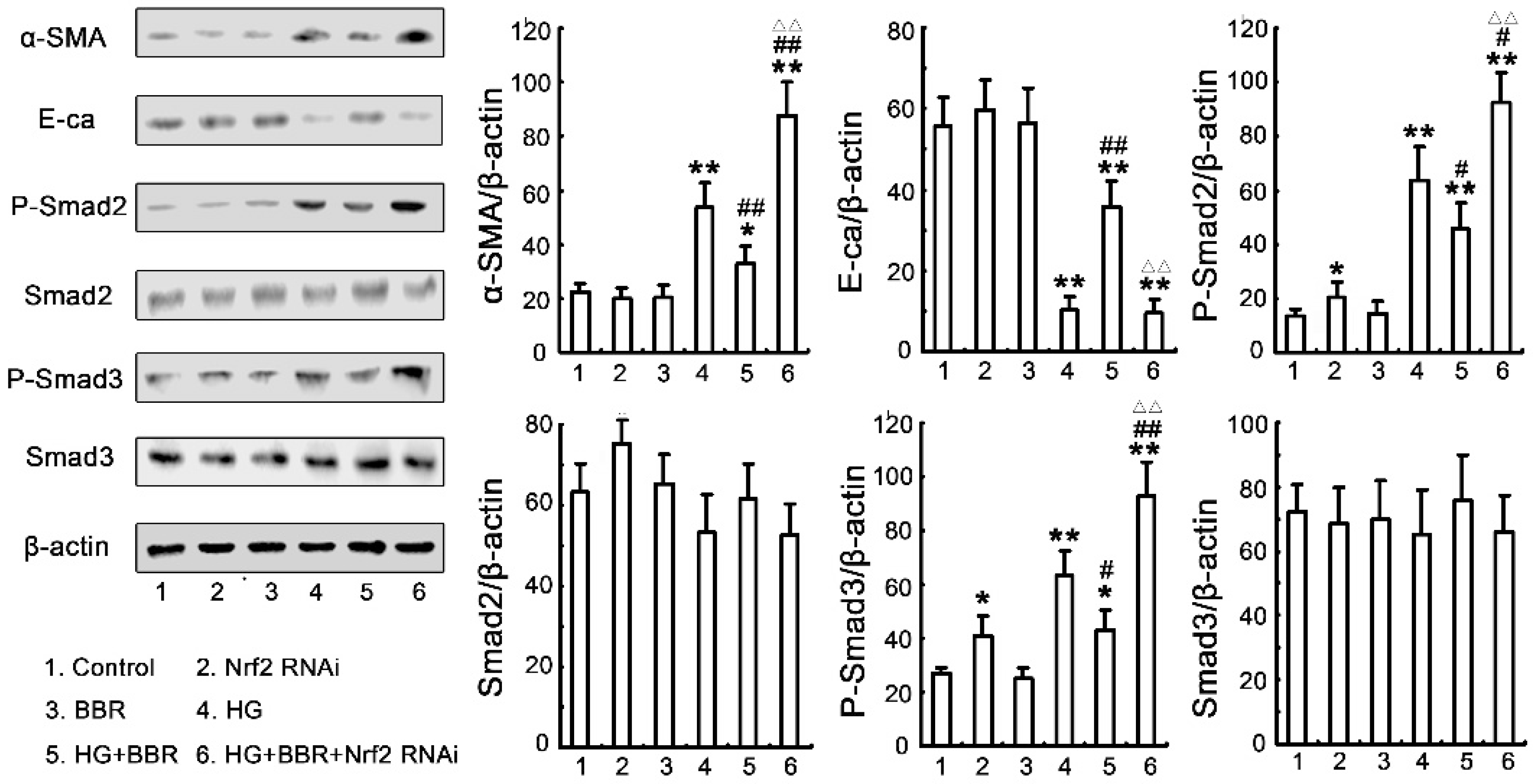
2.4. The Influence of BBR on HG-Induced EMT Markers in NRK-52E Cells
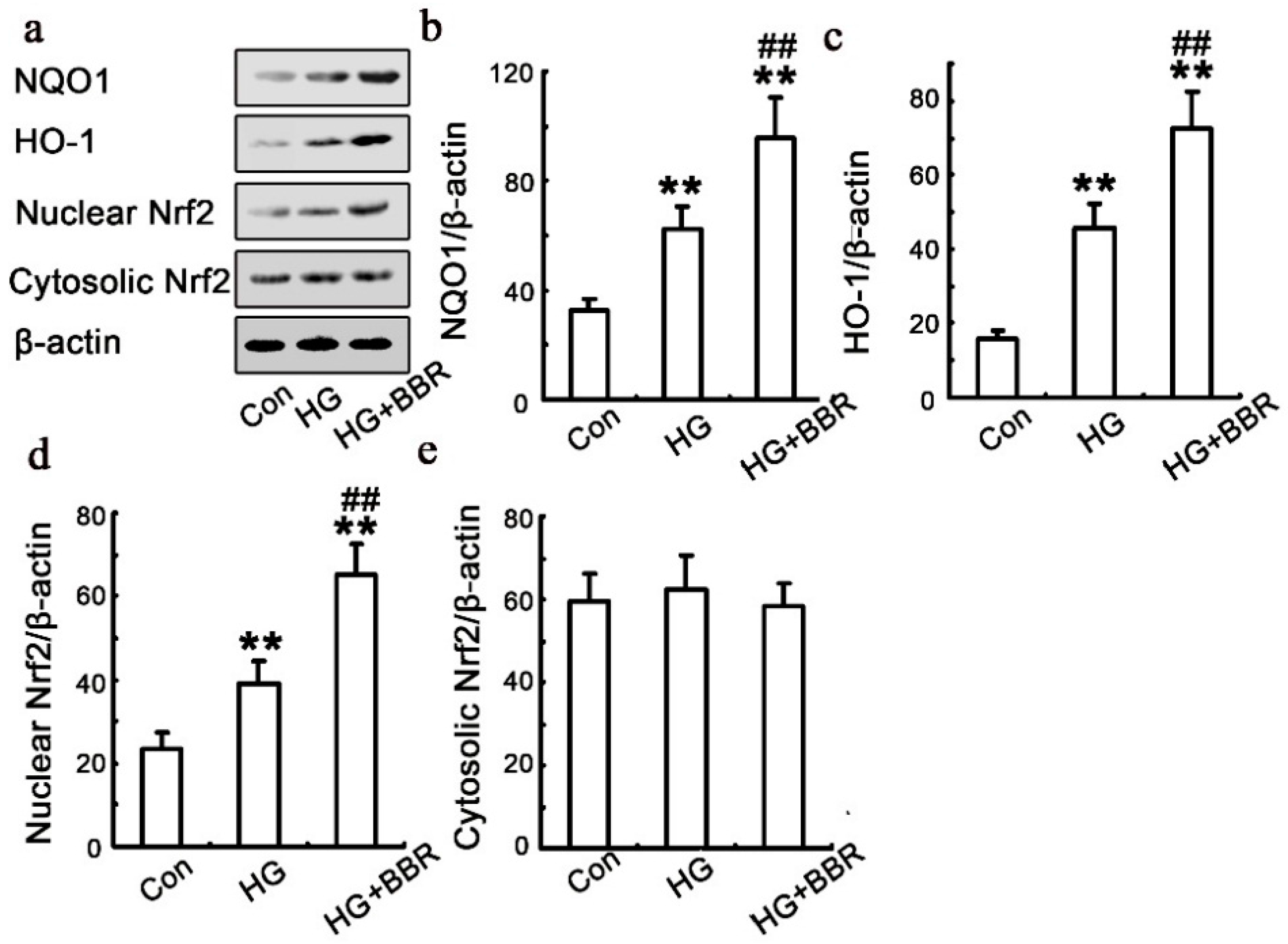
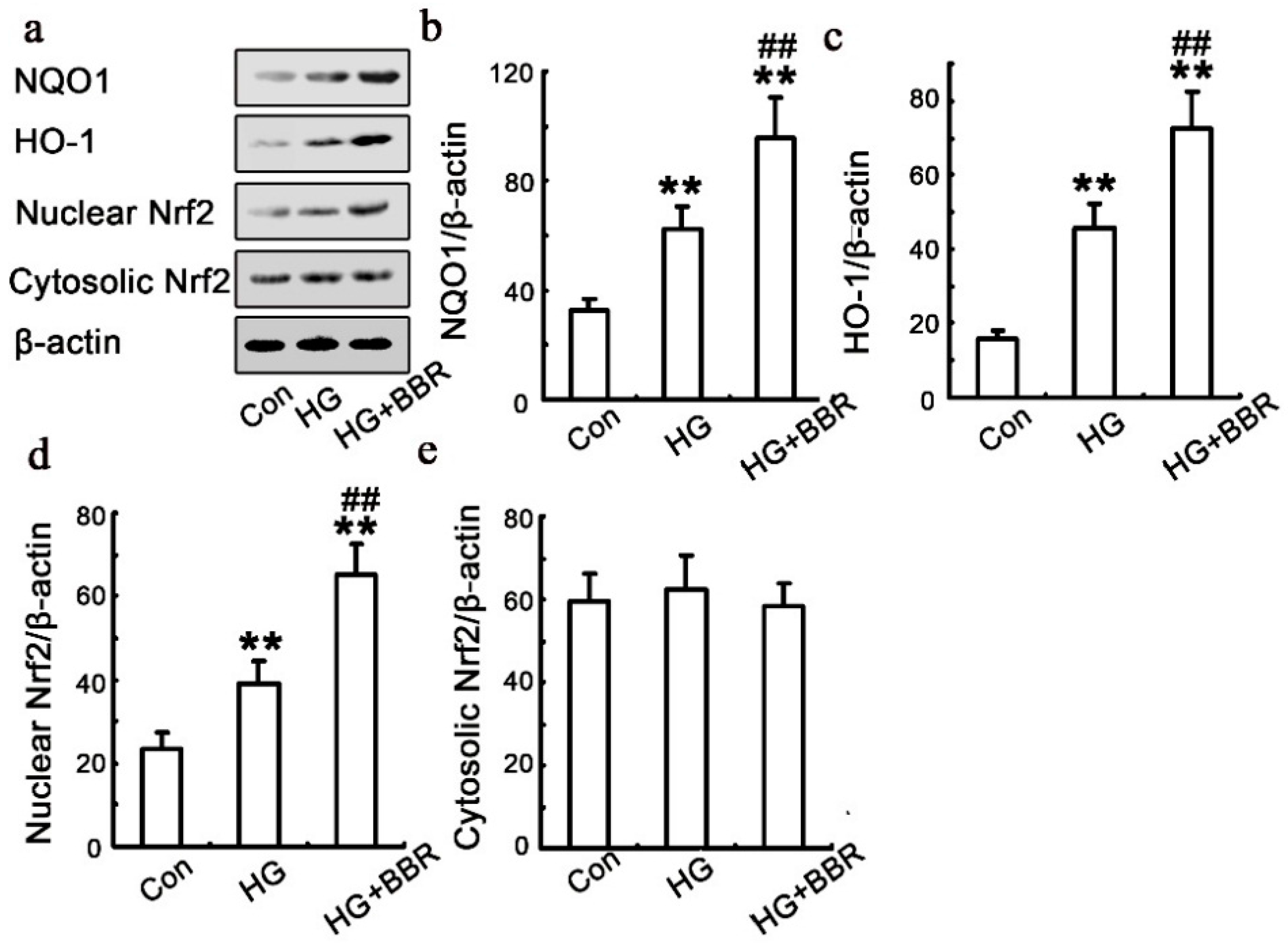
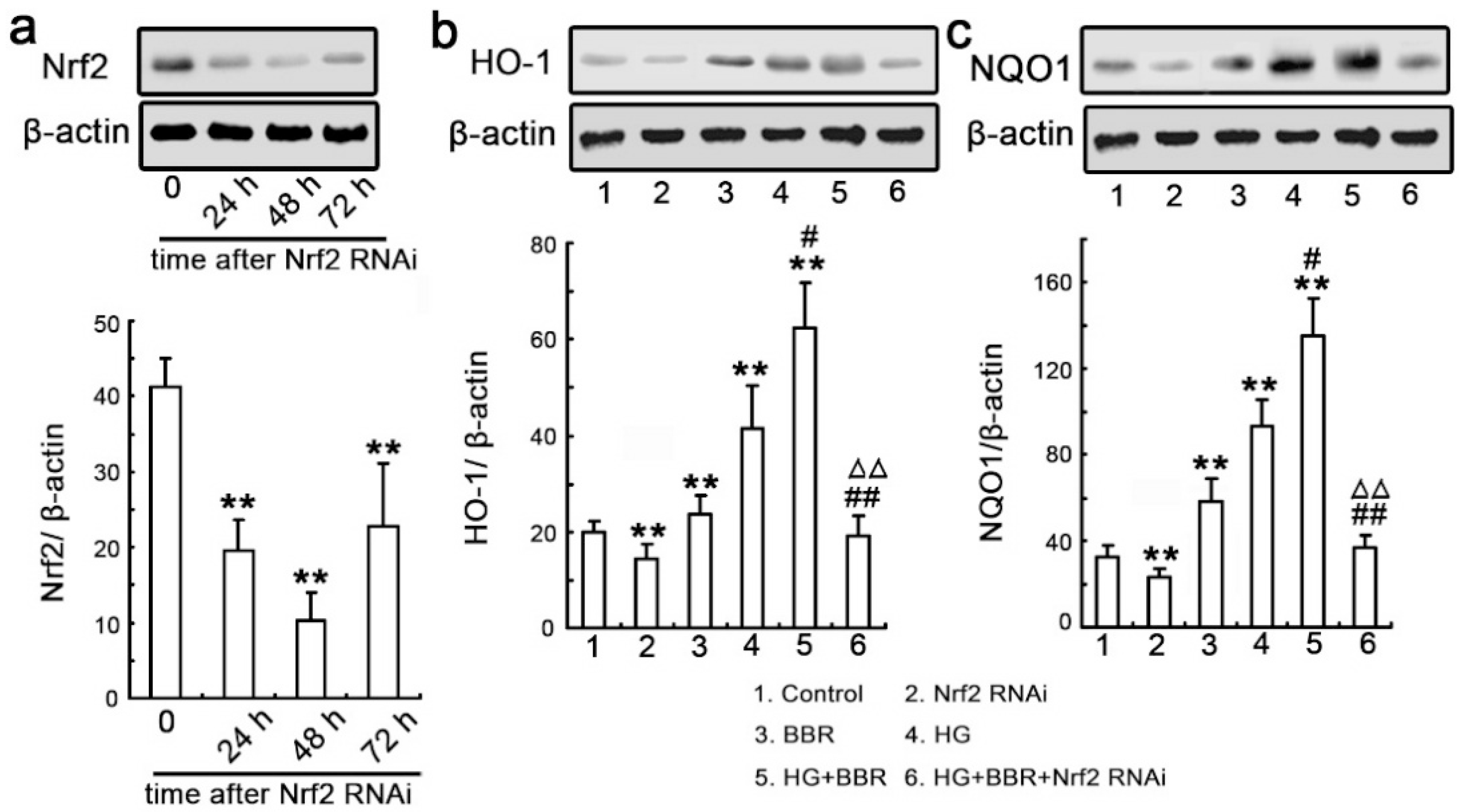
2.5. BBR can Affect HG-Induced Nrf2/HO-1 Signaling in NRK-52E Cells
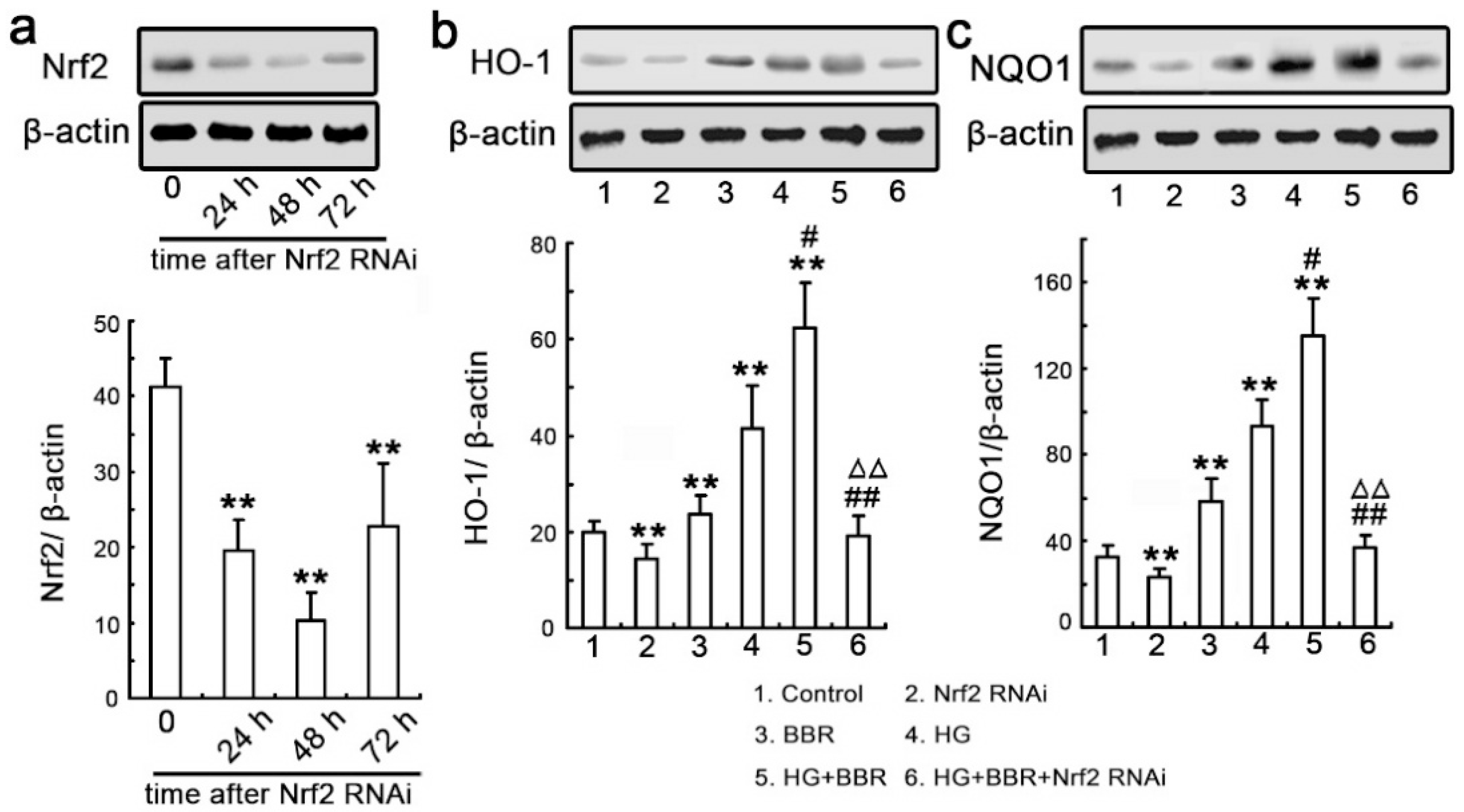
2.6. Knockdown Nrf2 Abrogates BBR-Induced NQO1 and HO-1 Expression
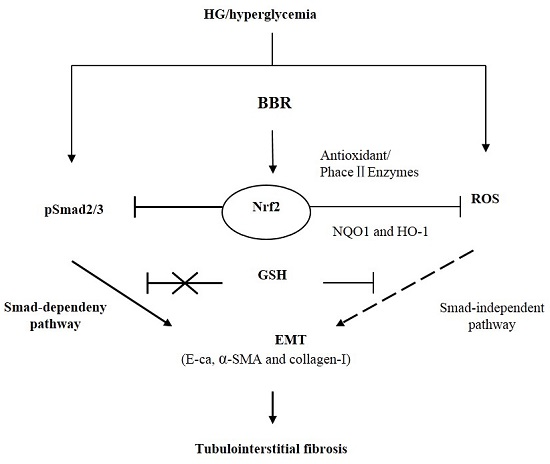
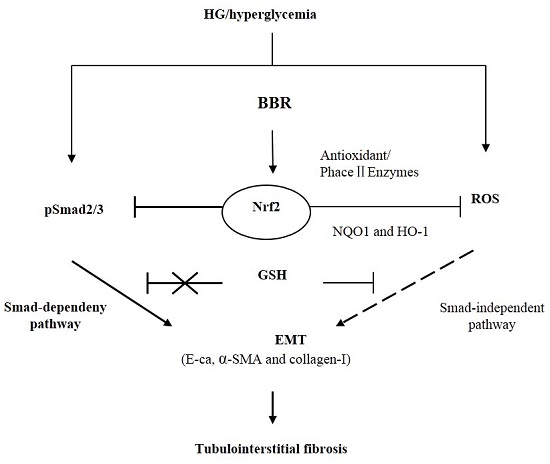
2.7. The Antifibrosis Effect of BBR on DN Is Depended on the Inhibition of Nrf2 Mediated the TGF-β/Smad Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Animal Treatment
4.2. Cell Culture
4.3. Cell Viability
4.4. Transient Transfection with Nrf2-Small Interfering RNA (siRNA)
4.5. Immunofluorescence Staining
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Williamson, J.R.; Tilton, R.G.; Chang, K.; Kilo, C. Basement membrane abnormalities in diabetes mellitus: Relationship to clinical microangiopathy. Diabetes Metab. Rev. 1988, 4, 339–370. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Cox, A.; Wu, L.L.; Allen, T.J.; Hulthen, U.L.; Jerums, G.; Cooper, M.E. Expression of transforming growth factor-beta1 and type IV collagen in the renal tubulointerstitium in experimental diabetes: Effects of ACE inhibition. Diabetes 1998, 47, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Martins, D.; Norris, K. Combating diabetic nephropathy with drug therapy. Curr. Diabet. Rep. 2001, 1, 148–156. [Google Scholar] [CrossRef]
- Border, W.A.; Yamamoto, T.; Noble, N.A. Transforming growth factor beta in diabetic nephropathy. Diabet. Metab. Rev. 1996, 12, 309–339. [Google Scholar] [CrossRef]
- Simonson, M.S. Phenotypic transitions and fibrosis in diabetic nephropathy. Kidney Int. 2007, 71, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Cooper, M.E. The tubulointerstitium in progressive diabetic kidney disease: More than an aftermath of glomerular injury? Kidney Int. 1999, 56, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Ziyadeh, F.N.; Han, D.C.; Cohen, J.A.; Guo, J.; Cohen, M.P. Glycated albumin stimulates fibronectin gene expression in glomerular mesangial cells: Involvement of the transforming growth factor-beta system. Kidney Int. 1998, 53, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Xia, L.; Goldberg, H.J.; Lee, K.W.K.; Shah, A.; Stavar, L.; Masson, E.A.Y.; Momen, A.; Shikatani, E.A.; John, R.; et al. Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes 2013, 62, 3874–3886. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Ziyadeh, F.N. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes 1995, 44, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Chatziantoniou, C.; Dussaule, J.C. Insights into the mechanisms of renal fibrosis: Is it possible to achieve regression? Am. J. Physiol. Ren. Physiol. 2005, 289, F227–F234. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Lee, H.B. Reactive oxygen species and matrix remodeling in diabetic kidney. J. Am. Soc. Nephrol. 2003, 14, S246–S249. [Google Scholar] [CrossRef] [PubMed]
- Rhyu, D.Y.; Yang, Y.; Ha, H.; Lee, G.T.; Song, J.S.; Uh, S.-T.; Lee, H.B. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J. Am. Soc. Nephrol. 2005, 16, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Qaisiya, M.; Coda Zabetta, C.D.; Bellarosa, C.; Tiribelli, C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell Signal. 2014, 26, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-H.; Park, H.-M.; Jung, K.-A.; Choi, H.-G.; Kim, J.-A.; Kim, D.-D.; Kim, G.K.; Kang, K.W.; Ku, S.K.; Kensler, T.W.; et al. The NRF2-heme oxygenase-1 system modulates cyclosporin A-induced epithelial-mesenchymal transition and renal fibrosis. Free Radic. Biol. Med. 2010, 48, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Pokharel, Y.R.; Lim, S.C.; Lim, S.C.; Han, H.-K.; Ryu, C.S.; Kim, S.K.; Kwak, M.K.; Kang, K.W. Inhibition of liver fibrosis by solubilized coenzyme Q10: Role of Nrf2 activation in inhibiting transforming growth factor-beta1 expression. Toxicol. Appl. Pharmacol. 