
Characterizing Aciniform Silk Repetitive Domain Backbone Dynamics and Hydrodynamic Modularity

 ,
,
Abstract
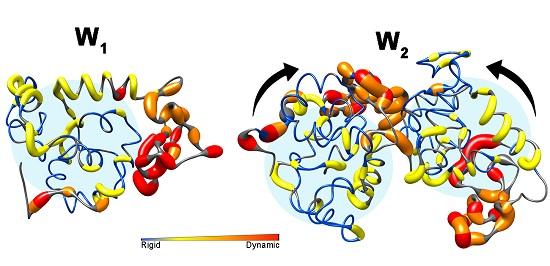
:
1. Introduction
2. Results
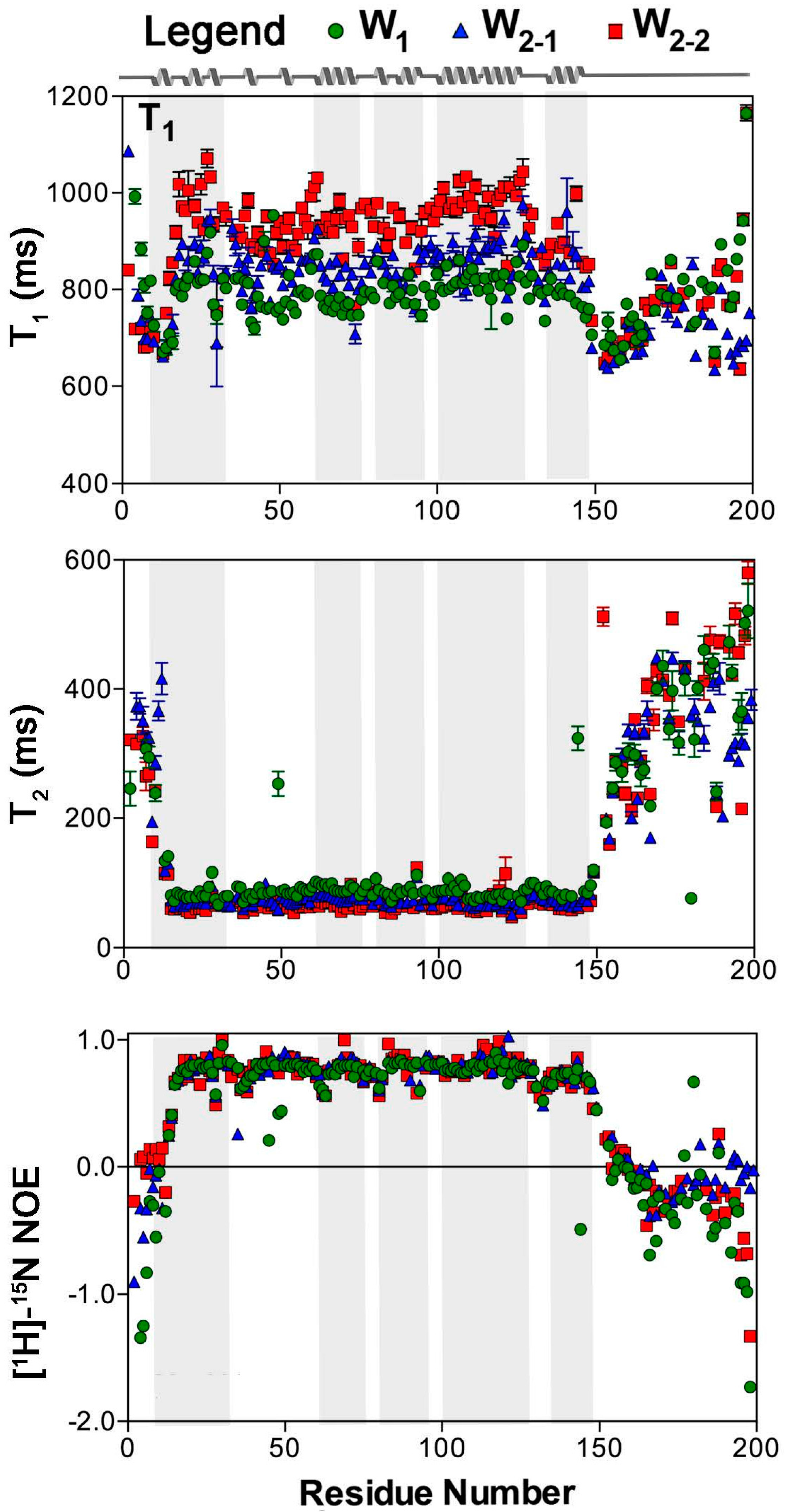
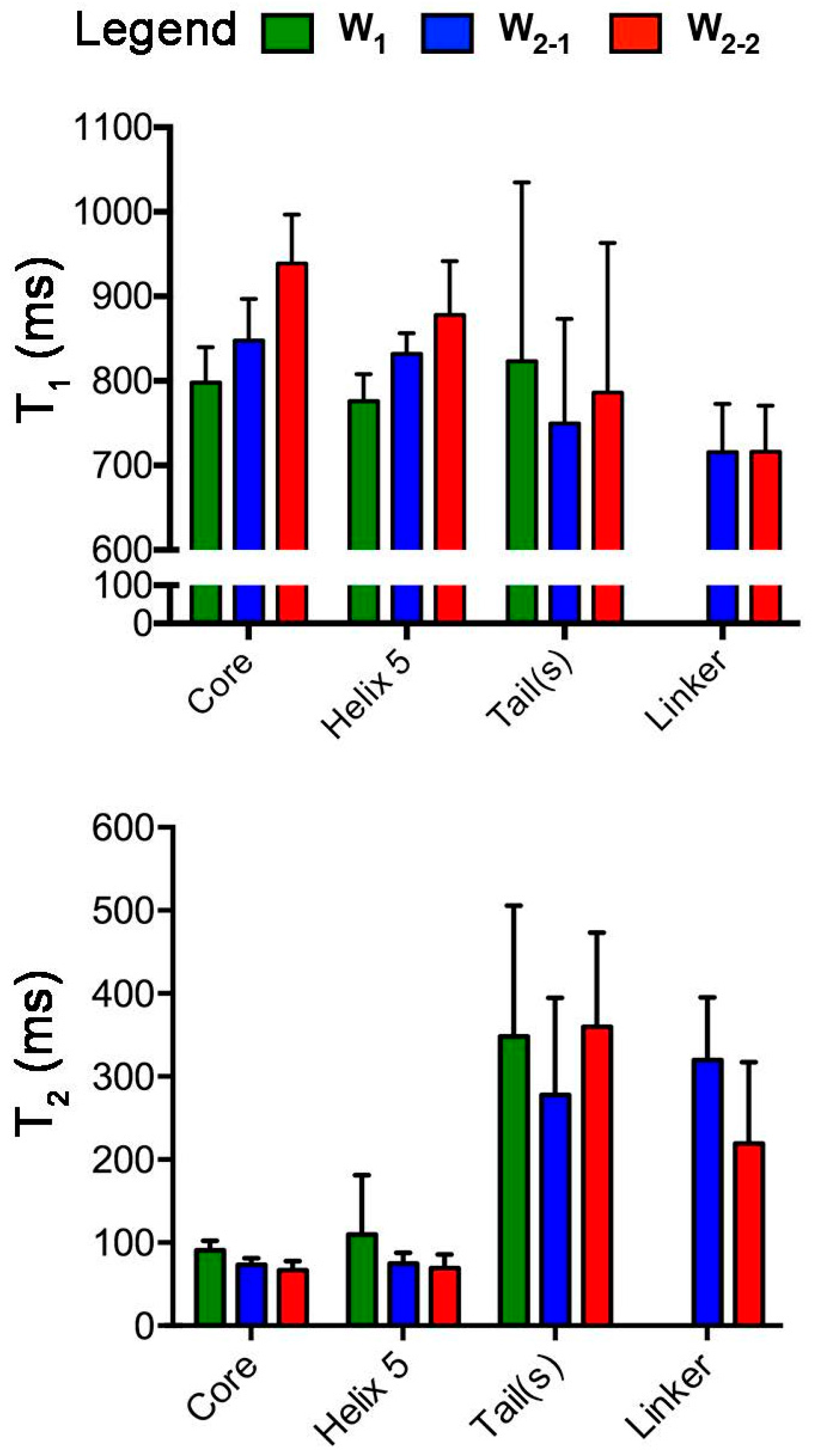
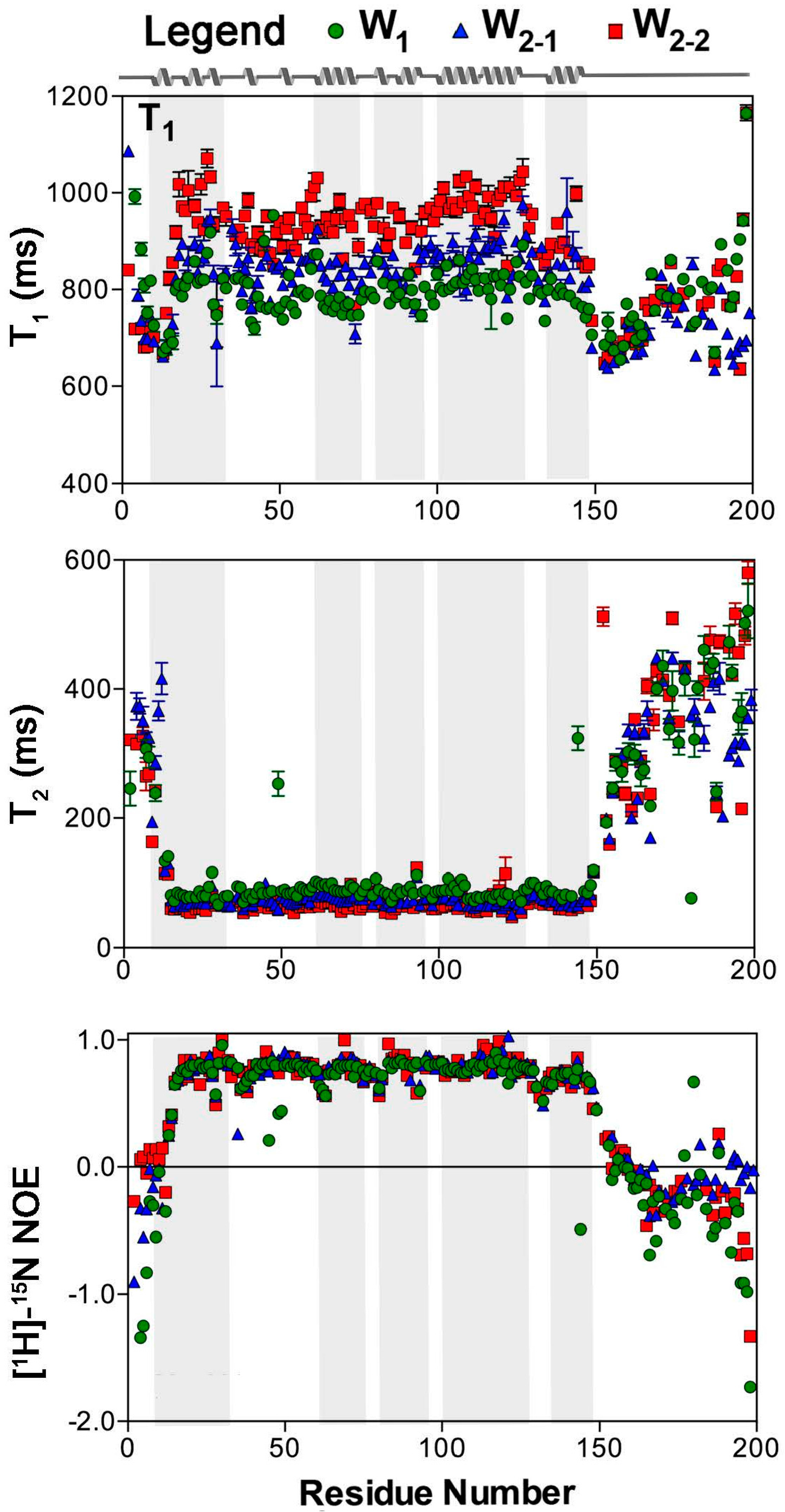
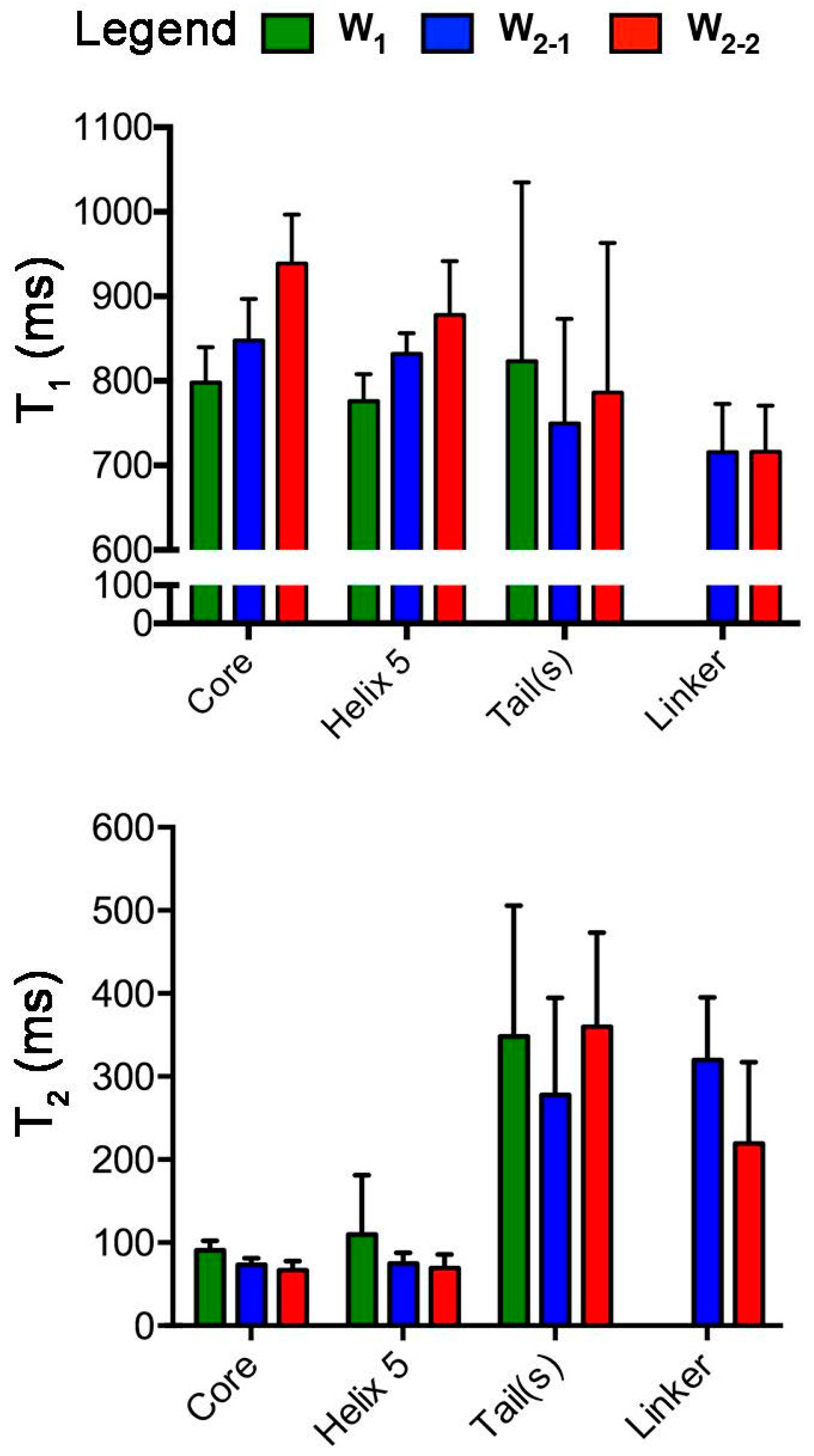
2.1. Nuclear Spin Relaxation Parameters
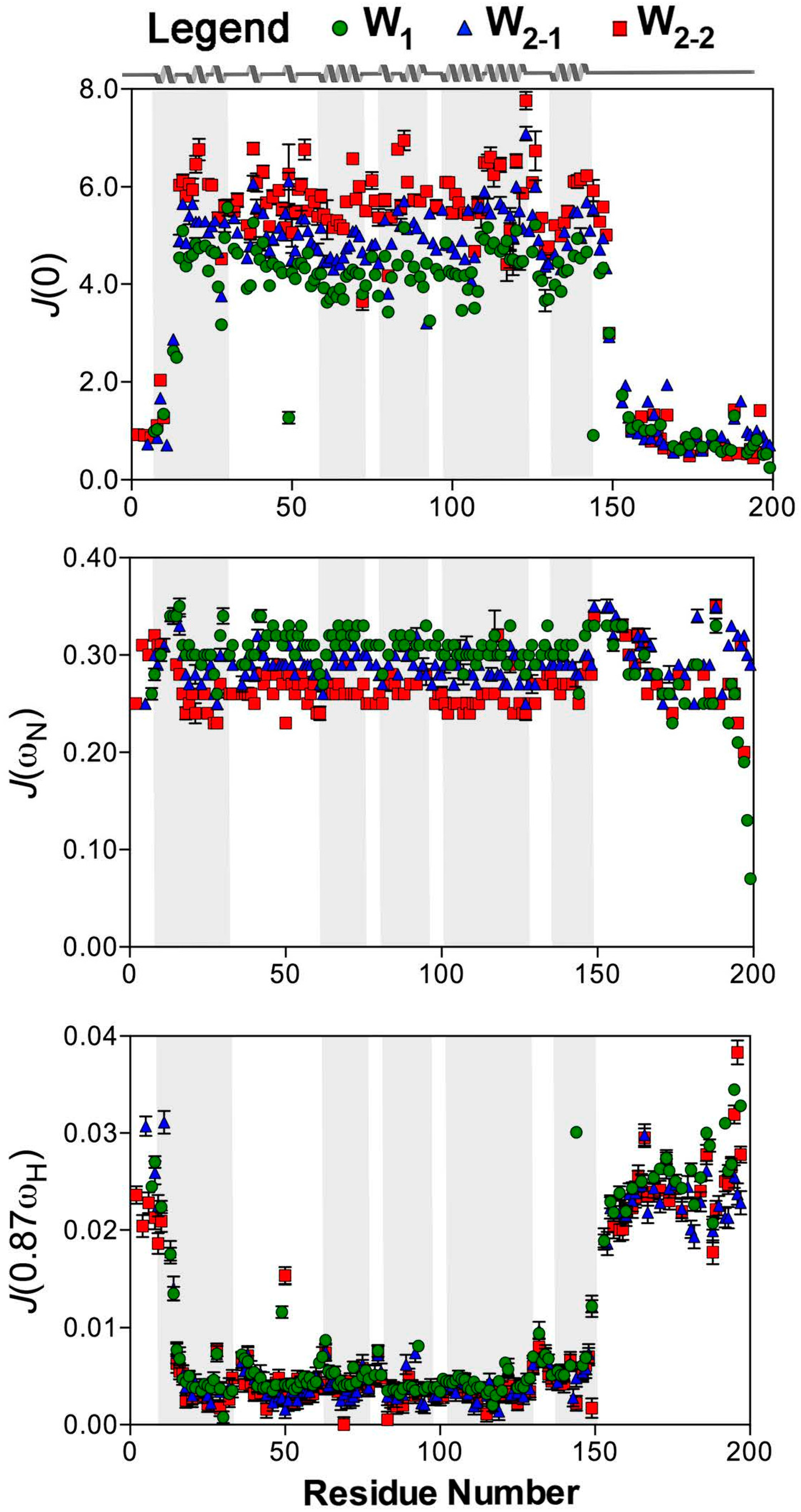
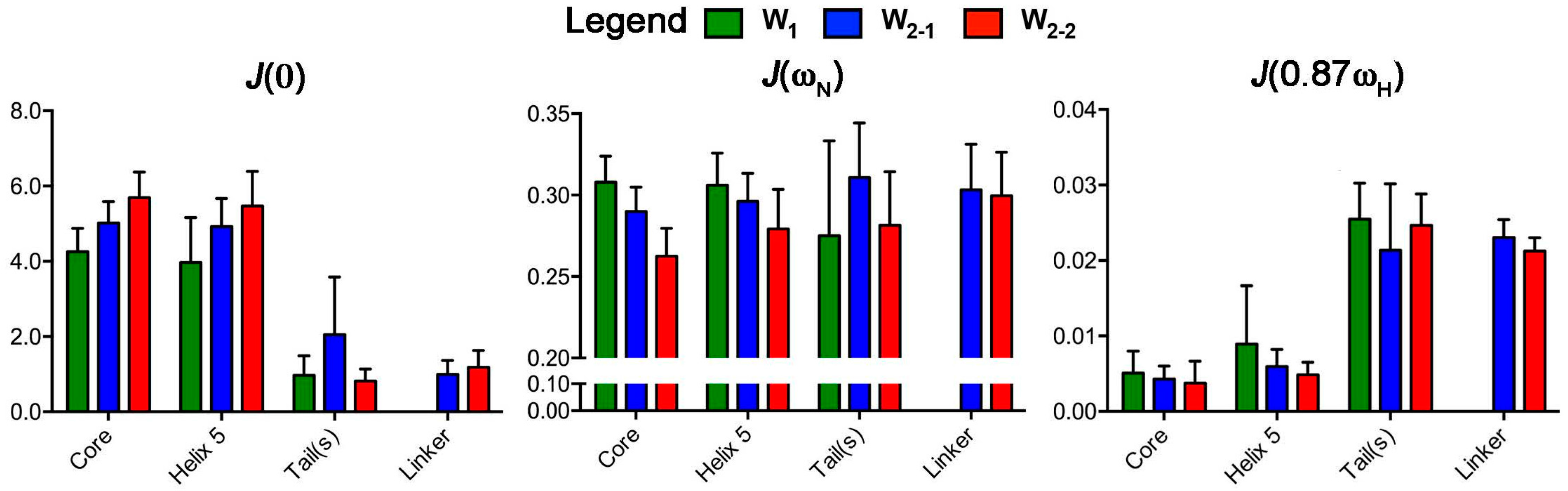
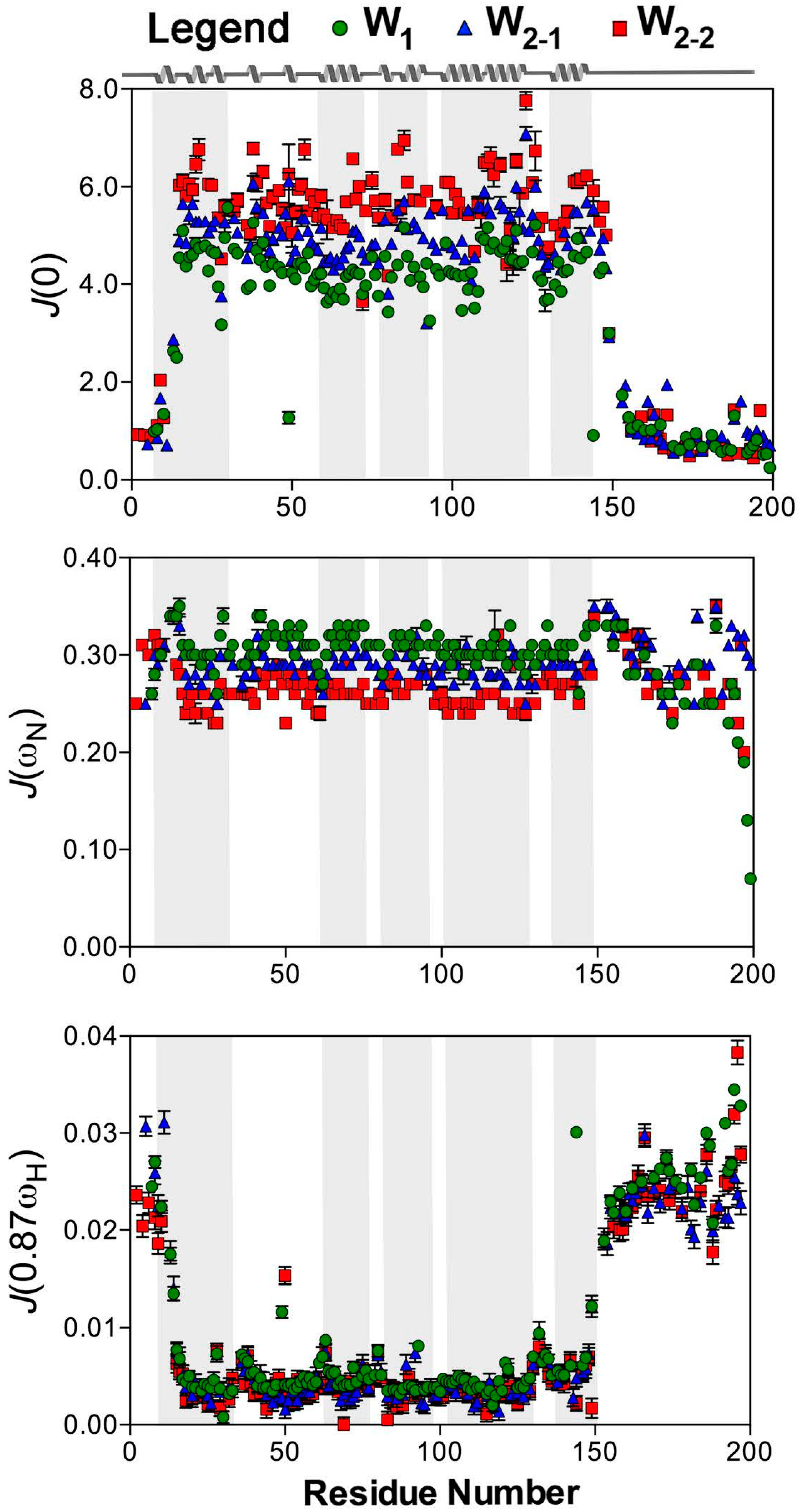
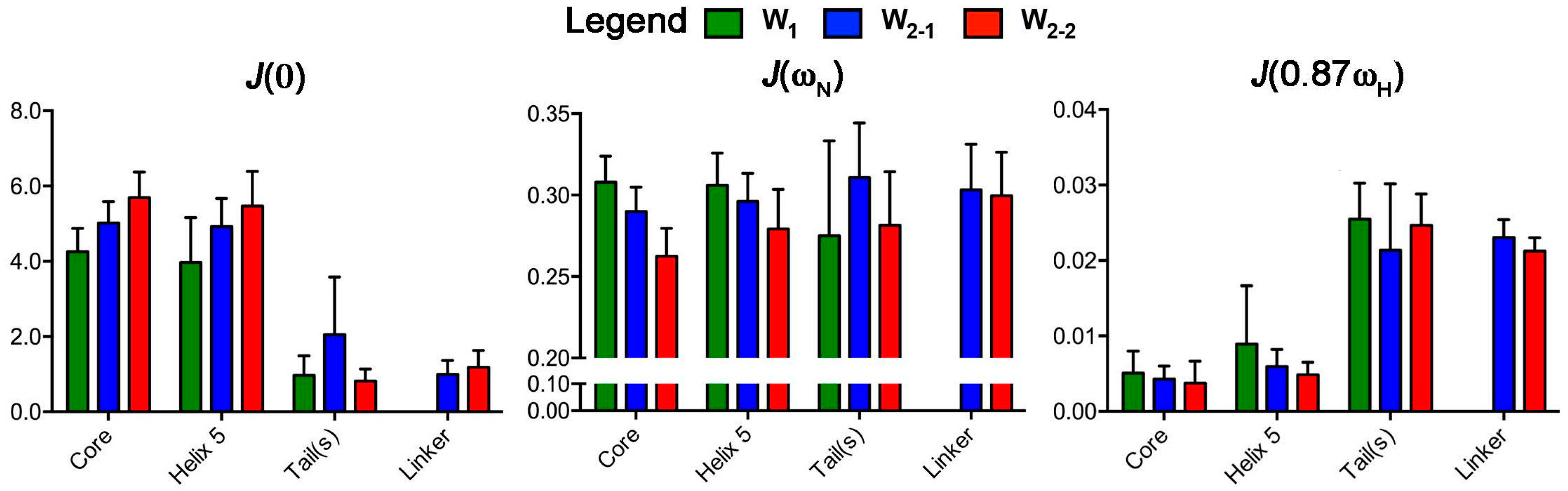
2.2. Reduced Spectral Density Mapping
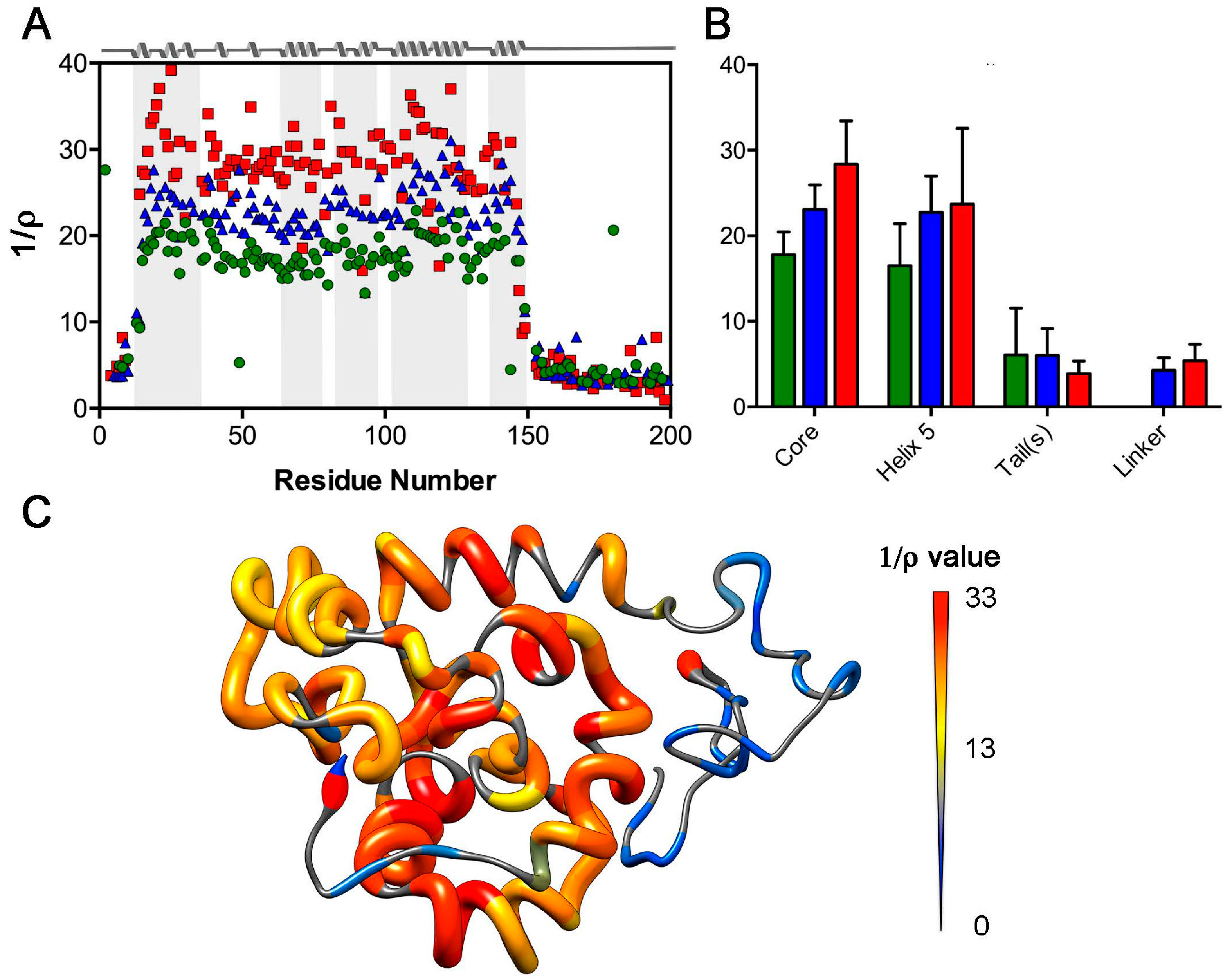
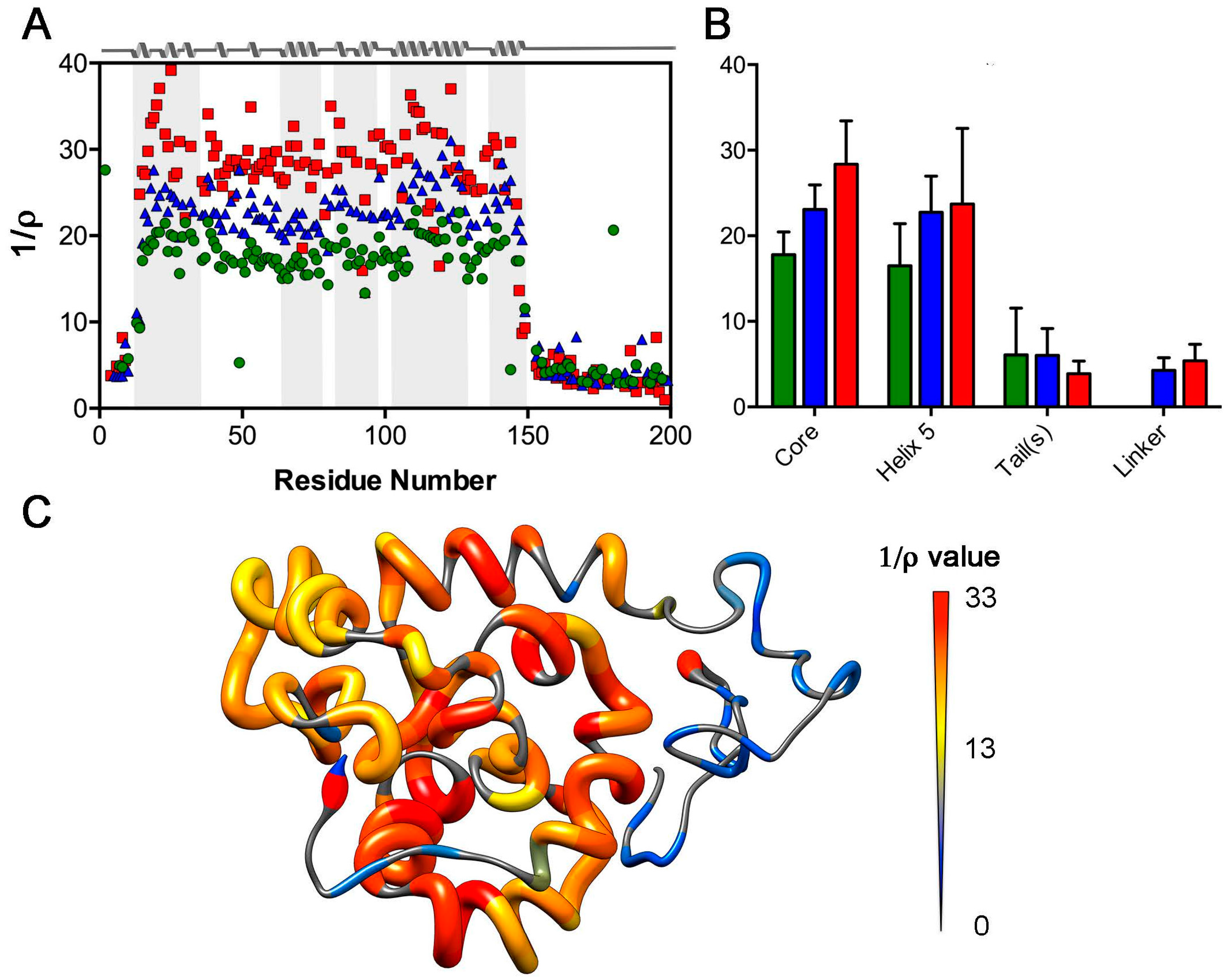
2.3. Analysis of Rotational Diffusion
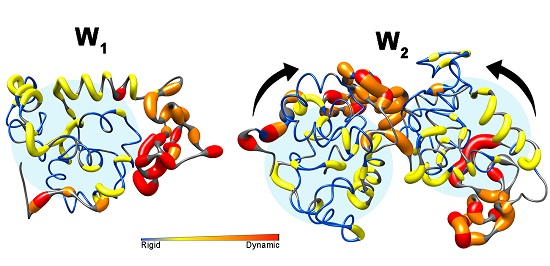
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Spin-Relaxation NMR Experiments
4.3. Determination of Spin Relaxation Parameters and Reduced Spectral Density mapping
4.4. Viscosity Determination
4.5. Analysis of Rotational Diffusion
4.6. Estimations of Rotational Correlation Time
4.7. Statistical Tests
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lewis, R.V. Spider silk: Ancient ideas for new biomaterials. Chem. Rev. 2006, 106, 3762–3774. [Google Scholar] [CrossRef] [PubMed]
- Yigit, S.; Dinjaski, N.; Kaplan, D.L. Fibrous proteins: At the crossroads of genetic engineering and biotechnological applications. Biotechnol. Bioeng. 2016, 113, 913–929. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.Z.; Confalonieri, F.; Medina, N.; Zivanovic, Y.; Esnault, C.; Yang, T.; Jacquet, M.; Janin, J.; Duguet, M.; Perasso, R.; et al. Fine organization of Bombyx mori fibroin heavy chain gene. Nucleic Acids Res. 2000, 28, 2413–2419. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, C.Y.; Blackledge, T.A.; Lewis, R.V. Molecular and mechanical characterization of aciniform silk: Uniformity of iterated sequence modules in a novel member of the spider silk fibroin gene family. Mol. Biol. Evol. 2004, 21, 1950–1959. [Google Scholar] [CrossRef] [PubMed]
