
Neuroimmunological Implications of AQP4 in Astrocytes

Abstract
:
1. Introduction
1.1. Aquaporin-4 (AQP4)
1.2. Astrocytes
1.2.1. Glial Fibrillary Acidic Protein
1.2.2. Oxidative Stress in Astrocytes
1.2.3. Calcium Signaling in Astrocytes
1.2.4. Astrocytes and Immune Cells
2. Neuroimmunological Role of AQP4
2.1. AQP4 in Neuromyelitis Optica (NMO)
2.2. AQP4 in Alzheimer’s Disease
2.3. AQP4 in Parkinson’s Disease
2.4. AQP4 in Depression
2.5. AQP4 in Blood-Retinal Barrier Breakdown
2.6. AQP4 in Traumatic Brain Injury
2.7. AQP4 in Ischemia
3. AQP4 in Reactive Astrocytes
4. AQP4 in Neural Stem Cells
5. AQP4 in Microglia
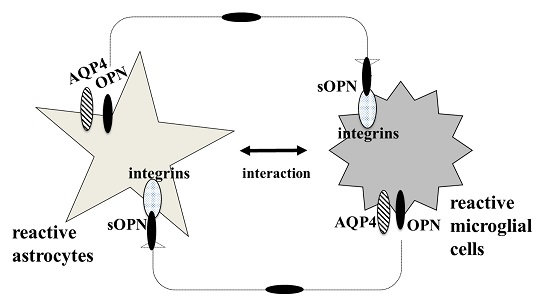
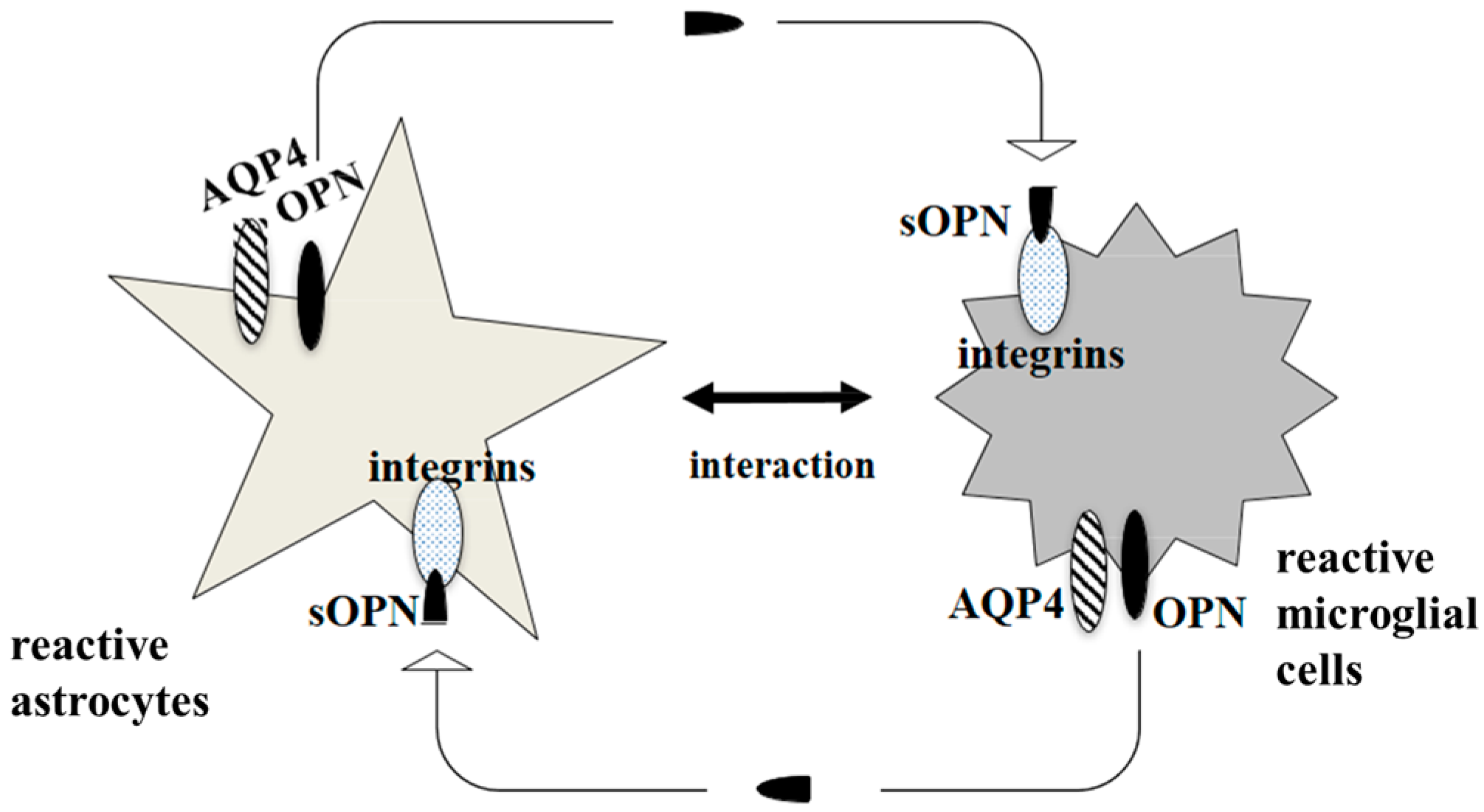
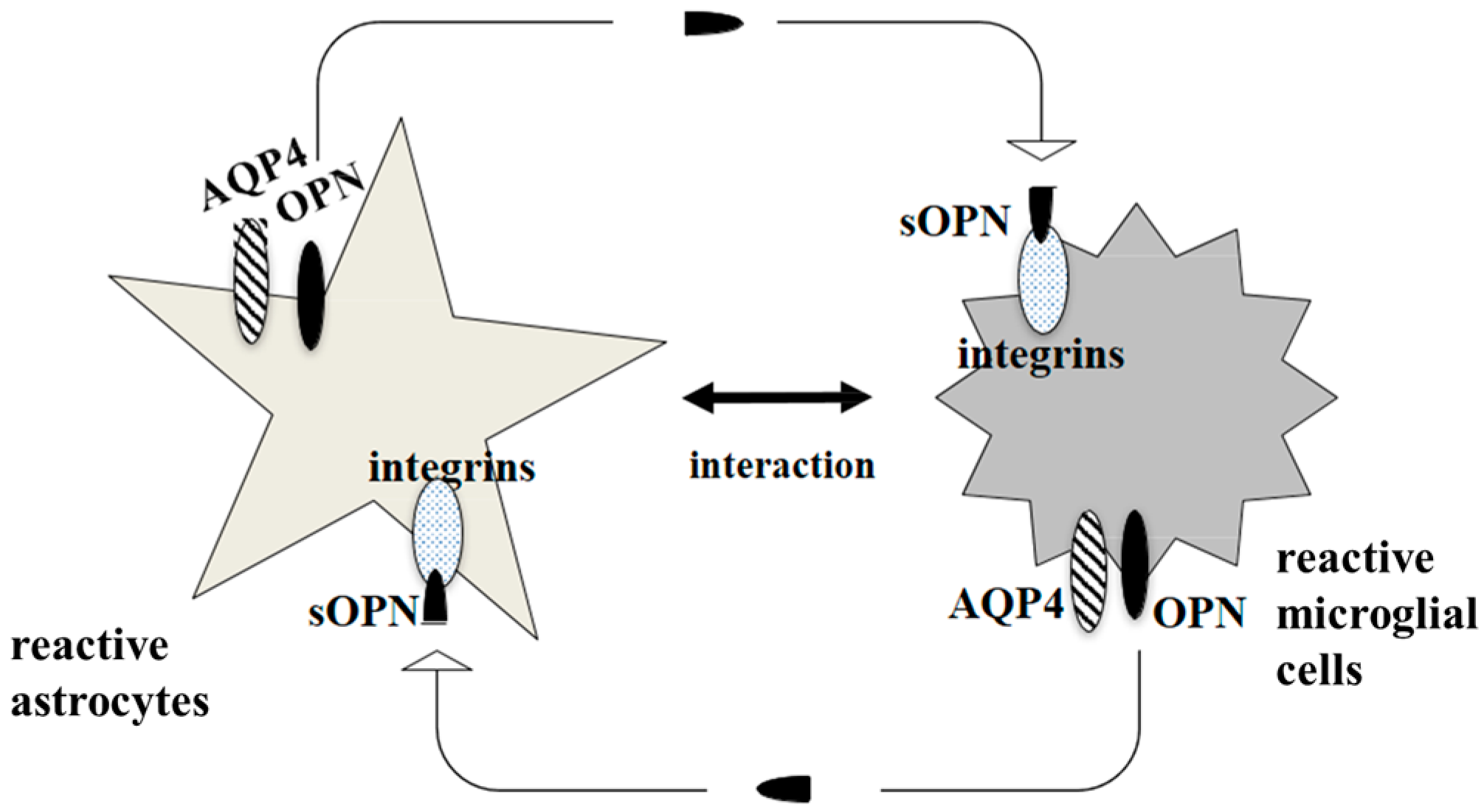
6. AQP4 Function in Astrocyte and Microglial Communication
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [PubMed]
- Camassa, L.M.; Lunde, L.K.; Hoddevik, E.H.; Stensland, M.; Boldt, H.B.; de Souza, G.A.; Ottersen, O.P.; Amiry-Moghaddam, M. Mechanisms underlying AQP4 accumulation in astrocyte endfeet. Glia 2015, 63, 2073–2091. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Fukuda, A.M.; Jullienne, A.; Petry, K.G. Aquaporin and brain diseases. Biochim. Biophys. Acta 2014, 1840, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Yukutake, Y.; Tsuji, S.; Hirano, Y.; Adachi, T.; Takahashi, T.; Fujihara, K.; Agre, P.; Yasui, M.; Suematsu, M. Mercury chloride decreases the water permeability of aquaporin-4-reconstituted proteoliposomes. Biol. Cell 2008, 100, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Gunnarson, E.; Axehult, G.; Baturina, G.; Zelenin, S.; Zelenina, M.; Aperia, A. Lead induces increased water permeability in astrocytes expressing aquaporin 4. Neuroscience 2005, 136, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Musa-Aziz, R.; Chen, L.M.; Pelletier, M.F.; Boron, W.F. Relative CO2/NH3 selectivities of AQP1, AQP4, AQP5, AmtB, and RhAG. Proc. Natl. Acad. Sci. USA 2009, 106, 5406–5411. [Google Scholar] [CrossRef] [PubMed]
- Seifert, G.; Schilling, K.; Steinhauser, C. Astrocyte dysfunction in neurological disorders: A molecular perspective. Nat. Rev. Neurosci. 2006, 7, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Wallace, G.C.t.; Holmes, C.; McDowell, M.L.; Smith, J.A.; Marshall, J.D.; Bonilha, L.; Edwards, J.C.; Glazier, S.S.; Ray, S.K.; et al. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 2012, 220, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Day, R.E.; Taylor, L.H.; Salman, M.M.; Bill, R.M.; Conner, M.T.; Conner, A.C. Identification and molecular mechanisms of the rapid tonicity-induced relocalization of the aquaporin 4 channel. J. Biol. Chem. 2015, 290, 16873–16881. [Google Scholar] [CrossRef] [PubMed]
- Nicchia, G.P.; Nico, B.; Camassa, L.M.; Mola, M.G.; Loh, N.; Dermietzel, R.; Spray, D.C.; Svelto, M.; Frigeri, A. The role of aquaporin-4 in the blood-brain barrier development and integrity: Studies in animal and cell culture models. Neuroscience 2004, 129, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Frigeri, A.; Nicchia, G.P.; Corsi, P.; Ribatti, D.; Quondamatteo, F.; Herken, R.; Girolamo, F.; Marzullo, A.; Svelto, M.; et al. Severe alterations of endothelial and glial cells in the blood-brain barrier of dystrophic mdx mice. Glia 2003, 42, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Tait, M.J.; Reza, A.; Davies, D.C.; Bell, B.A.; Verkman, A.S.; Papadopoulos, M.C. AQP4 gene deletion in mice does not alter blood-brain barrier integrity or brain morphology. Neuroscience 2009, 161, 764–772. [Google Scholar] [CrossRef] [PubMed]
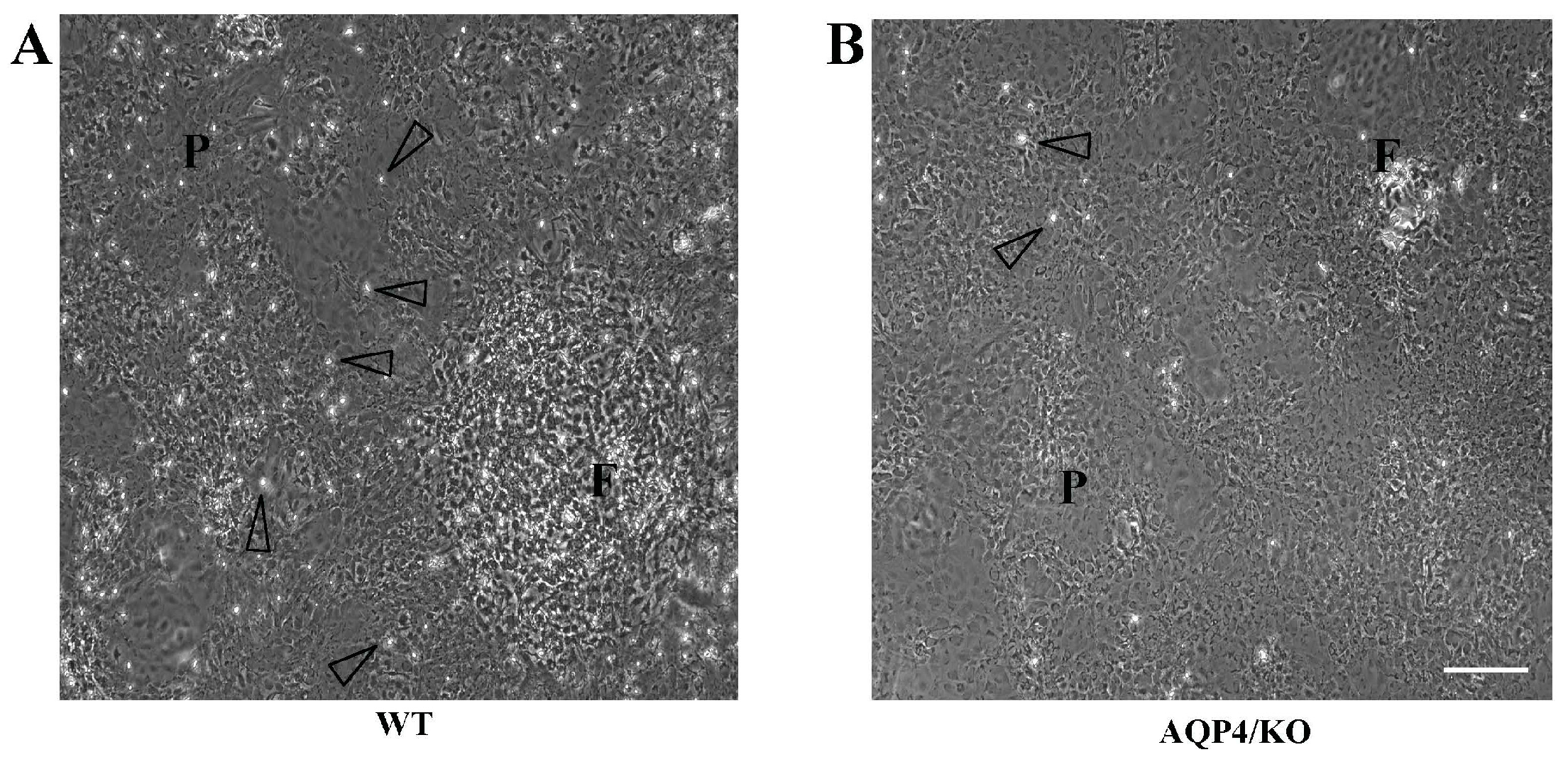
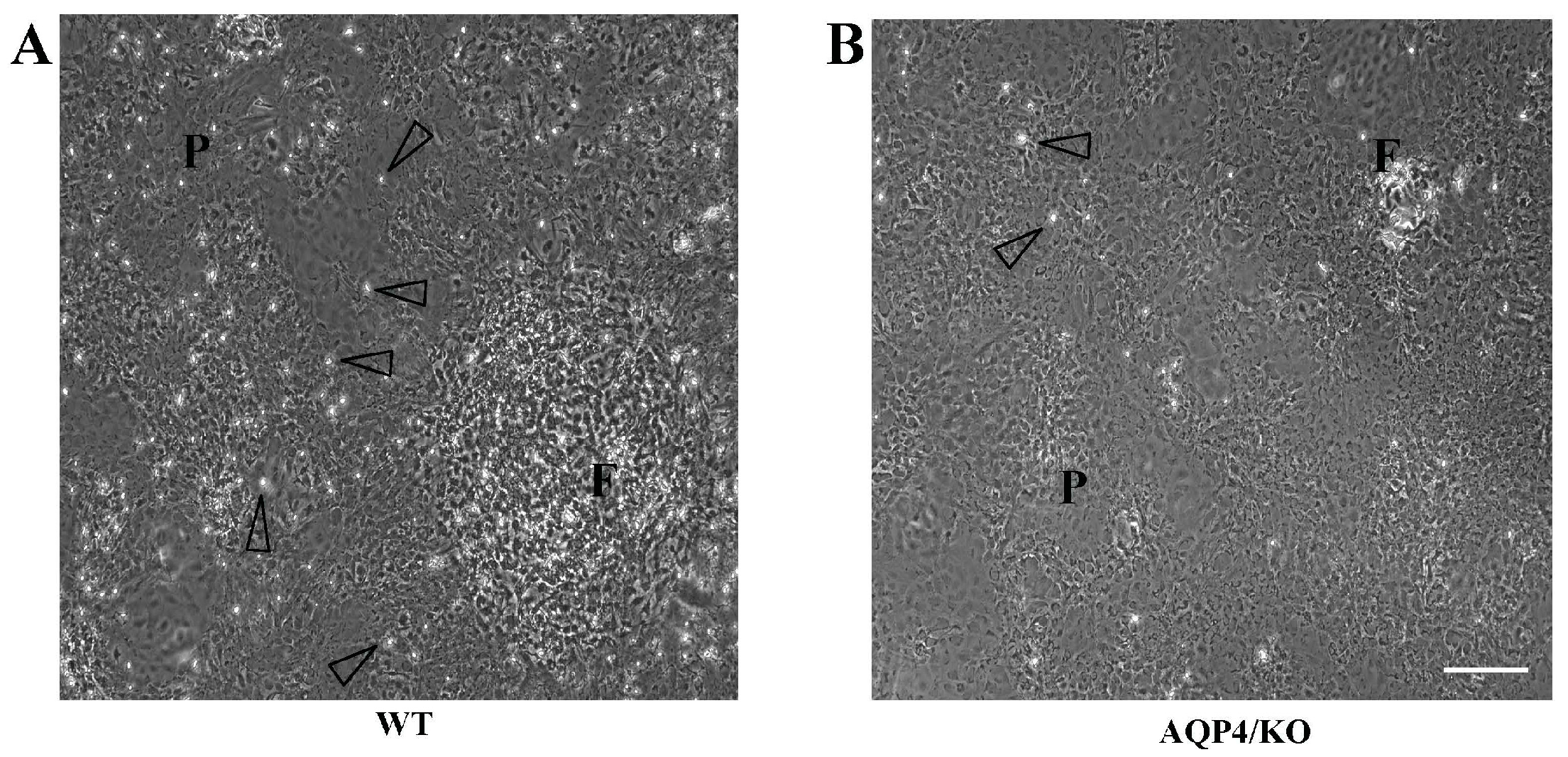