2009, 240, 377–384. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Kan, H.; Cai, L.; Ma, Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J. Mol. Cell. Cardiol. 2009, 46, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Leonard, M.O.; Kieran, N.E.; Howell, K.; Burne, M.J.; Varadarajan, R.; Dhakshinamoorthy, S.; Porter, A.G.; O’Farrelly, C.; Rabb, H.; Taylor, C.T. Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. FASEB J. 2006, 20, 2624–2626. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.Y.; Ka, S.M.; Chao, T.K.; Chang, J.-M.; Lin, S.-H.; Li, C.-Y.; Kuo, M.-T.; Chen, P.; Chen, A. Antroquinonol reduces oxidative stress by enhancing the Nrf2 signaling pathway and inhibits inflammation and sclerosis in focal segmental glomerulosclerosis mice. Free Radic. Biol. Med. 2011, 50, 1503–1516. [Google Scholar] [CrossRef] [PubMed]
- Aleksunes, L.M.; Goedken, M.J.; Rockwell, C.E.; Thomale, J.; Manautou, J.E.; Klaassen, C.D. Transcriptional regulation of renal cytoprotective genes by Nrf2, its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J. Pharmacol. Exp. Ther. 2010, 335, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.J.; Kim, J.Y.; Min, A.K.; Park, K.G.; Harris, R.A.; Kim, H.J.; Lee, I.K. Sulforaphane attenuates hepatic fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-beta/Smad signaling. Free Radic. Biol. Med. 2012, 52, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Qian, Q.; Adaikalakoteswari, A.; Rabbani, N.; Babaei-Jadidi, R.; Thornalley, P.J. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes 2008, 57, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Leng, S.H.; Lu, F.E.; Xu, L.J. Therapeutic effects of berberine in impaired glucose tolerance rats and its influence on insulin secretion. Acta Pharmacol. Sin. 2004, 25, 496–502. [Google Scholar] [PubMed]