- Pickford, A.R.; Campbell, I.D. NMR studies of modular protein structures and their interactions. Chem. Rev. 2004, 104, 3557–3566. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Batey, S.; Nickson, A.A.; Teichmann, S.A.; Clarke, J. The folding and evolution of multidomain proteins. Nat. Rev. Mol. Cell Biol. 2007, 8, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M. Nature of the protein universe. Proc. Natl. Acad. Sci. USA 2009, 106, 11079–11084. [Google Scholar] [CrossRef] [PubMed]
- Frueh, D.P.; Goodrich, A.C.; Mishra, S.H.; Nichols, S.R. NMR methods for structural studies of large monomeric and multimeric proteins. Curr. Opin. Struct. Biol. 2013, 23, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Ravera, E.; Salmon, L.; Fragai, M.; Parigi, G.; Al-Hashimi, H.; Luchinat, C. Insights into domain-domain motions in proteins and RNA from solution NMR. Acc. Chem. Res. 2014, 47, 3118–3126. [Google Scholar] [CrossRef] [PubMed]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Skiniotis, G.; Sherman, D.H. Architecture of the polyketide synthase module: surprises from electron cryo-microscopy. Curr. Opin. Struct. Biol. 2015, 31, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, M.; Barlow, P.N.; Tjandra, N. Decoding the components of dynamics in three-domain proteins. J. Comput. Chem. 2014, 35, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Ryabov, Y.E.; Fushman, D. A model of interdomain mobility in a multidomain protein. J. Am. Chem. Soc. 2007, 129, 3315–3327. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.J.; Stacey, M.M.; Liu, B.A.; Pawson, T. Molecular mechanisms of SH2- and PTB-domain-containing proteins in receptor tyrosine kinase signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a008987. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.H.; Sticht, H. Synthetic protein scaffolds based on peptide motifs and cognate adaptor domains for improving metabolic broductivity. Front. Bioeng. Biotechnol. 2015, 3, 191. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, C.A.; Spasser, L.; Bavikar, S.N.; Brik, A.; Fushman, D. Segmental isotopic labeling of ubiquitin chains to unravel monomer-specific molecular behavior. Angew. Chem. Int. Ed. 2011, 50, 11210–11214. [Google Scholar] [CrossRef] [PubMed]