- Ikeshima-Kataoka, H.; Abe, Y.; Yasui, M. Aquaporin 4-dependent expression of glial fibrillary acidic protein and tenascin-C in activated astrocytes in stab wound mouse brain and in primary culture. J. Neurosci. Res. 2015, 93, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Szu, J.I.; Binder, D.K. The role of astrocytic aquaporin-4 in synaptic plasticity and learning and memory. Front. Integr. Neurosci. 2016, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Skucas, V.A.; Mathews, I.B.; Yang, J.; Cheng, Q.; Treister, A.; Duffy, A.M.; Verkman, A.S.; Hempstead, B.L.; Wood, M.A.; Binder, D.K.; et al. Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J. Neurosci. 2011, 31, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Liu, M.; Wu, X.; Wang, F.; Ding, J.; Chen, J.; Hu, G. Aquaporin-4 promotes memory consolidation in Morris water maze. Brain Struct. Funct. 2013, 218, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, M.X.; Luo, Y.; Chen, T.; Liu, J.; Fang, P.; Jiang, B.; Hu, Z.L.; Jin, Y.; Chen, J.G.; et al. Chronic ceftriaxone treatment rescues hippocampal memory deficit in AQP4 knockout mice via activation of GLT-1. Neuropharmacology 2013, 75, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Haj-Yasein, N.N.; Jensen, V.; Ostby, I.; Omholt, S.W.; Voipio, J.; Kaila, K.; Ottersen, O.P.; Hvalby, O.; Nagelhus, E.A. Aquaporin-4 regulates extracellular space volume dynamics during high-frequency synaptic stimulation: A gene deletion study in mouse hippocampus. Glia 2012, 60, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Eng, L.F.; Ghirnikar, R.S. GFAP and astrogliosis. Brain Pathol. 1994, 4, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Dotti, M.T.; Buccoliero, R.; Lee, A.; Gorospe, J.R.; Flint, D.; Galluzzi, P.; Bianchi, S.; D’Eramo, C.; Naidu, S.; Federico, A.; et al. An infantile case of Alexander disease unusual for its MRI features and a GFAP allele carrying both the p.Arg79His mutation and the p.Glu223Gln coding variant. J. Neurol. 2009, 256, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M. Role of GFAP in CNS injuries. Neurosci. Lett. 2014, 565, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, W.; Edelmann, W.; Bieri, P.L.; Chiu, F.C.; Cowan, N.J.; Kucherlapati, R.; Raine, C.S. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron 1996, 17, 607–615. [Google Scholar] [CrossRef]
- Yuasa, S. Development of astrocytes in the mouse embryonic cerebrum tracked by tenascin-C gene expression. Arch. Histol. Cytol. 2001, 64, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, S. Development of astrocytes in the mouse hippocampus as tracked by tenascin-C gene expression. Arch. Histol. Cytol. 2001, 64, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Ikeshima-Kataoka, H.; Yuasa, S. Selective ablation of an astroglial subset by toxic gene expression driven by tenascin promoter. Neurol. Res. 2008, 30, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Ikeshima-Kataoka, H.; Saito, S.; Yuasa, S. Tenascin-C is required for proliferation of astrocytes in primary culture. In Vivo 2007, 21, 629–633. [Google Scholar] [PubMed]
- Ikeshima-Kataoka, H.; Shen, J.S.; Eto, Y.; Saito, S.; Yuasa, S. Alteration of inflammatory cytokine production in the injured central nervous system of tenascin-deficient mice. In Vivo 2008, 22, 409–413. [Google Scholar] [PubMed]