- Domitrovic, R.; Cvijanovic, O.; Pernjak-Pugel, E.; Skoda, M.; Mikelic, L.; Crncevic-Orlic, Z. Berberine exerts nephroprotective effect against cisplatin-induced kidney damage through inhibition of oxidative/nitrosative stress, inflammation, autophagy and apoptosis. Food Chem. Toxicol. 2013, 62, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Sheng, M.; Xu, R.; Yu, J.; Cui, K.; Tong, J.; Shi, L.; Ren, H.; Du, H. Berberine protects human renal proximal tubular cells from hypoxia/reoxygenation injury via inhibiting endoplasmic reticulum and mitochondrial stress pathways. J. Transl. Med. 2013, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.Y.; Tseng, Y.T.; Lo, Y.C. Berberine, a natural antidiabetes drug, attenuates glucose neurotoxicity and promotes Nrf2-related neurite outgrowth. Toxicol. Appl. Pharmacol. 2013, 272, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Liu, W.; Xie, X.; Huang, K.; Peng, J.; Huang, J.; Shen, X.; Liu, P.; Yang, H.; Huang, H. Berberine suppresses high glucose-induced TGF-beta1 and fibronectin synthesis in mesangial cells through inhibition of sphingosine kinase 1/AP-1 pathway. Eur. J. Pharmacol. 2012, 697, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Liu, P.; Wu, X.; Liu, W.; Shen, X.; Lan, T.; Xu, S.; Peng, J.; Xie, X.; Huang, H. Berberine attenuates lipopolysaccharide-induced extracelluar matrix accumulation and inflammation in rat mesangial cells: Involvement of NF-kappaB signaling pathway. Mol. Cell. Endocrinol. 2011, 331, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tang, F.; Deng, Y.; Li, X.; Lan, T.; Zhang, X.; Huang, H.; Liu, P. Berberine reduces fibronectin and collagen accumulation in rat glomerular mesangial cells cultured under high glucose condition. Mol. Cell. Biochem. 2009, 325, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, X.; Liu, P.; Shen, X.; Lan, T.; Li, W.; Jiang, Q.; Xie, X.; Huang, H. Effects of berberine on matrix accumulation and NF-kappa B signal pathway in alloxan-induced diabetic mice with renal injury. Eur. J. Pharmacol. 2010, 638, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sun, L.N.; Nie, H.B.; Wang, X.L.; Guan, G.J. Berberine improves kidney function in diabetic mice via AMPK activation. PLoS ONE 2014, 9, e113398. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Wang, Y.; Li, J.; Su, C.; Wu, F.; Xia, W.H.; Yang, Z.; Yu, B.-B.; Qiu, Y.-X.; Tao, J. Berberine improves endothelial function by reducing endothelial microparticles-mediated oxidative stress in humans. Int. J. Cardiol. 2013, 167, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H.; Allen, T.J. Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology 2007, 12, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liang, D.; Chi, Z.H.; Chu, Q.; Zhao, C.; Ma, R.-Z.; Zhao, Y.; Li, H. Effect of zinc on high glucose-induced epithelial-to-mesenchymal transition in renal tubular epithelial cells. Int. J. Mol. Med. 2015, 35, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Lai, K.N. The pathogenic role of the renal proximal tubular cell in diabetic nephropathy. Nephrol. Dial. Transpl. 2012, 27, 3049–3056. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; Mcrobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.J.; Kim, J.Y.; Choi, Y.K.; Kim, H.J.; Jeong, J.Y.; Bae, K.H.; Park, K.G.; Lee, I.K. Dimethylfumarate attenuates renal fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-β/Smad signaling. PLoS ONE 2012, 7, e45870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.J.; Xu, D.; Guo, Y.; Ping, J.; Chen, L.B.; Wang, H. Protection by and anti-oxidant mechanism of berberine against rat liver fibrosis induced by multiple hepatotoxic factors. Clin. Exp. Pharmacol. Physiol. 2008, 35, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Domitrovic, R.; Jakovac, H.; Marchesi, V.V.; Blazekovic, B. Resolution of liver fibrosis by isoquinoline alkaloid berberine in CCl4-intoxicated mice is mediated by suppression of oxidative stress and upregulation of MMP-2 expression. J. Med. Food 2013, 16, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Chitra, P.; Saiprasad, G.; Manikandan, R.; Sudhandiran, G. Berberine attenuates bleomycin