- Fushman, D.; Varadan, R.; Assfalg, M.; Walker, O. Determining domain orientation in macromolecules by using spin-relaxation and residual dipolar coupling measurements. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 44, 189–214. [Google Scholar] [CrossRef]
- Walsh, J.D.; Meier, K.; Ishima, R.; Gronenborn, A.M. NMR studies on domain diffusion and alignment in modular GB1 repeats. Biophys. J. 2010, 99, 2636–2646. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.L.; Xu, L.; Lefevre, T.; Sarker, M.; Orrell, K.E.; Leclerc, J.; Meng, Q.; Pezolet, M.; Auger, M.; Liu, X.Q.; et al. Spider wrapping silk fibre architecture arising from its modular soluble protein precursor. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Rainey, J.K.; Meng, Q.; Liu, X.Q. Recombinant minimalist spider wrapping silk proteins capable of native-like fiber formation. PLoS ONE 2012, 7, e50227. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, T.; Boudreault, S.; Cloutier, C.; Pezolet, M. Diversity of molecular transformations involved in the formation of spider silks. J. Mol. Biol. 2011, 405, 238–253. [Google Scholar] [CrossRef] [PubMed]
- Sarker, M.; Orrell, K.E.; Xu, L.; Tremblay, M.L.; Bak, J.J.; Liu, X.Q.; Rainey, J.K. Tracking transitions in spider wrapping silk conformation and dynamics by 19F nuclear magnetic resonance spectroscopy. Biochemistry 2016, 55, 3048–3059. [Google Scholar] [CrossRef] [PubMed]
- Mita, K.; Ichimura, S.; James, T.C. Highly repetitive structure and its organization of the silk fibroin gene. J. Mol. Evol. 1994, 38, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Weatherbee-Martin, N.; Xu, L.; Hupe, A.; Kreplak, L.; Fudge, D.S.; Liu, X.Q.; Rainey, J.K. Identification of wet-spinning and post-spin stretching methods amenable to recombinant spider aciniform silk. Biomacromolecules 2016, 17, 2737–2746. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ladizhansky, V. Recent advances in magic angle spinning solid state NMR of membrane proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 82, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Ortega, G.; Pons, M.; Millet, O. Protein functional dynamics in multiple timescales as studied by NMR spectroscopy. Adv. Protein Chem. Struct. Biol. 2013, 92, 219–251. [Google Scholar] [PubMed]