- Fernandez-Fernandez, S.; Almeida, A.; Bolanos, J.P. Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J. 2012, 443, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Genis, L.; Davila, D.; Fernandez, S.; Pozo-Rodrigalvarez, A.; Martinez-Murillo, R.; Torres-Aleman, I. Astrocytes require insulin-like growth factor I to protect neurons against oxidative injury. F1000Research 2014, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Pannicke, T.; Wurm, A.; Iandiev, I.; Hollborn, M.; Linnertz, R.; Binder, D.K.; Kohen, L.; Wiedemann, P.; Steinhauser, C.; Reichenbach, A.; et al. Deletion of aquaporin-4 renders retinal glial cells more susceptible to osmotic stress. J. Neurosci. Res. 2010, 88, 2877–2888. [Google Scholar] [CrossRef] [PubMed]
- Nase, G.; Helm, P.J.; Enger, R.; Ottersen, O.P. Water entry into astrocytes during brain edema formation. Glia 2008, 56, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Thrane, A.S.; Rappold, P.M.; Fujita, T.; Torres, A.; Bekar, L.K.; Takano, T.; Peng, W.; Wang, F.; Rangroo Thrane, V.; Enger, R.; et al. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc. Natl. Acad. Sci. USA 2011, 108, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Patel, S.; Khakh, B.S. Probing the complexities of astrocyte calcium signaling. Trends Cell Biol. 2016, 26, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Nuriya, M.; Hirase, H. Involvement of astrocytes in neurovascular communication. Prog. Brain Res. 2016, 225, 41–62. [Google Scholar] [PubMed]
- Tian, G.F.; Takano, T.; Lin, J.H.; Wang, X.; Bekar, L.; Nedergaard, M. Imaging of cortical astrocytes using 2-photon laser scanning microscopy in the intact mouse brain. Adv. Drug Deliv. Rev. 2006, 58, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Van Neerven, S.; Nemes, A.; Imholz, P.; Regen, T.; Denecke, B.; Johann, S.; Beyer, C.; Hanisch, U.K.; Mey, J. Inflammatory cytokine release of astrocytes in vitro is reduced by all-trans retinoic acid. J. Neuroimmunol. 2010, 229, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Barcia, C., Sr.; Mitxitorena, I.; Carrillo-de Sauvage, M.A.; Gallego, J.M.; Perez-Valles, A.; Barcia, C., Jr. Imaging the microanatomy of astrocyte-T-cell interactions in immune-mediated inflammation. Front. Cell Neurosci. 2013, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Neves, J.; Zhu, J.; Sousa-Victor, P.; Konjikusic, M.; Riley, R.; Chew, S.; Qi, Y.; Jasper, H.; Lamba, D.A. Immune modulation by MANF promotes tissue repair and regenerative success in the retina. Science 2016, 353, aaf3646. [Google Scholar] [CrossRef] [PubMed]
- Senecal, V.; Deblois, G.; Beauseigle, D.; Schneider, R.; Brandenburg, J.; Newcombe, J.; Moore, C.S.; Prat, A.; Antel, J.; Arbour, N. Production of IL-27 in multiple sclerosis lesions by astrocytes and myeloid cells: Modulation of local immune responses. Glia 2016, 64, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.G.; Guo, Y.; Jentoft, M.E.; Parisi, J.E.; Lennon, V.A.; Pittock, S.J.; Weinshenker, B.G.; Wingerchuk, D.M.; Giannini, C.; Metz, I.; et al. Diagnostic utility of aquaporin-4 in the analysis of active demyelinating lesions. Neurology 2015, 84, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Jacob, A.; Matiello, M.; Wingerchuk, D.M.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. Neuromyelitis optica: Changing concepts. J. Neuroimmunol. 2007, 187, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, S.; Mori, M.; Okuta, A.; Kamegawa, A.; Fujiyoshi, Y.; Yoshiyama, Y.; Mitsuoka, K.; Ishibashi, K.; Sasaki, S.; Hattori, T.; et al. Neuromyelitis optica and anti-aquaporin-4 antibodies measured by an enzyme-linked immunosorbent assay. J. Neuroimmunol. 2008, 196, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Marignier, R.; Nicolle, A.; Watrin, C.; Touret, M.; Cavagna, S.; Varrin-Doyer, M.; Cavillon, G.; Rogemond, V.; Confavreux, C.; Honnorat, J.; et al. Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain 2010, 133, 2578–2591. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, K.; Misu, T.; Takai, Y.; Sato, D.K.; Takahashi, T.; Abe, Y.; Iwanari, H.; Ogawa, R.; Nakashima, I.; Fujihara, K.; et al. Severely exacerbated neuromyelitis optica rat model with extensive astrocytopathy by high affinity anti-aquaporin-4 monoclonal antibody. Acta Neuropathol. Commun. 2015, 3, 82. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Abe, Y.; Iwanari, H.; Suzuki, Y.; Kikuchi, T.; Ito, T.; Kato, J.; Kusano-Arai, O.; Takahashi, T.; Nishiyama, S.; et al. Establishment of monoclonal antibodies against the extracellular domain that block binding of NMO-IgG to AQP4. J. Neuroimmunol. 2013, 260, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki-Komine, K.; Takai, Y.; Huang, P.; Kusano-Arai, O.; Iwanari, H.; Misu, T.; Koda, K.; Mitomo, K.; Sakihama, T.; Toyama, Y.; et al. High avidity chimeric monoclonal antibodies against the extracellular domains of human aquaporin-4 competing with the neuromyelitis optica autoantibody, NMO-IgG. Br. J. Pharmacol. 2016, 173, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Zeka, B.; Hastermann, M.; Hochmeister, S.; Kogl, N.; Kaufmann, N.; Schanda, K.; Mader, S.; Misu, T.; Rommer, P.; Fujihara, K.; et al. Highly encephalitogenic aquaporin 4-specific T cells and NMO-IgG jointly orchestrate lesion location and tissue damage in the CNS. Acta Neuropathol. 2015, 130, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.P.; Ritchie, A.; Rossi, A.; Schaller, K.; Wemlinger, S.; Schumann, H.; Shearer, A.; Verkman, A.S.; Bennett, J.L. Mutagenesis of the aquaporin 4 extracellular domains defines restricted binding patterns of pathogenic neuromyelitis optica IgG. J. Biol. Chem. 2015, 290, 12123–12134. [Google Scholar] [CrossRef] [PubMed]
- Rosales, D.; Kister, I. Common and rare manifestations of neuromyelitis optica spectrum disorder. Curr. Allergy Asthma Rep. 2016, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- De Seze, J.; Kremer, L.; Collongues, N. Neuromyelitis optica spectrum disorder (NMOSD): A new concept. Rev. Neurol. 2016, 172, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Tradtrantip, L.; Zhang, H.; Saadoun, S.; Phuan, P.W.; Lam, C.; Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann. Neurol. 2012, 71, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Phuan, P.W.; Asavapanumas, N.; Tradtrantip, L. Biology of AQP4 and anti-AQP4 antibody: Therapeutic implications for NMO. Brain Pathol. 2013, 23, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Hoedemaekers, A.C.; van Breda Vriesman, P.J.; de Baets, M.H. Myasthenia gravis as a prototype autoimmune receptor disease. Immunol. Res. 1997, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Paul, F.; Franciotta, D.; de Seze, J.; Munch, C.; Salvetti, M.; Ruprecht, K.; Liebetrau, M.; Wandinger, K.P.; Akman-Demir, G.; et al. Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: Ten new aquaporin-4 antibody positive cases and a review of the literature. Mult. Scler. 2012, 18, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.I.; Coutinho, E.; Lana-Peixoto, M.; Apostolos, S.; Waters, P.; Sato, D.; Melamud, L.; Marta, M.; Graham, A.; Spillane, J.; et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: A multicenter study of 16 patients. Neurology 2012, 78, 1601–1607. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of Alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Guénette, S.Y. Astrocytes: A cellular player in Aβ clearance and degradation. Trends Mol. Med. 2003, 9, 279–280. [Google Scholar] [CrossRef]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wu, Q.; Yuan, C.; Gao, J.; Xiao, M.; Gu, M.; Ding, J.; Hu, G. Aquaporin-4 mediates astrocyte response to beta-amyloid. Mol. Cell. Neurosci. 