induced pulmonary toxicity and fibrosis via suppressing NF-kappaB dependant TGF-beta activation: A biphasic experimental study. Toxicol. Lett. 2013, 219, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Liu, W.; Lan, T.; Xie, X.; Peng, J.; Huang, J.; Wang, S.; Shen, X.; Liu, P.; Huang, H. Berberine reduces fibronectin expression by suppressing the S1P-S1P2 receptor pathway in experimental diabetic nephropathy models. PLoS ONE 2012, 7, e43874. [Google Scholar]
- Xie, X.; Chang, X.; Chen, L.; Huang, K.; Huang, J.; Wang, J.; Shen, X.; Liu, P.; Huang, H. Berberine ameliorates experimental diabetes-induced renal inflammation and fibronectin by inhibiting the activation of RhoA/ROCK signaling. Mol. Cell. Endocrinol. 2013, 381, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Li, P.; Murphy, T.H. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J. Neurosci. 2005, 25, 10321–10335. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Reddy, S.P.; Yamamoto, M.; Kleeberger, S.R. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004, 18, 1258–1260. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Hirayama, A.; Ishizaki, K.; Yamada, A.; Takeuchi, M.; Yamagishi, S.; Morito, N.; Nakano, T.; Ojima, M.; Shimohata, H.; Itoh, K.; et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells 2008, 13, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.Y.; Chen, C.S.; Wu, S.N.; Jong, Y.J.; Lo, Y.C. Berberine activates Nrf2 nuclear translocation and protects against oxidative damage via a phosphatidylinositol 3-kinase/Akt-dependent mechanism in NSC34 motor neuron-like cells. Eur. J. Pharm Sci. 2012, 46, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Ren. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [PubMed]