- Kay, L.E.; Torchia, D.A.; Bax, A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 1989, 28, 8972–8979. [Google Scholar] [CrossRef] [PubMed]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Clore, G.M.; Szabo, A.; Bax, A.; Kay, L.E.; Driscoll, P.C.; Gronenborn, A.M. Deviations from the simple 2-parameter model-free approach to the interpretation of N-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 1990, 112, 4989–4991. [Google Scholar] [CrossRef]
- Farrow, N.A.; Zhang, O.; Szabo, A.; Torchia, D.A.; Kay, L.E. Spectral density function mapping using 15N relaxation data exclusively. J. Biomol. NMR 1995, 6, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Skrisovska, L.; Schubert, M.; Allain, F.H. Recent advances in segmental isotope labeling of proteins: NMR applications to large proteins and glycoproteins. J. Biomol. NMR 2010, 46, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, G.; Iwai, H. Protein trans-splicing and its use in structural biology: Opportunities and limitations. Mol. Biosyst. 2010, 6, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Vila-Perello, M.; Muir, T.W. Biological applications of protein splicing. Cell 2010, 143, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.C., Jr.; Martin, D.; Kolly, R.; Panne, D.; Sun, L.; Ghosh, I.; Chen, L.; Benner, J.; Liu, X.Q.; Xu, M.Q. Protein trans-splicing and cyclization by a naturally split intein from the dnaE gene of Synechocystis species PCC6803. J. Biol. Chem. 2000, 275, 9091–9094. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.P.; Abel-Santos, E.; Wall, M.; Wahnon, D.C.; Benkovic, S.J. Production of cyclic peptides and proteins in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 13638–13643. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, G.; Murphy, P.W.; Rowland, E.E.; Cronan, J.E., Jr.; Liu, X.Q.; Blouin, C.; Byers, D.M. Intein-mediated cyclization of bacterial acyl carrier protein stabilizes its folded conformation but does not abolish function. J. Biol. Chem. 2010, 285, 8605–8614. [Google Scholar] [CrossRef] [PubMed]