2012, 49, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.N.; Sun, X.L.; Gao, L.; Fan, Y.; Ding, J.H.; Hu, G. Aquaporin-4 deficiency down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol. Cell. Neurosci. 2007, 34, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.L.; Zou, S.; Chen, J.J.; Zhao, J.; Li, S. The neuroprotective effect of the association of aquaporin-4/glutamate transporter-1 against alzheimer’s disease. Neural Plast. 2016, 2016, 4626593. [Google Scholar] [CrossRef] [PubMed]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding dopaminergic cell death pathways in parkinson disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liang, R.; Yang, B.; Zhou, Y.; Liu, M.; Fang, F.; Ding, J.; Fan, Y.; Hu, G. Aquaporin-4 mediates communication between astrocyte and microglia: Implications of neuroinflammation in experimental Parkinson’s disease. Neuroscience 2016, 317, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, B.; Sun, H.; Zhou, Y.; Liu, M.; Ding, J.; Fang, F.; Fan, Y.; Hu, G. Aquaporin-4 deficiency diminishes the differential degeneration of midbrain dopaminergic neurons in experimental Parkinson’s disease. Neurosci. Lett. 2016, 614, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Fan, Y.; He, L.; Liu, W.; Wen, X.; Zhou, S.; Wang, X.; Zhang, C.; Kong, H.; Sonoda, L.; et al. Novel role of aquaporin-4 in CD4+ CD25+ T regulatory cell development and severity of Parkinson’s disease. Aging Cell 2011, 10, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Hughes, J.; Stockmeier, C.; Miguel-Hidalgo, J.J.; Maciag, D. Incomplete coverage of blood vessels by AQP4-IR astrocytic endfeet in orbitofrontal cortex in major depression. Biol. Psychiatry 2013, 73, 154s–154s. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Stockmeier, C.A. Astrocyte pathology in major depressive disorder: Insights from human postmortem brain tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Liu, Y.; Zhang, Z.; Lu, Y.; Wang, Y.; Huang, J.; Li, Y.; Chen, X.; Gu, X.; Wang, Y.; et al. Mesenchymal stem cells maintain blood-brain barrier integrity by inhibiting aquaporin-4 upregulation after cerebral ischemia. Stem Cells 2014, 32, 3150–3162. [Google Scholar] [CrossRef] [PubMed]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Ma, Y.; Li, W.; Liu, X.; Ying, G.; Fu, L.; Gu, F. Role of aquaporin-4 in the regulation of migration and invasion of human glioma cells. Int. J. Oncol. 2011, 38, 1521–1531. [Google Scholar] [PubMed]
- Klaassen, I.; van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Nicchia, G.P.; Pisani, F.; Simone, L.; Cibelli, A.; Mola, M.G.; dal Monte, M.; Frigeri, A.; Bagnoli, P.; Svelto, M. Glio-vascular modifications caused by aquaporin-4 deletion in the mouse retina. Exp. Eye Res. 2016, 146, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Krizaj, D. TRPV4-AQP4 interactions “turbocharge” astroglial sensitivity to small osmotic gradients. Channels 2016, 10, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, A.; Signoretti, S.; Fatouros, P.P.; Portella, G.; Aygok, G.A.; Bullock, M.R. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J. Neurosurg. 2006, 104, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Masson, F.; Thicoipe, M.; Aye, P.; Mokni, T.; Senjean, P.; Schmitt, V.; Dessalles, P.H.; Cazaugade, M.; Labadens, P. Aquitaine Group for Severe Brain Injuries Study. Epidemiology of severe brain injuries: A prospective population-based study. J. Trauma 2001, 51, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, C.R.; Liang, X.; Abidi, N.H.; Parveen, S.; Taya, K.; Henderson, S.C.; Young, H.F.; Filippidis, A.S.; Baumgarten, C.M. Selective vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP, V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 2014, 1581, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S.; Kim, S.M.; Farook, J.M.; Mir, S.; Saha, R.; Sen, N. Foxo3a transcriptionally upregulates AQP4 and induces cerebral edema following traumatic brain injury. J. Neurosci. 