- Anwar, A.A.; Li, F.Y.; Leake, D.S.; Ishii, T.; Mann, G.E.; Siow, R.C. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: Role of mitogen-activated protein kinases and Nrf2. Free Radic. Biol. Med. 2005, 39, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Wang, L.; Zhang, J.; Numazawa, S.; Tang, H.; Tang, X.; Han, X.; Li, J.; Yang, M.; Wang, Z.; et al. The crosstalk between Nrf2 and AMPK signal pathways is important for the anti-inflammatory effect of berberine in LPS-stimulated macrophages and endotoxin-shocked mice. Antioxid. Redox Signal. 2014, 20, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Kie, J.H.; Kapturczak, M.H.; Traylor, A.; Agarwal, A.; Hill-Kapturczak, N. Heme oxygenase-1 deficiency promotes epithelial-mesenchymal transition and renal fibrosis. J. Am. Soc. Nephrol. 2008, 19, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Hung, C.C.; Hsu, H.H.; Jing, Y.H.; Yang, C.W.; Chen, J.K. Resveratrol ameliorates early diabetic nephropathy associated with suppression of augmented TGF-beta/smad and ERK1/2 signaling in streptozotocin-induced diabetic rats. Chem. Biol. Interact. 2011, 190, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Lin, H.; Wu, T.; Wang, T.; Wang, W.; Xie, H.; Zhang, J.; Feng, Z. Inhibition of TGF-beta1-receptor posttranslational core fucosylation attenuates rat renal interstitial fibrosis. Kidney Int. 2013, 84, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Yuan, H.; Xu, Z.G.; Lanting, L.; Li, S.-L.; Wang, M.; Hu, M.C.-T.; Reddy, M.A.; Natarajan, R. Role of the Akt/FoxO3a pathway in TGF-beta1-mediated mesangial cell dysfunction: A novel mechanism related to diabetic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 3325–3335. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Mora, C.; Muros, M.; Garcia, J. Urinary tumour necrosis factor-alpha excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol. Dial. Transplant. 2006, 21, 3428–3434. [Google Scholar] [CrossRef] [PubMed]
- Schmid, H.; Boucherot, A.; Yasuda, Y.; Henger, A.; Brunner, B.; Eichinger, F.; Nitsche, A.; Kiss, E.; Bleich, M.; Gröne, H.-J.; et al. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes 2006, 55, 2993–3003. [Google Scholar] [CrossRef] [PubMed]