- Buskirk, A.R.; Ong, Y.C.; Gartner, Z.J.; Liu, D.R. Directed evolution of ligand dependence: small-molecule-activated protein splicing. Proc. Natl. Acad. Sci. USA 2004, 101, 10505–10510. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, M.; Wright, R.C.; Date, A.; Tullman, J.; Ostermeier, M. Protein switch engineering by domain insertion. Methods Enzymol. 2013, 523, 369–388. [Google Scholar] [PubMed]
- Mootz, H.D.; Blum, E.S.; Tyszkiewicz, A.B.; Muir, T.W. Conditional protein splicing: a new tool to control protein structure and function in vitro and in vivo. J. Am. Chem. Soc. 2003, 125, 10561–10569. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Kaihara, A.; Sato, M.; Tachihara, K.; Umezawa, Y. Split luciferase as an optical probe for detecting protein-protein interactions in mammalian cells based on protein splicing. Anal. Chem. 2001, 73, 2516–2521. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, L.; Russomanno, A.; Romanelli, A.; D’Andrea, L.D. Semi-synthesis of labeled proteins for spectroscopic applications. Molecules 2013, 18, 440–465. [Google Scholar] [CrossRef] [PubMed]
- Giriat, I.; Muir, T.W. Protein semi-synthesis in living cells. J. Am. Chem. Soc. 2003, 125, 7180–7181. [Google Scholar] [CrossRef] [PubMed]
- Muir, T.W. Studying protein structure and function using semisynthesis. Biopolymers 2008, 90, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Buchinger, E.; Aachmann, F.L.; Aranko, A.S.; Valla, S.; Skjak-Braek, G.; Iwai, H.; Wimmer, R. Use of protein trans-splicing to produce active and segmentally 2H, 15N labeled mannuronan C5-epimerase AlgE4. Protein Sci. 2010, 19, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Busche, A.E.; Aranko, A.S.; Talebzadeh-Farooji, M.; Bernhard, F.; Dotsch, V.; Iwai, H. Segmental isotopic labeling of a central domain in a multidomain protein by protein trans-splicing using only one robust DnaE intein. Angew. Chem. Int. Ed. 2009, 48, 6128–6131. [Google Scholar] [CrossRef] [PubMed]