2013, 33, 17398–17403. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [PubMed]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Ikeshima-Kataoka, H.; Abe, Y.; Abe, T.; Yasui, M. Immunological function of aquaporin-4 in stab-wounded mouse brain in concert with a pro-inflammatory cytokine inducer, osteopontin. Mol. Cell. Neurosci. 2013, 56, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Rittling, S.R.; Singh, R. Osteopontin in immune-mediated diseases. J. Dent. Res. 2015, 94, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Kariya, Y.; Kariya, Y.; Saito, T.; Nishiyama, S.; Honda, T.; Tanaka, K.; Yoshida, M.; Fujihara, K.; Hashimoto, Y. Increased cerebrospinal fluid osteopontin levels and its involvement in macrophage infiltration in neuromyelitis optica. BBA Clin. 2015, 3, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Ota, K.; Ikeguchi, R.; Kubo, S.; Kabasawa, C.; Uchiyama, S. Plasma osteopontin levels are associated with disease activity in the patients with multiple sclerosis and neuromyelitis optica. J. Neuroimmunol. 2013, 263, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Katada, R.; Akdemir, G.; Asavapanumas, N.; Ratelade, J.; Zhang, H.; Verkman, A.S. Greatly improved survival and neuroprotection in aquaporin-4-knockout mice following global cerebral ischemia. FASEB J. 2014, 28, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Akdemir, G.; Ratelade, J.; Asavapanumas, N.; Verkman, A.S. Neuroprotective effect of aquaporin-4 deficiency in a mouse model of severe global cerebral ischemia produced by transient 4-vessel occlusion. Neurosci. Lett. 2014, 574, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Nito, C.; Kamada, H.; Endo, H.; Narasimhan, P.; Lee, Y.S.; Chan, P.H. Involvement of mitogen-activated protein kinase pathways in expression of the water channel protein aquaporin-4 after ischemia in rat cortical astrocytes. J. Neurotrauma 2012, 29, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; Kimura-Kuroda, J.; Komuta, Y.; Yoshioka, N.; Li, H.P.; Kawamura, K.; Li, Y.; Raisman, G. Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res. 2012, 349, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Zhu, Q.; Zhang, Y.; Zhao, Y.; Cai, L.; Shields, C.B.; Cai, J. Reciprocal modulation between microglia and astrocyte in reactive gliosis following the CNS injury. Mol. Neurobiol. 2013, 48, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Tokunaga, H.; Nagashima, K.; Takeuchi, T. An autopsy case of minamata disease (methylmercury poisoning)—Pathological viewpoints of peripheral nerves. Toxicol. Pathol. 2002, 30, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Yasutake, A.; Kuwana, T.; Korogi, Y.; Akima, M.; Shimozeki, T.; Tokunaga, H.; Kaneko, Y. Methylmercury poisoning in common marmosets—A study of selective vulnerability within the cerebral cortex. Toxicol. Pathol. 2001, 29, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Takeya, M.; Ikeshima-Kataoka, H.; Yasui, M.; Kawasaki, Y.; Shiraishi, M.; Majima, E.; Shiraishi, S.; Uezono, Y.; Sasaki, M.; et al. Increased expression of aquaporin-4 with methylmercury exposure in the brain of the common marmoset. J. Toxicol. Sci. 2012, 37, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed]