- Hills, C.E.; Squires, P.E. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am. J. Nephrol. 2010, 31, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liang, D.; Guo, B.; Deng, W.; Chi, Z.-H.; Cai, Y.; Wang, L.; Ma, J. Zinc transporter 5 and zinc transporter 7 induced by high glucose protects peritoneal mesothelial cells from undergoing apoptosis. Cell Signal. 2013, 25, 999–1010. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Control | DM | DM + BBR |
|---|---|---|---|
| Blood glucose (mM) | 5.15 ± 0.12 | 26.32 ± 4.43 ** | 15.48 ± 4.97 **,## |
| Body weight (g) | 29.3 ± 1.15 | 20.35 ± 2.58 * | 24.92 ± 2.16 |
| Kidney weight (g) | 0.33 ± 0.03 | 0.31 ± 0.02 | 0.32 ± 0.04 |
| KW/BW (%) | 1.13 ± 0.02 | 1.52 ± 0.08 * | 1.28 ± 0.05 # |
| BUN (mM) | 6.12 ± 0.53 | 41.39 ± 5.33 ** | 19.63 ± 2.87 **,## |
| Cr (µM) | 28.54 ± 1.68 | 87.57 ± 11.61 ** | 45.82 ± 6.17 **,## |
| Albuminuria (µg/24 h) | 9.7 ± 0.58 | 42.3 ± 3.86 ** | 25.9 ± 2.17 **,## |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; He, H.; Liang, D.; Jiang, Y.; Liang, W.; Chi, Z.-H.; Ma, J. Protective Effects of Berberine on Renal Injury in Streptozotocin (STZ)-Induced Diabetic Mice. Int. J. Mol. Sci. 2016, 17, 1327. https://doi.org/10.3390/ijms17081327
Zhang X, He H, Liang D, Jiang Y, Liang W, Chi Z-H, Ma J. Protective Effects of Berberine on Renal Injury in Streptozotocin (STZ)-Induced Diabetic Mice. International Journal of Molecular Sciences. 2016; 17(8):1327. https://doi.org/10.3390/ijms17081327
Chicago/Turabian StyleZhang, Xiuli, Hui He, Dan Liang, Yan Jiang, Wei Liang, Zhi-Hong Chi, and Jianfei Ma. 2016. "Protective Effects of Berberine on Renal Injury in Streptozotocin (STZ)-Induced Diabetic Mice" International Journal of Molecular Sciences 17, no. 8: 1327. https://doi.org/10.3390/ijms17081327
APA StyleZhang, X., He, H., Liang, D., Jiang, Y., Liang, W., Chi, Z.-H., & Ma, J. (2016). Protective Effects of Berberine on Renal Injury in Streptozotocin (STZ)-Induced Diabetic Mice. International Journal of Molecular Sciences, 17(8), 1327. https://doi.org/10.3390/ijms17081327