- Muona, M.; Aranko, A.S.; Iwai, H. Segmental isotopic labelling of a multidomain protein by protein ligation by protein trans-splicing. ChemBioChem 2008, 9, 2958–2961. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Teruya, K.; Uegaki, K.; Yamazaki, T.; Kyogoku, Y. Improved segmental isotope labeling of proteins and application to a larger protein. J. Biomol. NMR 1999, 14, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Otomo, T.; Oda, N.; Kyogoku, Y.; Uegaki, K.; Ito, N.; Ishino, Y.; Nakamura, H. Segmental isotope labeling for protein NMR using peptide splicing. J. Am. Chem. Soc. 1998, 120, 5591–5592. [Google Scholar] [CrossRef]
- Southworth, M.W.; Adam, E.; Panne, D.; Byer, R.; Kautz, R.; Perler, F.B. Control of protein splicing by intein fragment reassembly. EMBO J. 1998, 17, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Hu, Z.; Liu, X.Q. Protein trans-splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. USA 1998, 95, 9226–9231. [Google Scholar] [CrossRef] [PubMed]
- Spyracopoulos, L. A suite of Mathematica notebooks for the analysis of protein main chain 15N NMR relaxation data. J. Biomol. NMR 2006, 36, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Kaderavek, P.; Zapletal, V.; Rabatinova, A.; Krasny, L.; Sklenar, V.; Zidek, L. Spectral density mapping protocols for analysis of molecular motions in disordered proteins. J. Biomol. NMR 2014, 58, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Tjandra, N.; Feller, S.E.; Pastor, R.W.; Bax, A. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J. Am. Chem. Soc. 1995, 117, 12562–12566. [Google Scholar] [CrossRef]
- Walker, O.; Varadan, R.; Fushman, D. Efficient and accurate determination of the overall rotational diffusion tensor of a molecule from 15N relaxation data using computer program ROTDIF. J. Magn. Reson. 2004, 168, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.; Amoros, D.; Garcia de la Torre, J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 2011, 101, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.A.; Wilkins, D.K.; Smith, L.J.; Dobson, C.M. Characterisation of protein unfolding by NMR diffusion measurements. J. Biomol. NMR 1997, 10, 199–203. [Google Scholar] [CrossRef]