- Gotz, M.; Sirko, S.; Beckers, J.; Irmler, M. Reactive astrocytes as neural stem or progenitor cells: In vivo lineage, In vitro potential, and Genome-wide expression analysis. Glia 2015, 63, 1452–1468. [Google Scholar] [CrossRef] [PubMed]
- Dimou, L.; Gotz, M. Glial cells as progenitors and stem cells: New roles in the healthy and diseased brain. Physiol. Rev. 2014, 94, 709–737. [Google Scholar] [CrossRef] [PubMed]
- Cavazzin, C.; Ferrari, D.; Facchetti, F.; Russignan, A.; Vescovi, A.L.; La Porta, C.A.; Gritti, A. Unique expression and localization of aquaporin-4 and aquaporin-9 in murine and human neural stem cells and in their glial progeny. Glia 2006, 53, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Buylla, A.; Seri, B.; Doetsch, F. Identification of neural stem cells in the adult vertebrate brain. Brain Res. Bull. 2002, 57, 751–758. [Google Scholar] [CrossRef]
- Teeling, J.L.; Perry, V.H. Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: Underlying mechanisms. Neuroscience 2009, 158, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Nobuta, H.; Ghiani, C.A.; Paez, P.M.; Spreuer, V.; Dong, H.; Korsak, R.A.; Manukyan, A.; Li, J.; Vinters, H.V.; Huang, E.J.; et al. STAT3-mediated astrogliosis protects myelin development in neonatal brain injury. Ann. Neurol. 2012, 72, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Benveniste, E.N. Critical role of tumor necrosis factor-α and NF-κB in interferon-γ-induced CD40 expression in microglia/macrophages. J. Biol. Chem. 2002, 277, 13796–13803. [Google Scholar] [CrossRef] [PubMed]
- Norris, J.G.; Tang, L.P.; Sparacio, S.M.; Benveniste, E.N. Signal transduction pathways mediating astrocyte IL-6 induction by IL-1 β and tumor necrosis factor-α. J. Immunol. 1994, 152, 841–850. [Google Scholar] [PubMed]
- John, G.R.; Chen, L.; Rivieccio, M.A.; Melendez-Vasquez, C.V.; Hartley, A.; Brosnan, C.F. Interleukin-1β induces a reactive astroglial phenotype via deactivation of the Rho GTPase-Rock axis. J. Neurosci. 2004, 24, 2837–2845. [Google Scholar] [CrossRef] [PubMed]
- Tomas-Camardiel, M.; Venero, J.L.; de Pablos, R.M.; Rite, I.; Machado, A.; Cano, J. In vivo expression of aquaporin-4 by reactive microglia. J. Neurochem. 2004, 91, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, H.; Varrin-Doyer, M.; Zamvil, S.S.; Verkman, A.S. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. 2011, 25, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Oyarzabal, E.A.; Sung, Y.F.; Chu, C.H.; Wang, Q.; Chen, S.L.; Lu, R.B.; Hong, J.S. Microglial regulation of immunological and neuroprotective functions of astroglia. Glia 2015, 63, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tang, Y.; Feng, J. Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 2011, 89, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Noell, S.; Fallier-Becker, P.; Mack, A.F.; Wolburg-Buchholz, K. The disturbed blood-brain barrier in human glioblastoma. Mol. Asp. Med. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Mou, K.J.; Mao, Q.; Chen, M.N.; Xia, X.Q.; Ni, R.Y.; Wang, P.; Liu, Y.H. AQP4 expression in the brains of patients with glioblastoma and its association with brain edema. Sichuan Da Xue Xue Bao Yi Xue Ban 2009, 40, 651–654. [Google Scholar] [PubMed]