- Chaw, R.C.; Zhao, Y.; Wei, J.; Ayoub, N.A.; Allen, R.; Atrushi, K.; Hayashi, C.Y. Intragenic homogenization and multiple copies of prey-wrapping silk genes in Argiope garden spiders. BMC Evol. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.H.; Urquidi, J.; Singh, S.; Robinson, G.W. Thermal offset viscosities of liquid H2O, D2O, and T2O. J. Phys. Chem. B 1999, 103, 1991–1994. [Google Scholar] [CrossRef]
- Xu, L.; Tremblay, M.L.; Orrell, K.E.; Leclerc, J.; Meng, Q.; Liu, X.Q.; Rainey, J.K. Nanoparticle self-assembly by a highly stable recombinant spider wrapping silk protein subunit. FEBS Lett. 2013, 587, 3273–3280. [Google Scholar] [CrossRef] [PubMed]
- Halle, B.; Davidovic, M. Biomolecular hydration: from water dynamics to hydrodynamics. Proc. Natl. Acad. Sci. USA 2003, 100, 12135–12140. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Dyson, H.J.; Wright, P.E. Prediction of the rotational tumbling time for proteins with disordered segments. J. Am. Chem. Soc. 2009, 131, 6814–6821. [Google Scholar] [CrossRef] [PubMed]
- Rezaei-Ghaleh, N.; Klama, F.; Munari, F.; Zweckstetter, M. Predicting the rotational tumbling of dynamic multidomain proteins and supramolecular complexes. Angew. Chem. Int. Ed. 2013, 52, 11410–11414. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tremblay, M.L.; Meng, Q.; Liu, X.Q.; Rainey, J.K. 1H, 13C and 15N NMR assignments of the aciniform spidroin (AcSp1) repetitive domain of Argiope trifasciata wrapping silk. Biomol. NMR Assign. 2012, 6, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Cantor, C.R.; Schimmel, P.R. Techniques for the study of biological structure and function. In Biophysical Chemistry; W.H. Freeman and Company: San Francisco, CA, USA, 1980; Volume 2, pp. 549–570. [Google Scholar]
- Cavanagh, J.; Fairbrother, W.J.; Palmer, A.G., III; Skelton, N.J. Protein NMR Spectroscopy: Principles and Practice; Academic Press: San Diego, CA, USA, 1996; pp. 16–18. [Google Scholar]
- Venable, R.M.; Pastor, R.W. Frictional models for stochastic simulations of proteins. Biopolymers 1988, 27, 1001–1014. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein 1 | N–H Bonds 2 | D⊥ (×10−7 s−1) | D|| (×10−7 s−1) | Anisotropy | α (°) | β (°) | τc (ns) | χ2/df 3 |
|---|---|---|---|---|---|---|---|---|
| W1 | 102 | 2.01 ± 0.05 | 2.30 ± 0.08 | 1.14 ± 0.17 | 13 ± 52 | 43 ± 19 | 7.91 ± 0.03 | 1.409 |
| W2-1 | 104 | 1.82 ± 0.07 | 1.93 ± 0.04 | 1.06 ± 0.13 | 164 ± 87 | 138 ± 20 | 8.98 ± 0.03 | 1.889 |
| W2-2 | 93 | 1.78 ± 0.03 | 1.63 ± 0.03 | 0.92 ± 0.09 | 77 ± 37 | 155 ± 10 | 9.65 ± 0.04 | 1.906 |
| W2 | 197 | 1.87 ± 0.04 | 1.68 ± 0.04 | 0.90 ± 0.11 | 26 ± 31 | 20 ± 22 | 9.24 ± 0.05 | 2.309 |
| Protein | η (cP) 1 | τc (ns) | |||||
|---|---|---|---|---|---|---|---|
| Stokes (Ideal) 2 | Stokes (DOSY) 3 | Structure 4 | T1/T2 5 | T1′/T2′ 5 | ROTDIF 6 | ||
| W1 | 1.040 | 7.8–9.9 | 10.6 | 14.1 ± 0.3 7 9.4 ± 0.2 8 8.4 ± 0.1 9 | 7.9 | 8.0 | 7.9 ± 0.01 |
| W2-1 | 1.056 | - | - | - | 9.0 | 9.1 | 9.0 ± 0.01 |
| W2-2 | 1.056 | - | - | - | 9.5 | 9.6 | 9.6 ± 0.01 |
| W2 | 1.056 | 14.7–17.8 | 19.7 | 31.8 ± 0.7 | 9.3 | 9.4 | 9.3 ± 0.03 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tremblay, M.-L.; Xu, L.; Sarker, M.; Liu, X.-Q.; Rainey, J.K. Characterizing Aciniform Silk Repetitive Domain Backbone Dynamics and Hydrodynamic Modularity. Int. J. Mol. Sci. 2016, 17, 1305. https://doi.org/10.3390/ijms17081305
Tremblay M-L, Xu L, Sarker M, Liu X-Q, Rainey JK. Characterizing Aciniform Silk Repetitive Domain Backbone Dynamics and Hydrodynamic Modularity. International Journal of Molecular Sciences. 2016; 17(8):1305. https://doi.org/10.3390/ijms17081305
Chicago/Turabian StyleTremblay, Marie-Laurence, Lingling Xu, Muzaddid Sarker, Xiang-Qin Liu, and Jan K. Rainey. 2016. "Characterizing Aciniform Silk Repetitive Domain Backbone Dynamics and Hydrodynamic Modularity" International Journal of Molecular Sciences 17, no. 8: 1305. https://doi.org/10.3390/ijms17081305
APA StyleTremblay, M.-L., Xu, L., Sarker, M., Liu, X.-Q., & Rainey, J. K. (2016). Characterizing Aciniform Silk Repetitive Domain Backbone Dynamics and Hydrodynamic Modularity. International Journal of Molecular Sciences, 17(8), 1305. https://doi.org/10.3390/ijms17081305