- Noell, S.; Wolburg-Buchholz, K.; Mack, A.F.; Ritz, R.; Tatagiba, M.; Beschorner, R.; Wolburg, H.; Fallier-Becker, P. Dynamics of expression patterns of AQP4, dystroglycan, agrin and matrix metalloproteinases in human glioblastoma. Cell Tissue Res. 2012, 347, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, M.; Monda, A.; Takemoto, R.; Fujimoto, Y.; Sugitani, M.; Iwamura, T.; Hiroyasu, T.; Inoue, A. High-mobility group box 1 up-regulates aquaporin 4 expression via microglia-astrocyte interaction. Neurochem. Int. 2014, 75, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Jansson, D.; Rustenhoven, J.; Feng, S.; Hurley, D.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Faull, R.L.; Dragunow, M. A role for human brain pericytes in neuroinflammation. J. Neuroinflamm. 2014, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chen, Z.L.; Norris, E.H.; Strickland, S. Astrocytic laminin regulates pericyte differentiation and maintains blood-brain barrier integrity. Nat. Commun. 2014, 5, 3413. [Google Scholar] [CrossRef] [PubMed]
- Balabanov, R.; Dore-Duffy, P. Role of the CNS microvascular pericyte in the blood-brain barrier. J. Neurosci. Res. 1998, 53, 637–644. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Q.; Tang, J.; Feng, H.; Zhang, J.H. The evolving roles of pericyte in early brain injury after subarachnoid hemorrhage. Brain Res. 2015, 1623, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Trost, A.; Tempfer, H.; Bauer, H.C.; Bauer, H.; Rohde, E.; Reitsamer, H.A.; Franklin, R.J.; Aigner, L.; Rivera, F.J. Brain pericyte plasticity as a potential drug target in CNS repair. Drug Discov. Today 2013, 18, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Buback, F.; Renkl, A.C.; Schulz, G.; Weiss, J.M. Osteopontin and the skin: Multiple emerging roles in cutaneous biology and pathology. Exp. Dermatol. 2009, 18, 750–759. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Function | Gene Description | WT | AQP4/KO |
|---|---|---|---|
| immune response | secreted phosphoprotein 1 (Spp1) = osteopontin (OPN) | 59.83 | 4.94 |
| lipocalin 2 (Lcn2) | 11.79 | <1.5 | |
| macrophage expressed gene 1 (Mpeg1) | 8.89 | <1.5 | |
| chitinase 3-like 1 (Chi3l1) | 4.67 | <1.5 | |
| leukocyte immunoglobulin-like receptor, subfamily B, member 4 (Lilrb4) | 4.36 | <1.5 | |
| enzyme | heme oxygenase (decycling) 1 (Hmox1) | 23.19 | 11.49 |
| transglutaminase 1, K polypeptide (Tgm1) | 5.29 | <1.5 | |
| lysosomal function | lysozyme 2 (Lyz2) | 18.53 | 1.9 |
| compliment activation | complement component 3a receptor 1 (C3ar1) | 8.77 | <1.5 |
| complement component 1, q subcomponent, C chain (C1qc) | 6.14 | <1.5 | |
| complement component 1, q subcomponent, beta polypeptide (C1qb) | 5.31 | <1.5 | |
| cytoskeleton | vimentin (Vim) | 7.03 | <1.5 |
| glial fibrillary acidic protein (Gfap) | 4.69 | <1.5 | |
| lymphocyte cytosolic protein 1 (Lcp1) | 4.52 | <1.5 | |
| antigen expression | CD180 antigen (Cd180) | 6.38 | <1.5 |
| CD68 antigen (Cd68) | 6.21 | 2.96 | |
| lymphocyte antigen 86 (Ly86) | 5.19 | <1.5 | |
| adipose function | adipose differentiation related protein (Adfp) | 6.85 | 2.49 |
| chemokine | chemokine (C-C motif) ligand 3 (Ccl3) | 5.37 | <1.5 |
| signal transduction | membrane-spanning 4-domains, subfamily A, member 6C (Ms4a6c) | 5.32 | <1.5 |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikeshima-Kataoka, H. Neuroimmunological Implications of AQP4 in Astrocytes. Int. J. Mol. Sci. 2016, 17, 1306. https://doi.org/10.3390/ijms17081306
Ikeshima-Kataoka H. Neuroimmunological Implications of AQP4 in Astrocytes. International Journal of Molecular Sciences. 2016; 17(8):1306. https://doi.org/10.3390/ijms17081306
Chicago/Turabian StyleIkeshima-Kataoka, Hiroko. 2016. "Neuroimmunological Implications of AQP4 in Astrocytes" International Journal of Molecular Sciences 17, no. 8: 1306. https://doi.org/10.3390/ijms17081306
APA StyleIkeshima-Kataoka, H. (2016). Neuroimmunological Implications of AQP4 in Astrocytes. International Journal of Molecular Sciences, 17(8), 1306. https://doi.org/10.3390/ijms17081306